
Abstract
Gas chromatography-mass spectrometry (GC/MS) was employed for the determination of 30 widely used pesticides including various transformation products and alkylphenols in water and agricultural soils with the aim of assessing the impact of these compounds in agricultural soils and the underlying aquifer. The extraction, clean-up, and analytical procedures were optimized for both water and soil samples to provide a highly robust method capable of determining target analytes at the ppb–ppt level with high precision. For water samples, different solid-phase extraction cartridges and conditions were optimized; similarly, pressurized liquid extraction conditions were tested to provide interference-free extracts and high sensitivity. Instrumental LODs of 3–4 pg were obtained. The multi-residue extraction procedures were applied to the analysis of groundwaters and agricultural soils from the Ebro river basin (NE Spain). Most ubiquitous herbicides detected were triazines but some acetanilides and organophosphorus pesticides were also found; the pesticide additive tributylphosphate was found in all water samples. Levels varied between 0.57 and 5.37 μg/L in groundwater, whereas nonylphenol was the sole compound detected in soil. Alkylphenols are used as adjuvants in pesticide formulations and are present in sludges employed as soil fertilizers. Occurrence was found to be similar to other environmental studies.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pesticide formulations are applied worldwide to eliminate crop pests. Once applied, pesticides can be absorbed by the plant [1] although the majority are deposited on the soil surface where they can degrade [2–5], adsorb onto organic matter of soil or clay [6], or lixiviate [7]. In this respect, triazine, acetanilide, and organophosphorus pesticides were reported in groundwaters from Spain or Italy as a result of leaching from agricultural practices [8, 9]. These processes are highly dependent on the type of pesticide, soil, crop, climatic conditions, and application procedures, and thus the fate of pesticides is highly variable.
Groundwaters are of special interest because of their use in irrigation and they often constitute a main drinking water source in many urban areas. Contamination of groundwaters is directly linked to the transport of the pollutant within the soil column supporting the advective and diffusional flow system, the geochemistry of the groundwater, and the overall groundwater flow. Although the pesticide concentration in a groundwater cannot be directly linked only to the agricultural activity right above the well, it is the only way to assess the quality of groundwater. The chemical properties of the soil particles, their distribution and size, and the amount of organic matter will influence the capacity of the soil to retain more hydrophobic compounds. Likewise, irrigation practices and rainfall frequency and intensity will also influence the leachability of compounds; a higher water input will promote the pollutants transport to the subsoil and the aquifer.
On the other hand, the type of pesticide or adjuvant exposure, the duration of exposure, and the chemical nature of the compound also play important roles in determining the mechanisms of transport of pesticides within a soil matrix. In this sense, the physico-chemical interactions between the contaminant and the soil matrix are used to assess the fate of pesticides within the soil/water column (see Table 1) [10–13]. Hydrophobic compounds with a high organic carbon partition coefficient (K oc) will have a high affinity to be retained in soil and thus their lixiviation will only take place under conditions where pesticides have a very high half life. This effect is directly related to the ground ubiquity score (GUS) index, which is used to assess the leaching potential of a compound [14]. Some examples of GUS indexes can be found in the literature [15]. For a pesticide to be a potential leacher, the GUS index must be high (usually over 2.8), indicating that the compound will not be degraded nor will it be retained in the organic matter of the soil. However, the K oc and the half life of a compound are not the sole parameters which can be used to explain the transport of pesticides within the soil matrix and eventually to groundwater. Lixiviation is favored when the vapor pressure of the pesticide is low or its solubility in water is high.
However, to estimate the leaching potential of pesticides in agricultural soils, highly accurate and reproducible methods need to be used to determine a wide range of pesticides which are often applied together in pesticide formulations. Most studies report specific methods to determine target pesticides either in water or soil matrices, but none of them report a method that can be used to determine pesticides in both water and soils using fast automated methods. Water samples have been typically extracted using solid-phase extraction [16–18]. Soil samples have traditionally been extracted using mechanical shaking [19], ultrasonic [20, 21], or Soxhlet [22] extraction; although better recovery rates are obtained, these methods are arduous, time-consuming, and need high solvent volumes. More recent techniques like pressurized liquid extraction (PLE) [22, 23], supercritical fluid extraction (SFE) [19], or microwave-assisted extraction (MAE) [22] usually present lower recovery rates but are more efficient in the throughput of samples owing to their fully automated capacities that lead to a drastic reduction of extraction times, their low solvent consumption, and their ability to heat and pressurize samples thus permitting improved contact between the extracting solvent and the target analytes.
Thus, the objectives of this study were to develop a gas chromatography-mass spectrometry (GC-MS) multi-residue method capable of determining pesticides in groundwaters and soils with either automated solid-phase extraction (SPE) or pressurized liquid extraction (PLE) procedures. The list of studied compounds is based on the list of priority pesticides established by the EU [24] plus some others widely used in agriculture. We also analyzed these pesticides in agricultural soils and underneath groundwaters to determine the occurrence of target compounds in groundwaters. This study was performed in fields located along the Ebro river basin where grapes, corn, and fruit trees are mainly cultivated and where pesticides are widely used.
Materials and methods
Chemicals and reagents
Native compounds were purchased from Dr. Ehrenstorfer (Augsburg, Germany) at 100 μg/mL in ethyl acetate (see Table 2). Single isotopically labeled surrogates (desethyl-atrazine-D6, atrazine-D5, alachlor-D13, and parathion-ethyl-D10) and an internal standard (terbutylazine-D5) were purchased from Dr. Ehrenstorfer at 100 μg/mL in ethyl acetate or acetone (see Table 2). Standard working solutions were diluted from the commercial ones in hexane.
Oasis HLB 60-mg, 3-cc SPE extraction cartridges were from Waters (Milford, MA USA); Isolute ENV+200 mg cartridges were from International Sorbent Technologies Ltd. (Hengoed, UK); LiChrolut EN 500 mg and LiChrolut RP-18 500 mg cartridges were from Merck (Darmstadt, Germany). The first three have been designed for the retention of hydrophilic and lipophilic compounds; the fourth (RP-18) is a type of the widely used C18 sorbent. GC- and HPLC-quality solvents were from Merck. Florisil powder (0.150–0.250 mm of residue analysis quality) was bought, as already activated at 675 °C, from Merck and it is heated at 150 °C for 4 h to ensure its dryness. Activation with distilled water was tested (3% of water) but proved to be counterproductive at the reconstitution step where two phases (aqueous and organic) appeared in the PLE extracts making the extraction procedure more difficult. Neutral aluminum oxide (alumina) powder (0.063–0.200 mm of column chromatography quality) was from Merck; baked at 150 °C for 4 h. Hydromatrix was from Varian (Palo Alto, CA USA). Nitrogen of 99.995% purity used as drying stream was from Air Liquide (Paris, France).
Water extraction
Waters samples were filtered through 0.45-μm nylon filters. To establish quality parameters HPLC-grade water was spiked by means of a 10-μL syringe with target compounds to a concentration of 0.1 μg/L and with the surrogate solution to 0.3 μg/L. Previous work in our lab showed that no significant differences were found between using HPLC-grade water and groundwater for determining extraction quality parameters. For the preconcentration step, a BAKER vacuum system from J.T. Baker (Phillipsburg, NJ, USA) was used. Four types of cartridge were tested (Lichrolut RP-18 500 mg, Lichrolut EN 500 mg, Isolute ENV+200 mg, and OASIS HLB 60 mg). The cartridge giving the best performance was afterwards tested in triplicate to check the robustness of the method. Extraction conditions were the same despite the type of cartridge and in all cases, 200 mL water was extracted. Conditioning was performed by gravity with 4 mL dichloromethane (DCM), 4 mL ethyl acetate (EtAc), 4 mL methanol, and 2 mL water. Water samples were loaded onto the cartridges at a flow rate of 6 mL/min and these were finally rinsed with 2 mL water. The cartridges were dried under vacuum for 20 min and elution was performed with 4 mL dichloromethane/ethyl acetate (1:1) and 4 mL dichloromethane followed by 2 mL of pushing air, all at a rate 1 mL/min using an automated ASPEC XL system from Gilson (Middleton, WI USA). A blank sample was analyzed for each extraction procedure.
The resulting extracts were evaporated at room temperature under a nitrogen stream and reconstituted in 250 μL hexane in an amber glass vial. At this stage, the internal standard terbutylazine-D5 was added at 240 μg/L.
Soil extraction
Soil samples were frozen at −20 °C and then freeze-dried for 48 h at −40 °C under a 10−2 mbar vacuum. Samples were then sieved through 500- and 120-μm mesh to obtain a homogeneous sediment material. One gram of this last fraction was spiked using a 10-μL syringe with the target standards to 15 μg/kg and with the surrogate solution to 50 μg/kg and extracted using the pressurized liquid extraction (PLE) system ASE 200 from Dionex (Sunnyvale, CA USA). This system was optimized to perform the extraction and clean-up within the ASE cell in a single step. A combination of 2 extraction solvent mixtures (acetone/hexane (1:1) and acetone/DCM (1:1)) with 2 clean-up powders, Florisil and alumina, were tested. A blank was performed for each extraction condition.
For the extraction step, 22-mL ASE stainless steel cells were packed as follows: 2 g of clean-up powder was placed at the outflow side of the cell and another 5 g was mixed with the sample. The remaining space was filled with pressed hydromatrix.
In all cases, a heat-up time of 5 min was applied to the extraction cell. Temperature was adjusted to 130 °C and pressure was fixed to 1,500 psi (1 psi = 6,894.76 Pa). The solvent flow was of 60%. Two cycles of extraction were performed with 5 min in static mode. The purge time was of 60 s. Extracts were evaporated to nearly dryness using a TurboVap LV from Caliper LifeSciences (Hopkinton, MA USA), spiked with the internal standard terbutylazine-D5 at a concentration of 240 μg/L and reconstituted in 250 μL hexane into amber glass vials for gas chromatography.
Instrumental analysis
A Trace 2000 gas chromatograph from Thermo Electron (San Jose, CA USA) coupled to a mass spectrometer from Thermo Electron was employed with an electron ionization (EI) mode at 70 eV.
Compound separation was achieved using a capillary column HP-5MS of 30 m × 0.25-mm i.d. and a film thickness of 0.25 μm from J&W Scientific (Folsom, CA USA) with the following temperature program: from 60 °C (holding time 1 min) to 175 °C (holding time 4 min) at 6 °C/min to 235 °C at 3 °C/min and finally to 300 °C at 8 °C/min (holding time 5 min). Injection was achieved in the splitless mode keeping the split valve closed for 0.8 min. Helium was used as carrier gas at a flow of 1.2 mL/min. The injector, transfer, and ion source temperatures were set at 280 °C, 250 °C, and 200 °C, respectively and the detector voltage at 400 V. The injection volume was 2 μL. Acquisition was achieved in time scheduled selected ion monitoring (SIM) mode to increase sensitivity and selectivity (see Table 2). Identification and quantification were carried out automatically by the Xcalibur software, fine-tuning the identification parameters: view width of 0.20 min, maximum peak width of 18 s, and identification of the presence and correct abundance of the 3 most intense ions per compound. Internal standard quantification was performed using the base peak (indicated in Table 2) except for chlorpyrifos and parathion-ethyl that co-eluted with the same base peak, so the second more intense peak was used for each one. Isotopically labeled standards were identified with two ions using the base peak for quantification purposes (Table 2).
Environmental samples
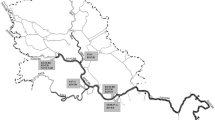
Sixteen groundwater samples and 9 corresponding agricultural soil samples were collected from the Ebro river basin (NE Spain). Sampling points were selected according to the agricultural areas paying special attention to areas with intense grape and corn production, distributing the sampling points along the middle and upper Ebro where these crops are dominant (see Fig. 1). When possible, a grab water sample was collected with an amber glass bottle placed inside a stainless steel cage. The collection was done at approximately 1 m under the water surface. In other cases, the water was pumped for 3 min or until constant conductivity prior to sample collection. Samples were collected in single-use PET amber bottles and were stored at +4 °C, for a period no longer than 10 days prior to extraction. Groundwaters sampled were from wells of 4- to 7-m depth plus G9 which was at 70-m depth. Temperature, pH, and conductivity were measured in situ by means of an integrated probe model 556MPS from YSI (Yellow Springs, OH USA).

Sampling points distribution in the Ebro river basin
Soil samples were collected in agricultural fields at no more than 50 m around their corresponding well. The final sample was a composite of 4 surface samples collected with a Dutch auger at 0- to 10-cm top soil collected randomly with at least 3 m between each other and 3 m away from the end of the field. Soil samples were then stored in glass jars with aluminum foils at +4 °C until arrival at the laboratory where they were frozen at −20 °C. Total organic carbon (TOC) and non-purgeable organic carbon (NPOC) were determined for soil and water samples, respectively. The list of samples is shown in Table 3 and their geographical distribution is represented in Fig. 1. This sampling campaign was performed throughout October 2004.
Results and discussion
Method performance
In this study it was important to provide a reliable method to determine a wide range of pesticides in both groundwater and soils. Whereas groundwater is a matrix free of interferences, soils may contain a varying amount of organic matter which might interfere with the detection of target analytes. In addition, due to the fact that both matrices may contain variable amounts of contaminants, we provide in this paper robust methods capable of determining a wide range of concentrations in both waters and soils. Emphasis is also given to report highly sensitive and reproducible methods. Table 4 provides the quality parameters of the GC-MS method and the recoveries of target compounds in both water and soil. Good linearity was obtained over a concentration range of 5–750 μg/L for nearly all compounds, as indicated by the linear regression constant (R) squared, except ometoate that was only linear from 70 μg/L. Instrumental detection limits calculated at a signal to noise ratio of 3 ranged from 0.5 to 5.7 pg, except for omethoate (see Table 4). This means that the GC-MS method is sensible enough to reach the ppt–ppb concentration level as long as the correct amount of sample is extracted. Good resolution and separation were obtained for most compounds and this was maintained in all samples processed, where no sample interferences appeared along the chromatogram. In addition, to check the ionization efficiency and matrix effect, the response of the IS was checked for each sample. On the other hand the surrogate standards used permitted is to verify the extraction efficiency for both waters and soils. The use of one surrogate standard per chromatographic window permitted us to check possible retention time shifts (<2 s) within chromatographic runs and in addition allowed us to precisely quantify all target compounds within each window. In this sense, the response of each compound in relation to the surrogate standard is indicated in Table 4. External standard quantification gave overestimated results for nearly all compounds showing the better suitability of the internal standard quantification.
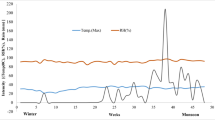
Among the SPE cartridges tested, LiChrolut EN and Isolute ENV+ usually gave overestimations due to a higher background noise in these extracts. LiChrolut RP18 and OASIS HLB gave good recoveries, although OASIS gave a more accurate response (see Fig. 2). Triplicate analysis using OASIS gave recoveries ranging from 56 to 127% with an RSD under 8.2%, which demonstrates the robustness and applicability of the extraction procedure (Table 4). Blank tests did not show any of the target analytes for any of the cartridges used.

Recoveries found for the SPE method
The somehow high recoveries obtained for nonylphenol and octylphenol were the result of a non-optimal internal calibration. Each mass spectrometric window contains from 6 to 10 target compounds and a surrogate which is used to carry out the internal calibration process. Due to the different nature of nonylphenol, octylphenol, and bisphenol A in relation to pesticides, their quantification was done using the surrogate 4-n-nonylphenol-D8. Unfortunately, the method could not put these four compounds in the same window. Due to the fact that the sensitivity between different mass spectrometric windows is different because of the nature and number of the selected ions of the SIM mode the quantification of nonylphenol and octylphenol was not optimal. The recovery rates would greatly improve if the four compounds were in the same chromatographic window [25] but this option would require a second injection just for the analysis of the three compounds. Because the objective was to develop a multiresidue analysis, a compromise between quality and effectiveness had to be taken and such a possibility was discarded.
For soils, several conditions were tested using PLE. Among sorbents used for internal clean-up, alumina proved to be very absorptive and most compounds were poorly recovered or not recovered at all, independently of the solvents used. However, Florisil using acetone/DCM (1:1) with provided recoveries ranging mainly from 48 to 144% with RSDs for three replicates under 8.8% (see Table 4). A similar behavior during the clean-up process has been previously described in the literature [26]. Other parameters from the ASE 200 system were tested with minor success: higher static time did not give better recoveries; lower extraction temperatures and pressure yielded poorer recoveries of most pollutants; a higher amount (9 g) of Florisil did not significantly influence the results. Soil extraction blanks were free of all target analytes.
However, out of thirty target compounds analyzed in soil, ten were not fully recovered (under 48% or over 144%); however, internal standard quantification allowed us to correct these differences. The high recoveries found for propanyl, dichlofenthion, parathion-methyl, and fenchlorphos are without doubt a problem of quantification with an inappropriate surrogate. The first four are in the same mass spectrometric window and then quantified with the same surrogate, alachlor-D13; this one is an acetanilide pesticide and thus different from the organophosphorus pesticides (and therefore has different properties) but was choose because of the importance of alachlor as a widely used herbicide and more probable contaminant of the environment.
Environmental levels
Table 3 shows the concentration of compounds identified in both groundwater and soil. Values encountered varied from 0.01 to 5.37 μg/L for water, being considered similar according to other American [27, 28], Canadian [29], and European [30] studies with concentrations of triazines around the 0.05 μg/L but reaching up to 11.0 μg/L. However, in this study, the sampling was carried out in the fall when the pesticide content in waters is low. Higher values have been reported in spring [31], attributed to the recent pre-seeding and post-seeding application of pesticides.
Considering the solubility of pesticides, a weak correlation was found with the concentration of pesticides, indicating that higher concentrations were found for more soluble compounds (R 2 = 0.47). However, no correlation was found between the NPOC and the concentration of pesticides. In general, the NPOC of these waters were standard to low values compared to other basins from Europe and North America where median values were around 3–4 mg/L. Parameters such as temperature (from 16.45 to 18.65 °C) and pH (from 5.5 to 6.5) were very homogeneous and did not induce differences in pesticide concentrations. The measured conductivity ranged from 518 to 3,739 μS/cm but this parameter would not affect pesticide concentration.
The most ubiquitous pesticides in groundwater samples were the group of the triazines (atrazine, simazine, terbutylazine) that were found in fourteen out of sixteen samples. The concentrations of these compounds were very similar, although in Spain atrazine is expected to be banned from 2007 and in the latter years is being substituted by terbutylazine. In some sites like G1 to G7, G13, and G14, DEA was not detected, suggesting that the application of atrazine was low or relatively recent. However, in other sites near the city of Zaragoza where high agricultural cultivations are found, atrazine was already detected at concentrations over 0.1 μg/L, i.e.,the ratio of desethyl-atrazine/atrazine was higher than 1, indicating a high microbial activity responsible for the degradation of atrazine [32]. Other herbicides detected were alachlor and metalachlor, detected in two sites, G15, a grape region and GA3, near Zaragoza. In these sites, herbicides like triazines and acetanilides are essentially applied in spring to prevent weed growth. Atrazine as well as alachlor are included as priority substances under the Water Framework Directive [24].
Among 14 organophosphorus studied, diazinon, fenitrothion, and azinphos-ethyl were only found in two samples, G8 and GA3, in the region around the city of Zaragoza. The concentrations of these three pesticides ranged from 0.02 to 0.57 μg/L, i.e., fenitrothion and azinphos-ethyl were the only two exceeding the EU maximum residual limit of 0.1 μg/L.
Surprisingly, tributylphosphate, used as a solvent in some commercial herbicides, was found in all water samples. This finding demonstrates that a source of tributylphosphate contamination in groundwaters is the pesticide formulation, and that it is prone to leaching, along with pesticides. As far as we know, there is no published data reporting positive levels of tributylphosphate in groundwaters. Tributylphosphate has a high solubility in water (see Table 1) that explains its leaching capacity. On the other hand, alkylphenols, which are also used as formulating agents, were not detected in any of the groundwater samples analyzed. Although nonylphenol was reported in earlier studies in groundwaters from agricultural, industrial, and urban areas [25], in the sampled area included in this monitoring, nonylphenol was not detected. Tributylphosphate and alkylphenols are used in pesticide formulations but not necessarily together so they can be detected individually within a water sample.
Contrary to groundwater, none of the target pesticides were detected in soils. Soil is a more stable matrix where more hydrophobic compounds would accumulate. Pesticides analyzed are generally of low to medium persistence (t 1/2 from a few days to a few months) and leaching or soil biodegradation would be preferential to adsorption. Nonylphenol was the only compound identified in 4 out of 9 samples at a concentration between 8.34 and 33.97 μg/kg. Nonylphenol is a highly lipophilic compound with a logK oc ranging from 5.24 to 5.76 [33], which tends to be adsorbed upon soil organic matter. Its low solubility in water makes lixiviation difficult. The presence of nonylphenol in soil is related to either its use as adjuvant in pesticide formulations or to the application of sludge as fertilizers.
Conclusions
The analytical method developed for the determination of different families of modern pesticides in water and soil samples provided good LODs, linearities, and recoveries for most of the compounds, permitting the use of such multi-residue methodology to monitor a wide range of pesticides in both water and soil matrices. The use of an automated SPE (ASPEC XL) and PLE (ASE 200) allowed the fast and efficient extraction of samples. Moreover, the use of multiple surrogates and internal standards permitted the precise and accurate quantification of all target compounds. Using these methods, some priority pollutants were detected in groundwater and soils in high agricultural areas in the Ebro river basin. The pesticides identified, their degradation products, or pesticide formulation adjuvants indicate a low level contamination in water with no absorption to soil. Although the monitoring campaign was performed in a period not characterized by pesticide application, once pesticide formulations are applied, these may persist in the environment for some period of time, thus being a potential risk for preserving groundwater quality.
References
Ripley BD, Ritcey GM, Harris CR, Denomme MA, Lissemore LI (2003) J Agric Food Chem 51:1328–1335
Thapar S, Bhushan R, Mathur RP (1995) Biomed Chromatogr 9:18–22
Singh BK, Walker A, Wright DJ (2002) Environ Toxicol Chem 21:2600–2605
Dagnac T, Jeannot R, Mouvet C, Baran N (2002) J Chromatogr A 957:69–77
Miller JL, Wollum III AG, Weber JB (1997) J Environ Qual 26:633–638
Turusov V, Rakitsky V, Tomatis L (2002) Environ Health Perspect 110:125–128
Vischetti C, Marucchini C, Leita L, Cantone P, Danuso F, Giovanardi R (2002) Eur J Agron 16:231–238
Garrido T, Fraile J, Ninerola JM, Figueras M, Ginebreda A, Olivella L (2000) Int J Environ Anal Chem 78:51–65
Guzzella L, Pozzoni F, Giuliano G (2006) Environ Pollut 142:344–353
Eichhorn P (2003) Occurrence and fate of surfactants in soil, subsoil and groundwater. In: Knepper TP, Barceló D, de Voogt P (eds) Analysis and fate of surfactants in the aquatic environment. Elsevier, Amsterdam, p 966
Krogh KA, Halling-Sørensen B, Mogensen BB, Vejrup KV (2003) Chemosphere 50:871–901
Comans RNJ, Roskam GD (2002) Leaching procedure for the availability of polycyclic aromatic hydrocarbons (PAHs) in contaminated soil and waste materials. In: Quevauviller P (ed) Methodologies for soil and sediment fractionation studies. Royal Society of Chemistry, Cambridge
Eichhorn P, López O, Barcelo D (2005) J Chromatogr A 1067:171–179
Gustafson DI (1989) Environ Toxicol Chem 8:339–357
Barceló D, Hennion MC (1997) Trace determination of pesticides and their degradation products in water. Elsevier, Amsterdam
Lacorte S, Molina C, Barceló D (1993) Anal Chim Acta 281:71–84
Patsias J, Papadopoulou-Mourkidou E (1996) J Chromatogr A 740:83–98
Pichon V (2000) J Chromatogr A 885:195–215
Rissato SR, Galhiane MS, Apon BM, Arruda MSP (2005) J Agric Food Chem 53:62–69
Gonçalves C, Alpendurada MF (2005) Talanta 65:1179–1189
Sanchez-Brunete C, Albero B, Tadeo JL (2004) J Agric Food Chem 52:1445–1451
Concha-Grana E, Turnes-Carou MI, Muniategui-Lorenzo S, Lopez-Mahia P, Fernandez-Fernandez E, Prada-Rodriguez D (2004) J Chromatogr A 1047:147–155
Dabrowski L, Giergielewicz-Mozajska H, Biziuk M, Gaca J, Namiesnik J (2002) J Chromatogr A 957:59–67
2455/2001/EC European Commission (2001) Official Journal of the European Communities
Latorre A, Lacorte S, Barceló D (2003) Chromatographia 57:111–116
Tekel J, Hatrfk S (1996) J Chromatogr A 754:397–410
Kolpin DW, Thurman ME, Goolsby DA (1996) Environ Sci Technol 30:335–340
Spalding RF, Watts DG, Snow DD, Cassada DA, Exner ME, Schepers JS (2003) J Environ Qual 32:84–91
Rudolph DL, Barry DAJ, Goss MJ (1998) J Contam Hydrol 32:295–311
van Maanen JMS, de Vaan MAJ, Veldstra AWF, HWPA M (2001) Environ Monit Assess 72:95
Lacorte S, Viana P, Guillamon M, Tauler R, Barceló D (2001) J Environ Monit 3:475–482
Erickson LE, Hee Lee K (1989) Crit Rev Environ Control 19:1–14
Patrolecco L, Capri S, De Angelis S, Pagnotta R, Polesello S, Valsecchi S (2006) Water Air Soil Pollut 172:151–166
Acknowledgements
This research project was founded by the European Union under the Global Change and Ecosystems (FP6) Water Cycle and Soil Related Aspects (AQUATERRA, Project number 505428 GOCE). This work reflects only authors’ views and the European Community is not liable for any use that may be made of the information contained. Financial support from Spanish Ministry of Education and Science (Project number CTM2005-25168-E) is also acknowledged. The authors also thank Waters Corporation and Merck for providing SPE cartridges, as well as Dr. Roser Chaler, Dori Fanjul, and Alicia Navarro for their assistance with GC-MS.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hildebrandt, A., Lacorte, S. & Barceló, D. Assessment of priority pesticides, degradation products, and pesticide adjuvants in groundwaters and top soils from agricultural areas of the Ebro river basin. Anal Bioanal Chem 387, 1459–1468 (2007). https://doi.org/10.1007/s00216-006-1015-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-006-1015-z