
Abstract
A study is performed to evaluate the occurrence of arsenic in polluted soils using acidic extractions and liquid chromatography–hydride generation–atomic fluorescence spectrometry (LC–HG–AFS) for speciation analysis. Seven soil samples were collected in an abandoned area polluted by mining in the Eastern Pyrenees (Spain), and two uncontaminated soils were taken for reference purposes. Moreover, the total arsenic content is evaluated in two different sieved fractions in order to obtain information on the possible particle-size-dependent association of arsenic with soil components. Soil samples were extracted with both phosphoric and ascorbic acids and the stabilities of the extracted species were studied. The arsenic species were determined by LC–HG–AFS. In addition, the ability of soil grinding to effect species change is also assessed. Arsenite and arsenate were found in the polluted soils, but only arsenate was found in the unpolluted soils. The quality of the results was assessed through a mass balance calculation and by analysing two soil Certified Reference Materials. Valuable information regarding arsenic occurrence in the studied soils is obtained from the speciation results. The presence of arsenite in the extracts can be attributed to arsenopyrite residues, whereas the presence of arsenate indicates release from weathered material.

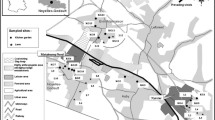
Abandoned mining polluted area in Eastern Pyrenees
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Arsenic concentrations in soils can be elevated due to anthropogenic activity or high natural abundance. Normal arsenic levels in soils range from 1 to 40 mg kg−1, although arsenic levels are significantly higher in mining zones [1, 2]. The groundwater used directly for human consumption and irrigation in agricultural zones could therefore be a source of contamination and toxicity in some instances. Arsenic is mainly present in inorganic forms (arsenate and arsenite) in soils. However, the organic compounds MMA and DMA may also be present in lower amounts [3].
The toxicity, biological availability and transport mechanisms involved depend upon the chemical form of arsenic in question. Inorganic arsenic species are known to be more toxic than organic arsenic species. Identification and quantification of the arsenic species are crucial tasks when elucidating the behaviour of this element in polluted soils [4]. Thus, the arsenic speciation of contaminated soils by means of extraction and measurement by hyphenated techniques is a research area that is currently attracting much interest.
Sample pretreatment is a critical step in speciation analysis, and one that could modify the integrity of the species in some cases. Although some authors have studied the effect of temperature during drying and storage, information in the literature about changes in species during sample pretreatment is scarce [5]. A common pretreatment practice involves grinding soil samples. However, no data is available on the transformation of arsenic species during this process.
Common extraction media used to speciate arsenic in soil samples include water, ammonium oxalate and sodium carbonate [6–9]. Phosphoric acid has also been proposed as a suitable extractant due to the chemical similarity of phosphorus to arsenic [5, 10–14].
The stability of the arsenic species in the extracts must be guaranteed, and this depends on the pH of the extraction media and on the other soil components extracted [6]. In general, arsenate is considered stable [15]. However, the arsenite in extracts from some soil samples was observed to be unstable [5, 13].
Most of the analytical techniques used for As speciation are based on hyphenated systems, such as liquid chromatography coupled to sensitive spectrometric detection [9, 11, 12, 16–18]. The coupling of HPLC with ICP–MS has also proved to be an effective technique [3, 6–8, 10, 14, 19, 20].
The aim of this study was to assess the relevance of the information obtained through arsenic chemical speciation when elucidating the behaviour of the original arsenic compounds in soils polluted by mining tailings that have been exposed to natural weathering.
Experimental
Apparatus
Block digester
A P/Selecta (Barcelona, Spain) Bloc Digest digester with a P/Selecta Model RAT 4000051 time and temperature regulator was used as the digestion system for pseudototal arsenic determination.
Microwave digestion
A Prolabo M301 (2.45 GHz; Paris, France) open microwave digester with a TX32 programmer was used for arsenic species extraction.
LC–HG–AFS system
A coupled LC–HG–AFS system was used for arsenic species measurements.
-
Chromatographic system: A PerkinElmer 250 LC binary pump (Shelton, CT, USA) was used, equipped with a Rheodyne 7125 injector (Cotati, CA, USA) with a 100 μl loop. A Hamilton PRP-X100 anion-exchange column (Reno, NV, USA) with spherical poly(styrene-divinylbenzene) trimethylammonium exchangers (particle size 10 μm; 250 mm × 4.1 mm i..d.) was also applied.
-
Hydride generation: A model 10.004 module from PS Analytical (Orpington, UK) was used. The sample channel was eliminated for speciation analysis, and a T-shape connector was employed to link the outlet from the LC system to the acid channel.
-
Detection: An Excalibur (PS Analytical) atomic fluorescence spectrometer equipped with a hollow cathode lamp (current intensities: primary = 27.5 mA, boost = 35.0 mA) was used, and a Perma Pure drying membrane (Perma Pure Products, Farmingdale, NJ, USA) was employed to dry the hydride generated. Nitrogen was used as drying gas. Measuring wavelength: 193.7 nm. Data acquisition was performed with a microcomputer and homemade software (Pendragon 1.0). Peak heights and peak areas were measured from custom-made software running in the Matlab language.
X-ray fluorescence
A Phillips (Eindhoven, The Netherlands) PW 2400 spectrometer was used to determine the X-ray fluorescence of each of the major components.
Elemental analysis
A model EA 1108 elemental analyzer (Carlo Erba/Thermo Finnigan, Milan, Italy) equipped with an AS200 autosampler was used along with a combustion oven, a Porapack GC column for gas separation, and a thermal conductivity detector (TCD) for elemental analysis.
ICP-AES
A PerkinElmer Optima 3000 PL inductively coupled plasma spectrometer with a “cross-flow” nebulizer was used to measure the total As in extracts of both the aqua regia and phosphoric/ascorbic acid.
Sample grinding
A tungsten carbide disc mill (Herzog, Osnabrück, Germany) was used to grind samples.
Standards, reagents and certified reference materials
Doubly deionized water (USF purelab plus, Ransbach Baumbach, Germany), with a resistivity of 18.2 MΩ cm−1, was used to prepare all the solutions.
Standards
Stock solutions (1,000 mg l−1 as As) of arsenic compounds were prepared as follows:
Arsenite: Prepared from As2O3 (NIST Oxidimetric Primary Standard 83d, 99.99%, MW 197.8414) dissolved in 4 g l−1 NaOH (Suprapure, Merck, Darmstadt, Germany).
Arsenate: Prepared from Na2HAsO4·7H2O (Carlo Erba, MW 312.0141) dissolved in water.
The stock solutions were kept at 4 °C and standardised against two arsenite solutions [As2O3 (Merck), As2O3 (NIST 83d)] by ICP–AES. Diluted solutions required for analysis were prepared daily.
Extraction reagents
1 mol l−1 o-phosphoric acid solution (85% H3PO4, Merck, pro analysi) with 0.5 mol l−1 L(+)-ascorbic acid solution (99.7%, Merck, pro analysi). These solutions were purged with an argon stream for about 15 min before the microwave extraction.
Mobile phase
Phosphate buffer at pH 6.0 prepared from 5 mmol l−1 and 100 mmol l−1 of NaH2PO4 and Na2HPO4 mixture (Merck, Suprapur), filtered through a 0.22-μm nylon membrane.
Hydride-generating reagents
HCl 2 mol l−1 prepared from hydrochloric acid solution (37% HCl, Merck, pro analysi). Sodium borohydride (NaBH4 tablets, Fluka, purum, purity >97%) at 1.5% in 0.2% NaOH (Merck, pro analysi).
Aqua regia extraction reagents
Nitric acid (70%, HNO3, Baker, Instra-analyzed) and hydrochloric acid (HCl 36.5–38%, Baker, Instra-analyzed) were used.
Certified Reference Materials
The following were used as quality controls: GBW 07405 soil from the NRCCRM (Beijing, PR China) with a certified total arsenic content of 412 ± 8 mg kg−1, and CRM 025–050 (Natural Matrix Certified Reference Material) from the Resource Technology Corporation (Laramie, WY, USA) which has a reference value for total arsenic content of 339 ± 51 mg kg−1.
Sampling and sample pretreatment
Vall de Ribes is located on the southern side of the Eastern Pyrenees (North Catalonia, Spain) along the north watercourse of the River Freser. The climate of Vall de Ribes can be considered to be temperate and wet, with an annual precipitation of approximately 900 mm and an annual average temperature of 11 °C. The region was an active mining district in the nineteenth century. As and Sb veins with smaller amounts of Cu, Pb and Ag were mined [21, 22]. Although the presence of gold and silver was also discovered at that time, the small thickness and the irregularity of the veins as well as the geographic location of the mines made them uneconomical, and they were closed by the end of the nineteenth century. Vegetation has now colonized the mining zones, and nearly all of the veins are located inside the forest. The geology around Queralbs can be considered to be very similar when viewed from an overall materials perspective but very varied when studied in detail. Minor minerals as pyrite and arsenopyrite can be found dispersed or concentrated in mm-thick layers. The mineralogy of Vall de Ribes has been described from a mineralogical point of view by Ayora et al. [21, 23] and Santanach [24]. Detailed mapping and lithostratigraphic and structural information on the lower part of the pre-Caradocian rocks around Queralbs, where most of the As and associated ores are located, can be found in another study by Ayora at al. [22].
Seven soil samples (P1 to P7) were collected from surface layers (0–15 cm depth) in October 2004 after selecting three different sampling sites. Samples P1 to P4 were taken from an area near Ribes de Freser, close to the “Ermita de Sant Antoni” area 5 km from Ribes de Freser, where four small Sb and Zn mines were located. Samples P5 and P6 were collected around the mine spoil from two old As and Sb mines next to Planoles (“Espinosa” mines), to the north-west of Ribes de Freser. Finally, sample P7 was collected near Queralbs, a village to the north of Ribes de Freser, where an abandoned As mine can be found. The sample was collected in the tailings from a vein called “Yellas”. Due to the dispersion of minor minerals in these veins, sampling points were selected accurately for the purposes of this study. All samples were collected less than 10 m from the mine tailings, where higher levels of arsenic were expected. Figure 1 shows a map of the Ripollés district in which the selected sampling points are located.

Map of the Ripollés district in the Eastern Pyrenees, Spain
In a second sampling in April 2005, two soil samples (B1 and B2) were collected from an uncontaminated area (Collada de Toses, see Fig. 1) that has similar mineralogical characteristics in terms of the materials present, but where there are no minor mineral veins. The aim of this sampling was to assess the natural background level of arsenic. These samples were used in the present study as reference samples. Sample B1 corresponds to a soil with similar characteristics to P1–P6. Sample B2 corresponds to a soil with similar characteristics to soil sample P7.
Sample pretreatment
Soil samples P1–P7 were air-dried separately at room temperature and divided into two portions. One of these portions was sieved through a 2 mm mesh and the other one through a 90 μm mesh. Soil samples B1 and B2 were also air-dried and sieved through a 2 mm mesh. The sieved subsamples were stored in plastic containers at room temperature until analysis. Aliquots of the 2 mm soil fraction of samples P1, P5, P7, B1 and B2 were ground to a fine powder with a tungsten carbide disc mill.
Procedures
Sample moisture determination
The moisture was determined gravimetrically, in duplicate, by drying 1 g of sample at 105 °C until the weight remained constant. All results described in the present study refer to dry mass.
X-ray fluorescence analysis
0.3 g of each ground and dried sample was weighed and mixed with 5.7 g of lithium tetraborate (1/20 dilution; procedure performed in duplicate). The sample was homogenised in a radiofrequency inductive oven (PERL′X-2, Philips) and X-ray fluorescence intensity was measured with a Phillips PW2400 spectrometer. Quantification of the elements was achieved by including 56 international geological reference samples in the calibration curve: for example, BCS375 and BCS376 from British Chemical Standards, and NBS278 from the National Institute of Standards and Technology (NIST). Kα lines were used to quantify major elements.
Elemental analysis
Sulfur and nitrogen were determined as follows. Twenty milligrams of a 90 μm fraction of dried soil were weighed, in duplicate, into tin capsules with Sn and V2O5 added as catalysts. The capsules were introduced into the instrument directly through the autosampler. The sample was introduced into the heater reactor (1,000 °C), and the resulting mixture containing the combustion gases was passed through the chromatography column (at 60 °C) to separate N2, CO2, H2O and SO2, before finally reaching the detector (also at 60 °C). Organic carbon was determined according to [25]. Sulfanilamide standard (Thermo Finnigan) with certified elemental contents of C, N and S was used for the calibration.
Aqua regia extraction
Soil samples were digested according to the ISO/CD 11466 standard from 1995 [26]. The soil subsample (3 g, performed in triplicate) was placed in the reflux vessel and the required volume of aqua regia was added. The soil suspension was maintained at room temperature for 16 h, and then the mixture was heated at 130 °C for 2 h. The resulting suspension was filtered through an ashless filter (Whatman 40) and the final solution was diluted to 100 ml and transferred to a polyethylene container, which was stored at 4 °C until analysis.
Arsenic determination by ICP–AES
The extracts with the highest arsenic contents (P6, P7, CRM GBW 07450 and CRM 025–050 extracts) were measured by ICP–AES, using an external curve prepared from As2O3 99.99% (NIST 83d) as a stock solution.
Arsenic determination by HG–AFS
This technique was used to quantify arsenic in samples with lower As concentrations (sample extracts P1–P5, B1 and B2). A prereduction step had to be applied before the measurement. Thus, an aliquot of the extract was diluted to 25 ml with the appropriate reagents to obtain a final concentration of 0.2% ascorbic acid, 1% potassium iodide, and 2 mol l−1 hydrochloric acid. The resulting solution was maintained at room temperature for at least one hour until measurements were performed in order to ensure that total reduction to As(III) occurred. The quantification was achieved using an external calibration curve. The measuring standards contained ascorbic acid, potassium iodide and hydrochloric acid at the same concentrations as in the sample extracts. Hydride generation was achieved using HCl 2 mol l−1 at a flow of 8.0 ml min−1, and NaBH4 1.5% (m/v) in NaOH 0.4% at a flow of 3.0 ml min−1.
Arsenic speciation
Extraction
Three aliquots of 0.1 g of both the 90 μm fraction and the 2 mm fraction ground to a fine powder, 15 ml of 1 mol l−1 phosphoric acid, and 0.5 mol l−1 ascorbic acid solution (purged with argon for about 15 min to slow down the kinetics of arsenite oxidation) were placed in the corresponding open reflux vessel and maintained at 60 W for 10 min in the microwave system. Once at room temperature, the mixture was filtered (Whatman 40) and diluted to 50 ml with doubly deionized water. Arsenic species quantification was carried out within 24 hours after extraction. More detailed information about the extraction procedure can be found in a previous publication in which the extraction efficiencies as well as the stabilities of the arsenic species were studied in depth [13].
HPLC-HG-AFS
The extract was filtered through a 0.22 μm nylon membrane and purged with argon for five minutes. Then, a 100 μl aliquot was injected into the chromatographic column. The separation was achieved at pH 6.0 with a concentration gradient of 5 mmol l−1 (solution A) and 100 mmol l−1 (solution B) phosphate buffer, at a flow rate of 1 ml min−1. The gradient program was: 100% A for 2 min; decreasing to 50% A in 0.1 min and maintained for 3 min; increasing to 100% A in 0.1 min and maintained for 9 min. The eluted arsenic species reached the hydride generation system and were measured by the AFS detector. Arsenic species were quantified (peak area) by the standard addition method.
Results and discussion
Characterisation of soil samples
Soil moisture was determined in the studied soils. Values ranging from 1.3 to 4.0% were obtained for the moisture content.
The concentrations of major soil components, the pH and the levels of organic C, S and N were determined. Table 1 shows the results obtained by X-ray fluorescence for all of the sampled soils. The major components in the 90 μm fractions of the P1–P7 soils and the 2 mm fractions of the P1, P5, P7, B1 and B2 soils are shown.
The soils were mainly siliceous. In addition, high percentages of Fe2O3 were found in the soils sampled close to the arsenic mines (P5, P6 and P7). Similar values were found when major components from the 2 mm and 90 μm fractions were compared. The exception was sample P7, in which higher percentages of iron were found in the smaller particle size fraction. The compositions of soils B1 and B2 were found to be similar to those of the P1–P7 soils.
Table 2 shows the pH values of the soils [27], as well as the results for the percentage nitrogen, sulfur and organic carbon, obtained by elemental analysis. Most of the soils presented acidic pH values, which correlate with the siliceous nature of those soils. A basic pH value was obtained for sample P7 due to the presence of carbonate in the sample (1.95% CaO, see Table 1).
Low percentages of nitrogen and sulfur were obtained for all samples. Higher sulfur concentrations were expected, due to the fact that pyrite and arsenopyrite are known to be present at these sites [22]. This low sulfur contents could be due to the oxidation of sulfide to sulfate, which is easily lixiviated and removed from the soil matrix [28]. Relatively high percentages of organic carbon were measured in most samples. This can be attributed to the sampling location: a forested area. Soil P7, which was sampled outside of the forest, shows the lowest %C.
Arsenic determination
Pseudototal arsenic content
The pseudototal arsenic content was determined by aqua regia extraction followed by both ICP–AES and HG–AFS measurements. Table 3 shows the results obtained for the soil samples (both the 2 mm and the 90 μm fractions). The results indicate that very high concentrations of arsenic are present in the samples collected near Queralbs and Planoles (P5, P6 and P7) compared to the reference samples (B1 and B2). Soils P1–P4 had lower arsenic contents. However, their As contents were still higher than those of B1 and B2. High arsenic values were obtained for the P5, P6 and P7 soils in the 90 μm fraction compared to those for the unground 2 mm fraction. This behaviour may be attributed to either a more effective extraction in the smaller particle fraction or the preferential association of arsenic with smaller particles. In order to ascertain the influence of the particle size on extraction, the 2 mm fractions of P1, P5 and P7 (belonging to every sampling zone selected) and B1 and B2 were ground with a tungsten carbide disc mill to a fine powder and digested in the same way. The results obtained are also reported in Table 3. The values agreed with those obtained for the unground 2 mm fraction. This shows the efficiency of aqua regia extraction in these samples, irrespective of particle size. Thus, from a geochemical perspective, the differences between the values in the first and the third columns of Table 3 could be attributable to the association of arsenic with smaller particles such as iron (hydr)oxides [29, 30]. This is supported by the analysis of the solid material by electron microprobe analysis, which shows the presence of significant amounts of arsenic on the surfaces of iron (hydr)oxide particles, together with smaller amounts of the original arsenopyrite.
Arsenic speciation
Table 4 reports the results for the arsenic species obtained from the phosphoric acid/ascorbic acid extracts of the 90 μm soil fraction and the CRMs, the sum of the two As species, and the total arsenic content in each extract. The presence of inorganic arsenic was only observed in the soil samples. Although the proposed technique is capable of determining methylated species in soil phosphoric extracts, they were not detected in the studied soils. Arsenite and arsenate were measured in most samples. However, only arsenate was measured in B1, B2 and the CRM GB W07450. Figure 2 shows, as an example, three chromatograms for soil sample extracts in which the presence of arsenic species was observed. In all cases, higher levels of arsenate were present than arsenite, irrespective of the total arsenic concentration in the extracts. The detection limits were calculated from three times the standard deviation of the baseline in the chromatogram (n = 70) for each extract divided by the corresponding calibration curve slope. The values obtained were: 94.4 ng As l−1 in solution (1.07 mg As kg −1 in dry mass) for arsenite; and 93.9 ng As l−1 in solution (1.44 mg As kg−1 in dry mass) for arsenate.

Example chromatograms for arsenic species in the 90 μm fraction soil extracts: a P2 soil sample; b P5 soil sample; and c P7 soil sample
Good agreement was also observed (Table 4) between the sum of the two As species and the total content in the extracts. In general, slightly higher extraction efficiencies were obtained when the total arsenic in the phosphoric acid–ascorbic acid extract was compared with the pseudototal arsenic content (see Table 3). However, when the CRMs were analysed, the values obtained for both extraction procedures were in agreement.
These results concur with previous findings reported in the literature. The main mechanism of arsenic release in the weathering of arsenopyrite is its oxidation to arsenate [31]. However, the soluble arsenate is adsorbed onto iron oxyhydroxides at pH values of around 4–5 [32]. Moreover, some kinetic studies on the oxidation of arsenopyrite in acidic solution show that arsenic is released as arsenite and that the rate of conversion to arsenate is relatively low. Thus, when extracting with phosphoric acid–ascorbic acid, the release of the arsenate associated with iron oxyhydroxides and arsenite from the remaining arsenopyrite would be expected [33]. When an extraction process is involved in the speciation analysis of soil samples, information gleaned about the arsenic compounds in the original sample can be inaccurate, because the forms of arsenic present in acidic solution are arsenite or arsenate, which do not correspond with the native compounds in the solid. However, the results from the present study are supported by the use of surface techniques in which all of the information about the original compounds of arsenic is obtained. Thus, complementary information can be obtained when both analytical speciation and surface techniques are used. This approach can be applied to soils with different characteristics in order to obtain more information about arsenic transformation and behaviour in soils contaminated by mining.
Sample pretreatment and stability studies
Pretreatment techniques must be considered an important part of the overall analysis in trace element determinations; they are a crucial step in speciation analysis. The lack of methods assessing the integrity of species during pretreatment and storage could lead to a serious risk of inaccurate results. Changes in humidity, temperature, redox potential and other parameters can produce important changes in the original mechanisms of binding and consequently in the release of species when a extraction procedure is applied. Although these aspects of sample handling are well known, no data on the influence of soil grinding on possible changes in the chemical species of arsenic are reported in the literature.
In the present study, in order to evaluate the effect of sample grinding on the speciation results, an analysis of the extracts was performed on subsamples obtained after grinding the original 2 mm fraction to a fine powder. For this, three replicates of samples P1, P5 and P7 were extracted with the proposed procedure and the extracts were analysed with LC–HG–AFS in the same way as for the unground samples. A significant decrease (t-test, 95% confidence level) in the arsenite/arsenate ratio was observed with respect to the unground fraction for the samples studied. In sample P1, even arsenite was not detected in the chromatogram from the unground fraction extract. As an example, Fig. 3 shows, for samples P1 and P7, a comparison of the species in the extracts from the corresponding ground and unground samples. This indicates that in the studied soils some transformation of the species in the soil occurred during the grinding procedure, despite the short grinding time (less than ten seconds). The importance of preserving the integrity of the species during sample pretreatment is highlighted by these results.

Example chromatograms for P1 (a) and P7 (b) extracts. a1 and b1 apply to the 90 μm fraction. a2 and b2 apply to the 2 mm fraction ground to a fine powder
On the other hand, several studies have investigated the stability of As species in solution in different media. Many of these studies show that most stability issues are associated with arsenite, because it is easily oxidised to arsenate [34]. Therefore, it is important to check the stability of arsenite in the soil extracts in order to obtain reliable results. In the present study, the stability of arsenite over time was checked for the phosphoric–ascorbic acid extracts. No decrease in the arsenite peak height was observed in any of the samples studied in the first 24 hours. As an example, Fig. 4 shows chromatograms from the extracts of two soil samples (P2 and P7) just after extraction, after 48 h and 120 h. To avoid mistakes during arsenite quantification, it is recommended that the analysis should be carried out within 24 hours of extraction, as stated in a previous work [13].

Species stability after extraction of samples P2 (a) and P7 (b)
Conclusions
The use of phosphoric acid–ascorbic acid extractant and LC–HG–AFS provides valuable information about the behaviour of arsenic in soils polluted by mining that are now exposed to natural weathering conditions. Thus, the presence of arsenite in the extracts can be attributed to its release from arsenopyrite residues in the studied soils.
The quantitative extraction of arsenic was assessed by analysing two Certified Reference Materials and using mass balance calculations. The stability of the extracted arsenite was also assessed within the first 24 hours after extraction.
From the high concentrations of arsenic obtained from the smaller particle size fraction and the information obtained by electron microprobe analysis, it can be concluded that arsenic is associated mainly with compounds yielding small particle sizes, such as iron (hydr)oxides.
New data was obtained on the influence of sample pretreatment. A decrease in the arsenite/arsenate ratio is observed when the soil sample is ground to fine powder.
This study has enhanced our understanding of the environmental behavior of arsenic. The results obtained provide relevant information for assessing the potential mobility of arsenic from polluted soils.
References
Cullen WR, Reimer KJ (1989) Chem Rev 89:713–764
Mandal BK, Suzuki KT (2002) Talanta 58:201–235
Yehl PM, Gurleyuk H, Tyson JF, Uden PC (2001) Analyst 126:1511–1518
Melamed D (2004) Monitoring arsenic in the environment. A review of science and technologies for field measurements and sensors (EPA 542/R–04/002). US EPA, Washington, DC (see http://www.epa.gov/tio/download/char/arsenic_paper.pdf, last accessed 4th November 2006)
Garcia-Manyes S, Jiménez G, Padró A, Rubio R, Rauret G (2002) Talanta 58:97–109
Bissen M, Frimmel FH (2000) Fresenius J Anal Chem 367:51–55
Pongratz R (1998) Sci Total Environ 224:133–141
Koellensperger G, Nurmi J, Hann S, Stingeder G, Fitz WJ, Wenzel WW (2002) J Anal Atom Spectrom 17:1042–1047
Guerin T, Molenat N, Astruc A, Pinel R (2000) Appl Organomet Chem 14:401–410
Thomas P, Finnie JK, Williams JG (1997) J Anal Atom Spectrom 12:1367–1372
Vergara-Gallardo M, Bohari Y, Astruc A, Poitin-Gaultier M, Astruc M (2001) Anal Chim Acta 441:257–268
Montperrus M, Bohari Y, Bueno M, Astruc A, Astruc M (2002) Appl Organomet Chem 16:347–354
Ruiz-Chancho MJ, Sabé R, López-Sánchez JF, Rubio R, Thomas P (2005) Microchim Acta 151:241–248
Pizarro I, Gómez M, Cámara C, Palacios MA (2003) Anal Chim Acta 495:85–98
Pantsar-Kallio M, Manninen PKG (1997) Sci Total Environ 204:193–200
Manning BA, Martens DA (1997) Environ Sci Technol 31:171–177
Gómez-Ariza JL, Sánchez-Rodas D, Giráldez I (1998) J Anal Atom Spectrom 13:1375–1379
Caballo-López A, Luque de Castro MD (2003) Anal Chem 75:2011–2017
Kahakachchi C, Uden PC, Tyson JF (2004) Analyst 129:714–718
Demesmay C, Ollé M (1997) Fresenius J Anal Chem 357:1116–1121
Ayora C, Phillips R (1981) Bull Mineral 104:556–564
Ayora C, Casas JM (1986) Mineral Deposita 21:278–287
Ayora C (1980) Les concentracions metalliques de la Vall de Ribes. Thesis, University of Barcelona, Barcelona, Spain
Santanach P (1972) Estudio tectónico del Paleozoico inferior del Pirineo entre la Cerdanya y el rio Ter. Thesis, University of Barcelona, Barcelona, Spain
ISO (1995) ISO10694: Soil quality, determination of organic and total carbon after dry combustion (elementary analysis). International Organization for Standardization (ISO), Geneva, Switzerland
ISO (1995) ISO11466: Extraction of trace elements soluble in aqua regia (international standard). International Organization for Standardization (ISO), Geneva, Switzerland
Department of Agriculture (1986) Spanish official analytical methods for soils, vol 3. Department of Agriculture, Madrid
McGrath S T, Zhao F, Blake-Kalff M (2002) Sulfur in soils: processes, behaviour and measurement (from Proc Int Fertilizer Soc). The International Fertilizer Society, York, UK, p 499
Jones CA, Inskeep WP, Neuman DR (1997) J Environ Qual 26:433–439
Sadiq M (1997) Water Air Soil Pollut 93:117–136
Dove PM, Rimstidt JD (1985) Am Mineral 70:838–844
Garcia-Sánchez A, Alvarez-Ayuso E (2003) J Geochem Explor 80:69–79
Yumnei Y, Yongxuan Z, Williams-Jones A E, Zhenmin G, Dexian L (2004) Appl Geochem 19:435–444
Francesconi KA, Huehnelt D (2004) Analyst 129:373–395
Acknowledgements
The authors thank DGICYT (Project number BQU2003-02951) for the financial help received in support of this study; C. Ayora and J. Cama (Institut de Ciencies de la Terra “Jaume Almera”-CSIC) for their invaluable support in sampling and their help in the interpretation of the electron microprobe analysis; and R. Miravet (Departament de Química Analítica, Universitat de Barcelona) for his help with sample pretreatment. M.J. Ruiz-Chancho wishes to thank the Universitat de Barcelona for their support, provided through a predoctoral grant.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ruiz-Chancho, M.J., López-Sánchez, J.F. & Rubio, R. Analytical speciation as a tool to assess arsenic behaviour in soils polluted by mining. Anal Bioanal Chem 387, 627–635 (2007). https://doi.org/10.1007/s00216-006-0939-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-006-0939-7