
Abstract
A new analytical method is proposed for simultaneous determination, by liquid chromatography, of the three main urinary thiols–cysteine, cysteinylglycine, and homocysteine. To measure the total amount of these thiols urine is reduced with sodium borohydride, to convert disulfides to thiols which are then derivatized with 2-chloro-1-methylquinolinium tetrafluoroborate. Separation and quantitation of the 2-S-quinolinium thiol derivatives formed were achieved by high-performance liquid chromatography with detection at 355 nm. Validation showed the method enabled reliable simultaneous determination of these aminothiols in urine. The calibration graphs for each analyte, obtained by use of normal urine spiked with increasing amounts of cysteine, cysteinylglycine, and homocysteine, were linear (R 2≥0.997) over the range covering most practical situations. The recovery of the assay was 98–100% and sensitivity was 0.12–0.25 μmol L−1. The method was applied to 91 different samples of normal urine to establish reference values for the aminothiols, normalized on creatinine.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Biological thiols are products of sulfur metabolism. They are reactive and ubiquitous and have been extensively studied in a variety of biological systems. Cysteine, glutathione, and homocysteine are the most important; other biological thiols are derivatives of these compounds. Despite their similar structures and common chemistry, the functions of thiols in organisms are remarkably different. The best known of these thiols is glutathione, which is present in large amounts in, for example, liver cells and erythrocytes, but is present extracellularly at much lower concentrations. The first step in glutathione breakdown in the γ-glutamyl cycle is catalysis by γ-glutamyltranspeptidase, which removes the γ-glutamyl moiety of glutathione [1]. The remaining cysteinylglycine can be split to cysteine and glycine. Homocysteine, a product of demethylation of methionine, may be converted to cysteine or remethylated to methionine. Disorders of cysteine metabolism [2] include cystinosis, an autosomal recessive disease caused by a defect in lysosomal transport, and cystinuria, an inherited disorder of transport of the amino acids cystine, ornithine, and arginine. This defect leads to high concentrations of these compounds in the urine. Because of the very low solubility of cystine in urine, kidney-stone formation is a clinical manifestation of classical cystinuria. In laboratory diagnosis of cystinuria, after a positive sodium nitroprusside test, quantitative analysis of the amino acids cysteine and homocysteine is required to differentiate between cystinuria and homocystinuria. Elevated plasma and urinary levels of cysteinylglycine and other aminothiols are observed in patients with rheumatoid arthritis [3, 4] and may be associated with the extent of inflammation. To elucidate the function of these important amino acids in biochemical and clinical practice their identification and determination in urine, which, to some extent, mirrors changes in plasma, is essential. As a consequence, and because of increased general interest in the determination of cysteine and metabolically related aminothiols in urine, several HPLC and CE methods have been reported in recent years [5–19]. To the best of our knowledge, however, only four papers [8, 10, 15, 16] offer procedures for simultaneous quantitation of cysteine, cysteinylglycine, and homocysteine, and results for these three aminothiols have been given in two papers only [8, 16]. Because of lack of structural features suitable for use of conventional detectors, and to block the –SH function, susceptible to oxidation during sampling and final analysis, most methods depend on derivatization [3–17]. In this report we describe a new UV–derivatization HPLC method for simultaneous determination of total cysteine, cysteinylglycine, and homocysteine in urine. The method has been used to estimate reference values for the total target aminothiols in human urine, normalized on creatinine.
Experimental
Reagents
L-cystine (CSSC) and L-cysteine (CSH) were from Reanal (Budapest, Hungary). D,L-homocystine (HCSSCH), D,L-homocysteine (HCSH), cysteinylglycine (CGSH), and creatinine (Crn) were from Sigma (St Louis, MO, USA). 2-Chloro-1-methylquinolinium tetrafluoroborate (CMQT) was prepared in this laboratory by a procedure described elsewhere [20]. Perchloric acid (PCA), hydrochloric acid (HCl), sodium hydroxide (NaOH), sodium hydrogen phosphate heptahydrate (Na2HPO4·7H2O), sodium dihydrogen phosphate dihydrate (NaH2PO4·2H2O), and HPLC-grade acetonitrile were from J.T. Baker (Deventer, The Netherlands). Trichloroacetic acid (TCA) and sodium borohydride (NaBH4) were from Merck (Darmstadt, Germany). All other reagents were HPLC or analytical-reagent grade and purchased from commercial sources. The pH of buffers was adjusted by potentiometric titration. The titration system was calibrated with standard pH solutions. All reagents were tested and found to be sufficiently stable for unattended analysis. The water used for all buffers was purified by use of a Millipore Milli-QRG system (Vienna, Austria).
Apparatus
HPLC analysis was performed with a Hewlett–Packard (Waldbronn, Germany) HP 1100 Series system equipped with quaternary pump, autosampler, thermostatted column compartment, vacuum degasser, and diode-array detector and controlled by HP ChemStation software. UV spectra were recorded on a Hewlett-Packard HP 8453 diode-array UV–visible spectrophotometer. A Hach One (Loveland, USA) pH meter was used for pH measurement.
Sample collection and pretreatment
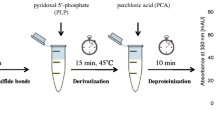
Urine was collected from healthy, ethnically homogenous volunteers and either analyzed without delay or stored at −20 °C until analysis. To 200 μL urine in a polypropylene vial 50 μL n-octanol, 100 μL NaBH4 (6 mol L−1) in 0.1 mol L−1 NaOH diluted 2:1 with dimethyl sulfoxide, and 60 μL HCl (3 mol L−1) were added. The mixture was vortex-mixed and, after 2 min, an additional 50 μL 3 mol L−1 HCl was added to decompose excess sodium borohydride (pH≈7.5, indicator paper). Next, 100 μL phosphoric buffer (pH 7.5, 0.2 mol L−1) and 30 μL CMQT reagent (0.1 mol L−1) were added. The mixture was vortex-mixed, put aside for 2 min, acidified with 100 μL TCA (3 mol L−1), then centrifuged (12000×g, 10 min). The solution (20 μL) was then analyzed by HPLC.
HPLC separation of 2-S-quinolinium derivatives
The final analytical solution (20 μL) was injected by use of an autosampler into a 4.6 mm×150 mm, 5 μm particle, Zorbax SB-C18 column (Agilent Technologies, Waldbronn, Germany). Separation of the 2-S-quinolinium derivatives of cysteine, cysteinylglycine, and homocysteine was achieved by use of a gradient prepared from 0.05 mol L−1, pH 3.2, TCA buffer prepared from 0.05 mol L−1 trichloroacetic acid and 0.05 mol L−1 lithium hydroxide, component A, and acetonitrile, component B. The gradient profile was: 0–4 min 11% B, 4–8 min 11–30% B, 8–12 min 30–11% B. The temperature was 25 °C, the flow rate 1.5 mL min−1 and the detector wavelength 355 nm. Identification of peaks was based on comparison of retention times and diode-array spectra, acquired during analysis, with corresponding data obtained for authentic compounds.
Calibration standards
Stock solutions of 10 mmol L−1 cysteine, cysteinylglycine, and homocysteine and their oxidized forms needed for method development and validation procedures were prepared by dissolving appropriate amounts of the compounds in 0.1 mol L−1 hydrochloric acid; the solutions were stored at 4 °C. Working standard solutions were prepared by dilution with water when required. To prepare calibration standards 200 μL urine was placed in polypropylene vials and spiked with increasing amounts of working standard solutions of the thiols in their disulfide form at six levels of concentration. Because 2 mol thiol are generated by reduction of 1 mol disulfide, all results are expressed in equivalents of thiol. The calibration ranges were: for cysteine 50–500 (50, 100, 200, 300, 400, 500) μmol L−1 urine, for cysteinylglycine 2.5–25 (2.5, 5, 10, 15, 20, 25) μmol L−1 urine, and for homocysteine 2–14 (2, 4, 6, 8, 10, 14) μmol L−1 urine. Calibration standards were subjected to all steps of the recommended analytical procedure.
Determination of creatinine
Creatinine (Crn) was determined by use of an HPLC method reported elsewhere [21] with some modification. In brief, 20 μL of a urine sample diluted 1:500 was injected on to the HPLC column (4.6 mm×150 mm, 5 μm Zorbax SB-Aq C-18; Agilent Technologies) for analysis. The mobile phase was 98:2 (v/v) 15 mmol L−1 pH 7.4 phosphate buffer–acetonitrile. The flow rate was 1 mL min−1, the temperature 25 °C, and the analytical wavelength 234 nm. Identification of the creatinine peak was based on comparison of retention time and diode-array spectra, acquired during analysis, with corresponding data obtained for the authentic compound. Peak heights were used for quantitation.
Results and discussion
Urine, plasma, and serum, the most commonly analyzed biofluids, present different analytical problems. Urine is usually free of proteins and lipids but contains very many components, the concentrations of which depend largely on diet, lifestyle, and ambient temperature. To facilitate comparison for different individuals the analytical results for urinary thiols can be normalized on creatinine. In common with most published procedures for total urinary thiols [5–11, 13–17], the proposed method depends on three essential steps–reduction of disulfide bonds, derivatization, and final separation and quantitation.
Reduction and derivatization
In general, aminothiols occur in urine in two forms; reduced thiols characterized by the presence of an −SH function, susceptible to oxidation and nucleophilic displacement reactions, and oxidized with disulfide bridges (−S−S−). The oxidized form encompasses symmetrical and mixed disulfides. During the reduction step disulfides are converted into their thiol derivatives with −SH functions accessible to thiol-specific derivatization reagents. For reduction we recommend sodium borohydride and for derivatization 2-chloro-1-methylquinolinium tetrafluoroborate (CMQT). Because of a strongly alkaline environment during reduction, homocysteine thiolactone is hydrolyzed to homocysteine. CMQT reacts with urinary thiols rapidly and quantitatively to form the 2-S-quinolinium derivatives of the thiols (Fig. 1). The CMQT reagent and its reaction with hydrophilic thiols have been described in detail elsewhere [20]. Briefly, the CMQT derivatization reaction leads to the rapid formation of 2-S-quinolinium derivatives in slightly alkaline aqueous solution. These stable thioethers have well-defined absorption maxima with high molar absorptivity at 350 nm.

Equation for derivatization of urinary thiols, represented by cysteinylglycine, with 2-chloro-1-methylquinolinium tetrafluoroborate
Chromatography of 2-S-quinolinium derivatives
To establish optimum HPLC conditions for separation of thiol-CMQT derivatives from each other, from excess derivatization reagent, and from unidentified urinary matrix components, several mobile phases of different composition, and pH, temperature, and flow-rate were tested to determine their effect on the peak heights, retention factors and resolution (details are not shown here). The chromatogram shown in Fig. 2 represents the final result of the optimization procedure and is indeed indicative of satisfactory distribution of the peaks throughout the chromatogram. Under optimum conditions, specified in the Experimental section, homocysteine, cysteine, and cysteinylglycine eluted after 2.89 (RSD 0.40%), 8.07 (RSD 0.17%), and 8.63 (RSD 0.15%) min, respectively.

Typical chromatogram obtained from endogenous thiols in human urine after reduction and then derivatization with CMQT. Peaks: 1, homocysteine 19.5 μmol L−1; 2, cysteine 174.8 μmol L−1; 3, cysteinylglycine 10.8 μmol L−1. Chromatographic conditions were as described in the Experimental section
Determination of creatinine
Because of fluctuation of the concentration of solutes, owing to variability in urine volume, it is reasonable to express analytical results relative to creatinine. Creatinine, an endogenous metabolite, is excreted by the kidney predominantly by filtration and in proportion to total excretion. Consequently, in this work each of 91 urine samples was subjected to HPLC analysis for creatinine. In the chromatographic procedure described in the Experimental section creatinine reached the detector after 2.83 (RSD 0.14%) min. Results are listed in Table 1.
Validation
Linearity and sensitivity
The linearity of relationships between thiol concentration and peak height of the respective analyte–CMQT derivative was determined by analysis of normal urine spiked with standard solutions of the thiols prepared as described in the Experimental section. Peak heights were plotted against analyte concentration and the curves were fitted by least-square linear regression analysis. Six-point standard plots showed the relationship between response and concentration was linear for cysteine, cysteinylglycine, and homocysteine within practical calibration ranges. The lower limit of detection (LLD) was determined for each analyte by analysis of aqueous samples containing low concentrations of the thiols. The lowest concentrations giving a signal-to-noise ratio of 3 was regarded as the LLD. Detailed method calibration data and lower limits of detection are listed in Table 2.
Precision and accuracy
Precision and recovery were studied by analysis of normal urine to which known amounts of target thiols had been added (Table 3). The system showed analytical recovery for cysteine, cysteinylglycine, and homocysteine was from 98.09 to 100.17% and intra-day imprecision (RSD, %) was from 2.57 to 6.41. Outliers were not excluded. For details see Table 3.
Application to analysis of real samples
When the analytical procedure had been validated it was applied to analysis of normal urine samples to measure total cysteine, cysteinylglycine, and homocysteine. The urine samples were donated by 91 apparently healthy volunteers (38 men and 53 women, 17–85 and 18–77 years old, respectively). Results from analysis of the target aminothiols are listed in Table 4. Values for urinary total cysteine, cysteinylglycine, and homocysteine were not significantly different for males and females (P≥0.05). To determine whether pH affects urinary excretion of the target aminothiols we examined the relationship between urinary concentration of particular thiols and pH. As shown in Fig. 3a–c there was no correlation between amounts of these thiols and urinary pH.

Relationship between urine pH and total concentration of (a) cysteine, (b) cysteinylglycine, and (c) homocysteine
Conclusion
We have developed a method enabling detection and quantitation of the three main urinary thiols cysteine, cysteinylglycine, and homocysteine in urine from normal humans. Reduction with a suitable compound, for example sodium borohydride, is essential for measurement of total thiols because these amino acids are mostly present as their disulfides, which are not accessible to the derivatization reagent. By use of this method we have determined normal ranges for total thiols in urine. Because of variation of the volume of urine excreted, the results are expressed as ratios to creatinine content. Our values for total cysteine and total homocysteine are consistent with the results of others [10, 14, 15]. Results for cysteinylglycine normalized on creatinine are not available in the literature. We found no correlation between pH and amount of any of the three target thiols in fresh morning urine. This observation is in accordance with the conclusions of other authors for urinary homocysteine [19]. Although we were not in possession of urine samples from patients suffering from disorders of sulfur amino acid metabolism, for example cystinosis, cystinuria, or homocystinuria, we believe this method would be useful in routine diagnosis of this diseases.
References
Meister A (1978) In: Sies H, Wendel A (eds) Function of glutathione in liver and kidney. Springer, Berlin Heidelberg New York, pp 43–59
Segal S, Thier SO (1995) The metabolic and molecular basis of inherited disease, 7th Edition. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) McGraw - Hill, New York
Rojkovich B, Nagy E, Prohle T, Poor G, Gergely P (1999) Clin Diagn Lab Immun 6:683–685
Hernanz A, Plaza A, MartinMola E, DeMiguel E (1999) Clin Biochem 32:65–70
Kagedal B, Kallberg M (1984) J Chromatogr 308:75–82
Stabler SP, Marcell PD, Podell ER, Allen R (1987) Anal Biochem 162:185–196
Birwe H, Hesse A (1991) Clin Chim Acta 31:33–42
Fiskerstrand T, Refsum H, Kvalheim G, Ueland PM (1993) Clin Chem 39:263–271
Wroński M (1996) J Chromatogr B 676:29–34
Pastore A, Massoud R, Motti C, Lo Russo A, Fucci G, Cortese C, Federici G (1998) Clin Chem 44:825–832
Kaniowska E, Chwatko G, Głowacki R, Kubalczyk P, Bald E (1998) J Chromatogr A 798:27–35
Zhao Ch, Zhang J, Song J (2001) Anal Biochem 297:170–176
Bald E, Głowacki R, Drzewoski J (2001) J Chromatogr A 913:319–329
Fermo I, Arcelloni C, Paroni R (2002) Anal Biochem 307:181–183
Lochman P, Adam T, Friedecky D, Hlidkova E, Skopkova Z (2003) Electrophoresis 24:1200–1207
Amarnath K, Amarnath V, Amarnath K, Valentine HL, Valentine WM (2003) Talanta 60:1229–1238
Proksch B, Jelesnianski S, Oberrauch W, Fux R, Gleiter ChH (2005) J Chromatogr B 828:122–125
Jakubowski H (2002) Anal Biochem 308:112–199
Chwatko G, Jakubowski H (2005) Clin Chem 51:408–415
Bald E, Głowacki R (2001) J Liq Chromatogr 24:1323–1339
Shi H, Ma Y, Ma Y (1995) Anal Chim Acta 312:79–83
Acknowledgements
The authors wish to thank the University of Lodz for financial support of this research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kuśmierek, K., Głowacki, R. & Bald, E. Analysis of urine for cysteine, cysteinylglycine, and homocysteine by high-performance liquid chromatography. Anal Bioanal Chem 385, 855–860 (2006). https://doi.org/10.1007/s00216-006-0454-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-006-0454-x