
Abstract
X-ray diffraction [crystal-truncation-rod (CTR)] studies of the surface structure of moisture-equilibrated hematite reveal sites for complexation not present on the bulk oxygen-terminated surface, and impose constraints on the types of inner-sphere sorption topologies. We have used this improved model of the hematite surface to analyze grazing-incidence EXAFS results for arsenate sorption on the c (0001) and r (10–12) surfaces measured in two electric vector polarizations. This work shows that the reconfiguration of the surface under moist conditions is responsible for an increased adsorption density of arsenate complexes on the (0001) surface relative to predicted ideal termination, and an abundance of “edge-sharing” bidentate complexes on both studied surfaces. We consider possible limitations on combining the methods due to differing surface sensitivities, and discuss further analysis possibilities using both methods.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The problem of determining sorption complex geometry: indirect and direct structural methods
When analyzing surface complexation it is crucial to determine the type of bonding interactions that hold the complex to the surface. The nature of the bonding affects the strength of the bond and the lability of the complex, and thus the probability of desorption and migration. Knowledge of the bonding also allows estimation of the possible density or sorption capacity of a given surface. Finally, the combination of bonding type and geometry dictate the stoichiometry of the complexation reaction, and this allows prediction of reaction products and estimation of kinetics [1].
However, determination of complexation bonding and geometry is limited by what might be called “indirect structural” methods, in other words those techniques that measure quantities related to structural details only indirectly, such as vibrational band frequencies or splitting, the energies of electronic spectral features, or chemical shifts in magnetic resonance spectra (direct structural techniques such as X-ray scattering and EXAFS determine bond lengths and coordination numbers explicitly). To determine the structural details of complexation, these methods must be standardized by comparing results with known compounds. However, even this can create ambiguities in interpretation, as vibrational states can be split not only by a coordination change or by distortion from a particular symmetry, but also by changes in protonation. Similarly, chemical shifts indicative of a certain coordination number can also be affected by the type of ligand, the spin state, or other structural parameters [2, 3].
Another way to estimate complexation bonding on an ambiguous surface is via complexation modeling and reaction stoichiometry. Both methods rely on assumptions about protonation and surface site types and concentrations [1, 4]. For example, if a surface is assumed to have attachment sites that are oxygens bonded to metal ions, and they are singly protonated (hydroxyl-terminated), then a complexation reaction that releases two protons will involve two surface oxygens. This result suggests a bidentate attachment site and an inner-sphere complex. However, for any given surface model there will be more than one type of bidentate complex geometry, and possibly very many. Similarly, complexation modeling can often be shown to achieve reasonable results from several different models of surface sites [5, 6]. Because of these limitations, one direct-structural method has been increasingly used to determine complexation topology: EXAFS. EXAFS has the advantage of being element-specific, so that it can be used to study a given complex in mixed chemical environments. It is also a local structure method, which means that it primarily affords bond distances and coordination numbers for the immediate neighbors and next nearest neighbors of a sorbed complex. A recent compendia of EXAFS results [7] demonstrates just how widespread EXAFS studies have become.
In our work we have used a surface-sensitive version of EXAFS, where the incident beam is applied to a single crystal sample at below the grazing-incidence angle for total external reflection. This method, GIXAFS, has improved sensitivity and the potential to extract more information than a standard EXAFS experiment [8].
Advantages of GIXAFS analysis
GIXAFS experiments (Figs. 1, 2) have three important advantages over bulk or powder EXAFS. First, as a well-characterized and prepared single crystal is used, the nature of the surface is better defined than with a powder experiment, where the grains may expose all types of surfaces to the sorption process. Second, the geometry allows for polarized experiments with the X-ray electric vector at any angle relative to the surface plane or a particular vector in the surface plane. For experiments where the propagation direction of the X-rays also matter (for example in measurements of XANES structure), this can also be varied, though not completely independently of the X-ray electric vector orientation. Third, the quality of the surface allows for near-complete reflection of the incident beam and very low bulk (and background) scattering. The superposition of incident and reflected fields can create an enhanced X-ray intensity (up to fourfold) at the surface, and when combined with the low background, can achieve much larger signal-to-noise ratios than are possible in powder experiments (typically 500×) [9]. Hence the method can provide structural information that is not averaged through all orientations, and do this with much greater sensitivity in the best cases. It is also useful to compare GIXAFS with surface EXAFS measured via electron-yield or “partial-yield” methods. In the latter experiments, the sample is kept under vacuum conditions to permit electrons to traverse the sample–detector distance. This produces high surface sensitivity, down to a single atomic layer, and depth selectivity when the energies of the detected electrons can be set independently. However, such experiments cannot be done with bulk water present, and typically have considerably less sensitivity than GIXAFS [10].

GIXAFS geometry. Top Cartoon showing addition of X-ray amplitudes for total external reflection. Middle Orientation of X-ray electric vector. Bottom Distribution of EXAFS photoelectron amplitude in S→P transition (K edge)

GIXAFS instrument at beamline 11-2 SSRL. Top 30 element detector position. Middle Sample cell with hematite sample. Bottom Arrangement of instrument in hutch. X-ray beam enters the square slits at the front right of the apparatus
In typical GIXAFS experiments, data is collected with the X-ray electric vector in the plane of the sample and normal to the plane. If the symmetry of the sample surface is lower than threefold, then two orthogonal measurements in the plane of the sample can be performed to yield additional independent data. Finally, measurements can be made at several angles between in-plane and normal. These measurements do not supply independent data, but are useful for unraveling the detailed spectral changes for the fitting process. A similar method has been devised for the EXAFS analysis of highly textured powder samples, and is termed P-EXAFS [11].
GIXAFS has been extremely useful in probing particular surface complexation reactions [8], but analysis using it proceeds under two important assumptions. First, the surface is ideal and represented by the bulk oxygen terminated structure in the orientation required. Second, complexation on this surface results in no surface relaxation or other adjustments. These assumptions are still far fewer than those needed to analyze powder EXAFS spectra, but they present a potentially serious limitation in their own right. We now believe that the combination of GIXAFS with the results from crystal truncation rod (CTR) surface diffraction can eliminate the need for either assumption in the analysis.
CTR surface diffraction
CTR surface diffraction is a remarkably straightforward technique for determining surface structure. The technique is based on the interaction of the diffraction effects from a terminated surface with those from the bulk of the sample, creating significant diffracted intensity between the Bragg peak positions in the direction normal to the surface termination [12]. These scattering regions are sensitive to the deviation of the surface structure from the ideal bulk structure, and to the presence of sorbed species. Figure 3 shows how a model calculation of a part of a CTR responds to changes in surface layer position and occupation. A significant change in phase occurs with a spacing change on the order of fractions of the layer spacing. Similarly, occupations of a surface layer are detectable at the 5% level. When we can collect CTR patterns for a range of nonequivalent orientations in diffraction space (data collected along different truncation rods), a great deal of information can be obtained about surface atom positions near the surface, allowing positions to be defined in three dimensions, and the site occupancy and disorder to be evaluated.

Model CTR calculation. After [31]. Curves show the effect of a change in surface lattice spacing of 40% (z=1.4c) which produces a phase change in the CTR, and the effect of an occupation change of 30% (f s/f uc=1.0:0.7) which only produces an intensity variation. Bottom idealized expression for CTR amplitude. F uc=structure factor due to substrate unit cell, F s= structure factor due to surface structure
The CTR analysis became a practical tool due to the advent of synchrotron radiation sources. It is clear from the model calculation shown in Fig. 3 and from the data shown in Fig. 4 that the intensity measured over the CTR scan can range over five or more orders of magnitude. Hence extremely high incident X-ray intensities are necessary to measure the signal in the weakest scattering parts of a rod scan precisely.

Fits to CTR data for a wet hematite (0001) surface. Black curve termination of bulk structure with double Fe octahedral layers. Green curve: termination by 100% occupied single Fe octahedral layer. Red curve: termination by 30% occupied single Fe octahedral layer. (hkl) designations refer to the direction in reciprocal space scanned to collect the rod, e.g. the (00L) scan is from (000) to (00 15)
CTR analysis has traditionally been applied to metal and semiconductor surfaces equilibrated in vacuum, but in the last few years they have been used by our group and others on surfaces equilibrated with aqueous solutions [13–15] (“wet” surfaces). Particularly noteworthy are the differences between the vacuum- and water solution-equilibrated surfaces of corundum and hematite, and the deviations of either of these types of surfaces from a hypothetical bulk-terminated model.
In the corundum (α-Al2O3) (0001) surface, the wet structure is oxygen-terminated, as opposed to the vacuum-equilibrated Al-metal termination. A relaxation of the metal ions in the two layers below the surface oxygens also occurs on the wet surface, and this appears to be in the direction of the gibbsite (α-Al(OH)3) bulk structure. Another feature of the (0001) surface is the existence of an ordered overlayer of water molecules above the surface termination oxygens.
Another CTR-type analysis has been done on a set of mineral surfaces by utilizing the single CTR of the specular reflection orientation, in other words the (001) rod [16]. These single rod scans are usually called “reflectivity” scans, and afford information on the z-position and occupation of surface species, but are insensitive to x, y changes in the atomic positions. However, in many cases, the specular rod will contain the most information (in terms of variations from a simple bulk model with ideal termination) of any rod scan. Hence reflectivity scans can provide significant information about surface structure.
Equilibrium wet hematite surfaces
The wet hematite (0001) surface shows a dramatic difference from the nominally isostructural α-Al2O3 (0001) surface [14]. The main difference is the depopulation of the near-surface metal layers. Fits to the CTR data are shown in Fig. 4. The uppermost Fe layer is completely missing, and the second Fe layer has an occupation of about 30%. As this result is an average over the full surface there are two interpretations: a statistical occupation of the second Fe layer, or domains (“islands”) of full occupany of the second layer (Fig. 5 shows both a full second layer and one at 30% occupancy). The missing uppermost Fe layer represents Fe ions whose coordination polyhedra share faces with the next lowest Fe layer. Hence, using traditional crystal-chemical rules (Pauling [17–19]), these Fe site should have relatively high energy, and would be expected to be depleted at a free surface. The next lowest layer does not share such polyhedra elements with still lower Fe polyhedra, and thus the occupation must be controlled by other factors.

Cartoon of the CTR-derived wet hematite (0001) surface structure. Top 100% occupied single Fe octahedral layer. Bottom CTR predicted result—30% occupied single Fe octahedral layer
The key feature of this surface is the relative number of inner-sphere complexation sites that are available relative to a surface with the wet equilibrated α-Al2O3 (0001) surface structure. This is shown via possible complexation geometries for arsenate attachment in Fig. 6. The fully occupied double Fe layer surface has no available sites for arsenate sorption, except as outer-sphere complexes, or at defect sites, such as at growth step edges. This is because all surface oxygens are bonded to two Fe atoms (and likely protonated), and cannot tolerate additional bonding to As5+. In contrast, the CTR-determined surface can have several types of monodentate, bidentate and tridentate attachment sites away from any step edges, allowing for a high possible concentration of sorbates.

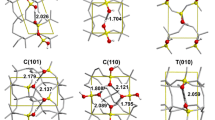
Possible arsenate complexation geometries on the wet hematite (0001) surface. Top from left to right, tridentate corner-sharing complex, edge-bicorner tridentate complex, monodentate complex and edge-sharing bidentate complex. Bottom appearance of same complexes in plan view of surface
The situation is somewhat different on the hematite r (10–12) surface. On this surface the uppermost Fe polyhedra do not share faces with the next lower Fe layer, and hence CTR analysis does not predict a depopulation of this layer. Hence we can approach analysis of the sorption of this surface by using a bulk oxygen-terminated structure (Fig. 7). The main feature of this surface are rows of two different orientations of FeO6 octahedra whose apical oxygens point outward from the (10–12) surface. Figure 7 shows several possible sorption positions for arsenate. By bridging the apical oxygens, arsenate can form inner-sphere bidentate complexes on the top of the surface layers, but there are no free Fe polyhedral edges for the formation of edge-sharing complexes. In contrast, along the borders of layers there are abundant free polyhedral edges for edge-sharing complexes. In about half of these sites the As-Fe interatomic vector would be almost in the surface plane, producing high polarization sensitivity in the GIXAFS experiment. In general, we expect hematite surfaces to be depopulated in Fe that would ordinarily inhabit a polyhedron that is face-sharing with a deeper polyhedron. For nanoparticulate hematite it is thus likely that a significant change in structure could result from this effect.

Possible arsenate complexation geometries for the hematite (10–12) wet surface. Top from top, edge-sharing complex at edge of layer, corner-corner sharing complex between Fe octahedral rows, edge-sharing complex at edge of layer, corner-corner sharing complex between octahedral rows. Bottom profile view of (10–12) surface consistent with CTR studies. Two of the complexation sites are shown
GIXAFS arsenate analysis
Grazing incidence EXAFS experiments were performed at SSRL beamline 11-2 using an apparatus dedicated to GIXAFS analysis [20]. The instrument consists of a carriage that holds the sample crystal stage, with motor drives for positioning the sample over 5 degrees of freedom (sample x, y, z translation and rotation angles θ and φ), and complete X-ray optics including several sets of collimator slits. This entire assembly is mounted so that it can be rotated about the incident X-ray beam (χ) to set the angle of the X-ray electric vector polarization plane with respect to the sample surface. Ionization chamber detectors are mounted both before and after the sample within the apparatus for transmission absorption data collection, and a large independently mounted multielement Ge detector can be positioned over the sample to collect fluorescence signals (Fig. 2).
The measurements are made by slitting down the incidence beam height in the sample z direction so that at the critical angle for X-ray total reflection (about 0.21° for arsenic K-edge [11.6 KeV] on hematite) the incident beam just covers the entire length of the sample. For a hematite sample of 2.5 cm in length, this width is about 85 microns. Excess beam width is not useful, as it strikes the front of the sample creating background scattering, or scatters off other parts of the apparatus at the rear of the sample. The incidence beam angle is selected to be 0.03° below the critical angle for total external X-ray reflection so that a slight decrease in this angle as a function of energy during an EXAFS scan will have negligible effect on the sample beam penetration [4]. Any situation which creates an incidence angle at or above the critical angle on any part of the sample, such as a sample curvature or a focused beam divergence irregularity, can produce strong Bragg diffraction. This oversaturates the detector electronics and produces severe “glitches” in GIXAFS spectra which often cannot be removed during analysis.
For our purpose we used “epi-quality” polished natural hematite single crystal samples that were equilibrated with 10−4 M sodium arsenate solutions at pH 5.0 for 2 h [14]. Sorption density was determined by using the fluorescence detector to measure the As signal at 10° incidence and 12.5 keV X-ray energy, and then comparing to a standard As sample (arsenate solution dried on a filter paper disc). All of the arsenate in the standard was presumed to contribute to the fluorescence signal. This measurement indicated that both samples had ~10% monolayer coverage of sorbed arsenate, corresponding to a surface density of ~0.6 μmol/m2. Measurements at grazing incidence were made at both 0 and 90° χ angle (electric vector in plane and normal to sample plane, respectively) and required an average of 12 EXAFS scans to achieve suitable signal/noise. During the experiment the surface of the crystals was kept moist by maintaining a constant flow of water-saturated He over the samples. No change in spectra was observed between the first EXAFS scan and the last scan over a period of about 3.5 h.
Results
C plane (0001) surface
The averaged EXAFS spectra and Fourier transforms are shown in Figs. 8 and 9. The in-plane χ=0° electric vector transform shows a large peak due to four first nearest neighbor oxygens about the arsenic, and a single small second shell peak due to next nearest neighbor Fe atoms. Fitting trials to the averaged raw data using EXAFSPACK [21] and SIXPACK [22] software from varied starting estimates yielded the same result: 4.2 (0.2) O neighbors at 1.69 (0.005) Å, 1.5 (0.2) Fe neighbors at 2.88 (0.02) Å and 0.75 (0.2) Fe neighbors at 3.25 (0.02) Å. Single As–Fe shells could not be fit acceptably. The As–Fe distances can immediately be assigned to edge-sharing bidentate and corner sharing bidentate or tridentate inner sphere complexation, respectively, based on the geometry of the CTR-refined (0001) surface and past analyses [23]. For the same sample with data collected with the e-vector normal to the sample surface plane (χ=90°), the transform shows a near-identical second peak due to As–Fe second neighbors, and a third peak due to As–Fe distances deeper into the sample surface. Refined fitting yields 3.9 (0.2) O neighbors about the As atom, 0.7 (0.1) Fe neighbors at 2.85 (0.02) Å, 0.87 (0.2) Fe neighbors at 3.26 (0.02) Å and 1.2 (0.3) Fe neighbors at 4.09 (0.05) Å. The As–Fe second neighbor distances agree with the same complexation geometry as in the χ=0° case. The change in the relative number of complexes with polarization orientation can be used to deduce the angle of the electric vector with the interatomic As–Fe vector direction, φ. The observed coordination numbers for a polarized EXAFS measurement are related to the isotropic (powder) EXAFS coordination numbers by: CNpol= CNiso.3cos2 φ. For the edge-sharing complexes we observe half as many second neighbors at χ=90° as at χ=0°, and this is consistent with φ=45°. For the bidentate complexes, φ appears to be larger at 57°. Using these values, the proportion of edge complexes is 71%. Allowing for the refinement errors in CN this value is 71(±9)%. Other systematic errors, which we cannot account for at this time, are differences in the Debye-Waller factors for the two complexes, and scaling factors related to fluorescence self-absorption (which should be small compared to other uncertainties). Fitting results are shown in Table 1.

GIXAFS raw data. Top 15 scan average for the GIXAFS data for (0001) surface at χ=0°. Quality equivalent to concentrated model compound scan. Bottom EXAFS extracted from GIXAFS data. Useable to k=15

GIXAFS analysis results for the hematite (0001)/arsenate sorption geometry. Top left Fourier transform (pair correlation function) showing two scattering vector orientations. Green e-vector normal to surface. Red e-vector along surface. Bottom Fit to extracted filtered (back-transformed) EXAFS from first and second shells. Top right Geometry of corner-sharing bidentate complex from fitting two As–Fe neighbors at about 3.25 Å and a single As–Fe neighbor at about 4 Å in the lower Fe partial layer. Fitting results listed in Table 1
The appearance of the longer As–Fe distance can be readily assigned to Fe atoms residing below the corner-sharing complexes (see structure diagram in Fig. 9). The As–Fe scattering vector would be nearly collinear with the electric vector at χ=90°, and thus would be strongly enhanced in the EXAFS. The anticipated As–Fe distance of about 3.95 Å is in good agreement with the observed distance within uncertainty. The predicted CN for these Fe neighbors based on the polarization dependence is 0.29×3=0.87, which is also in good agreement with the fitting results, within uncertainty. The edge-sharing complexes reside above empty Fe positions in the layer below and would contribute less scattering at a longer distance, for example 4.5–4.8 Å, to other Fe in that layer. We do observe an additional small peak in the transform in this region, which has the correct magnitude. However fitting this small peak requires both As–Fe and As–O phase contributions and is thus much more uncertain. Another interpretation is that the edge complexes are preferentially found binding to Fe polyhedra at the edges of crystallographic steps. In this case the nearest underlying Fe atoms may be much more distant. Summarizing, the results show that the corner-sharing complexes must reside within partially occupied Fe layers on the hematite surface, indicating that the model for this surface cannot assume only a step-ledge type disorder. The appearance of a second possible As–Fe longer distance shell confirms that most of the edge-sharing complexes also reside on a partially occupied Fe layer. It is also interesting that edge-sharing complexes greatly outnumber corner-sharing complexes, when this has not been observed in other related powder EXAFS studies (such as for ferrihydrite [23, 24]). The simplest explanation for this is the larger number of possible edge attachment sites if the topmost Fe octahedrons are relatively dispersed on the surface, and are not present as large densely populated islands. In the case of maximum dispersion, for N Fe atoms in the layer, there are 3N edge attachment positions, but only N double-corner attachment positions on average (and very few triple-corner attachment positions). Assuming that the energetics of sorption are similar and hence that the relative types of arsenate complexes are controlled by the availability of attachment sites, the observed 3:1 ratio of edge to corner sites is expected.
(10–12) surface
The GIXAFS spectra for this surface are more complex than on the c plane surface, and show more dramatic differences between electric vector polarization directions (Fig. 10). In particular, the first neighbor As–Fe shells change considerably, indicating that different types of complexes are preferentially observed at the two observation angles. For χ=0°, we observe an As–Fe peak at about 2.3 Å in the raw transform, with a weaker shoulder at 2.75 Å, while for χ=90° the 2.3 Å peak has disappeared and the 2.75 Å peak is much increased. With fitting, these peaks in the χ=0° spectra correspond to complexes having As–Fe distances of 2.75 with a CN of 1.4, and 3.26 Å with a CN of 0.8, respectively, equivalent to the edge and corner-sharing complexes we have already identified on the (0001) surface. For χ=90°, refinement gives only a single contribution of 2.2 Fe atoms at 3.24 Å. These distances are consistent with the corner and edge bidentate complexes observed on the (0001) surface, with 78(±10)% being the edge type. The loss of any contribution from the edge-sharing complexes in the χ=90° orientation indicates that these complexes have an As–Fe interatomic vector that is nearly in the plane of the surface. The observed coordination number for the corner sharing complexes in the χ=90° polarization of 2.2 is also in good agreement with the expected CN (2.0) if these complexes are “pointing out” from the surface above the rows of FeO6 octahedra predicted by the CTR measurements.

GIXAFS analysis results for the hematite (10–12)/arsenate sorption geometry. Top and center Back transform of second shell region of pair correlation function (As-X distances of from 2 to 3.5 Å), and theoretical fitting (solid lines). Top surface plane e-vector. The strong beat indicates two different As–Fe distances corresponding to edge- and corner-sharing complexes. Center surface normal e-vector. The single frequency indicates contributions from just one As–Fe distance, that of corner-sharing complexes. Fitting results listed in Table 1 Bottom Fourier transform (pair correlation function) showing two scattering vector orientations. Green e-vector normal to surface. Red e-vector along surface <01–10> direction.
The smaller peak seen in the Fourier transform of the χ=90° data at about 4.0 Å is consistent with an As–Fe distance of about 4.3 Å, and appears to be due to a summation of backscattering contributions from both the edge and corner-sharing complexes with Fe atoms deeper in the hematite surface. Further work with additional polarization directions is needed to clarify this contribution. Complete results from fitting all spectra are compiled in Table 1.
Sampling differences between GIXAFS and CTR analysis
The most important consideration in coupled GIXAFS-CTR analysis is the relative sensitivity to surface disorder. The CTR pattern arises because the periodic scattering function from the bulk interferes with the near-surface scattering. If the surface scattering is affected by a disorder of some type, the CTR intensities will be weakened or removed entirely (only bulk diffraction effects will be observed). For example, if the uppermost Fe ions in the hematite (0001) surface were positionally disordered by a sufficient amount, they would essentially make no contribution to the CTR. This means that if a surface has disordered regions, the CTR that is observed only arises from the remaining ordered regions, and this bias must be understood.
It can be readily shown [25–27] that surface roughness due to a poor polish can greatly affect CTR intensity. This can have an experimental advantage inasmuch as large scratches (optically visible) on the surface produce regions with no CTR contribution, and these do not affect CTR interpretation of the more regular surface regions. These considerations contrast somewhat with GIXAFS analysis, where all surface species within 2.5 nm or so of the surface contribute to the measured signal, as this is the X-ray penetration depth below the critical angle for total external reflection. Hence surface roughness at the 2.5 nm level, which would reduce CTR intensity considerably, would not inhibit a GIXAFS measurement. However, such an irregular surface would create other problems with GIXAFS. Reflectivity would be low and the sensitivity of the GIXAFS experiment would drop by up to 75% compared to an optimal surface. Other types of surface sites might also be present on an irregular surface, thus making the interpretation of the GIXAFS difficult. Finally, a “shadowing effect” due to beam transit through a larger amount of surface material will reduce the fluorescence intensity still further.
It is possible to consider the effects of 0.5, 2.5 and 20 nm rms surface roughness in both experiments in a semi-quantitative fashion. The 0.5 nm level of roughness is consistent with steps on the hematite (0001) surface that are one unit cell in height (1.5 nm), covering about a third of the surface. This type of surface would be consistent with the highest quality now attainable with “epi-quality” chemical-mechanical polishing. If the steps are regular (the termination is in the unit cell size regime), or on some crystallographically regular aspect of the subcell, then the fitting of the CTR needs only to include those termination alternatives. In the ROD program for fitting CTR intensities [28, 29] we include surface terminations for each alternative, reflecting the lower symmetry of the average surface cell. What remains are irregularities in the surface from displaced or missing atoms, impurity atoms, thermal disorder, and disorder due to defects such as stacking faults and screw dislocations. It is difficult to separate out the effects from each of these sources, so as an approximation the disorder is handled by means of an occupancy factor β, where the surface roughness σ rms is given by: σ rms=β 1/2 d/(1−β) [24]. Here d is the surface lattice spacing. As the CTR intensity between Bragg peaks scales roughly as (1−β)2 [24, 29], for roughness on the order of several lattice spacings there is considerable diminution of the CTR intensity. For our case, a roughness of 0.5 nm on the hematite (1000) surface, equivalent to the best surface now readily achievable, β is equal to about 0.1 and (1−β)2 is about 0.81. This value of roughness will allow good sensitivity to surface structure. At a roughness of 2.5 nm, β is equal to about 0.6 and (1−β)2 is about 0.16, which is significant. “Optical quality” polishes would produce β values of 0.95 or more, and (1−β)2 factors below 0.0025, so CTR intensity would be negligible.
For the GIXAFS experiment, increased surface roughness degrades the reflectivity and the measured amplitude of the XAS signal, but neglecting fluorescence self-absorption effects there is no other effect on the amplitude of the measured XAS function, and no alteration in the phase of the function. What is significant is the relative sensitivity between highly perfect areas, where reflectivity boosts the fluorescence signal by as much as fourfold, and very rough areas (such as those with only an optical polish), where the fluorescence intensity would only be that due to the incident beam. As the rough areas may have a larger variation in type of surface complex, we end up with decreased contributions from these variant sites. Hence in GIXAFS, as with CTR measurements, we will be preferentially probing the highest quality parts of the sample surfaces. To gauge the effect experimentally, GIXAFS spectra could be obtained at various angles of incidence. However, this is not usually attempted as the experiment is performed because of the need to maximize sensitivity. For the present then, it is necessary to prepare surfaces of the highest quality for both techniques.
CTR analysis differs from GIXAFS analysis in another important way. The CTR intensity is a measure of the occupations and positions of ordered species on a surface, in the sense of Bragg-Williams site-specific ordering. GIXAFS, in contrast, is a local measurement that does not require site-specific widespread order. To emphasize this difference, consider a highly damaged hematite surface with sorbed arsenate. Suppose that all of the complexes bond to the edges of a Fe octahedron, but that the relative positions of the arsenates are highly varied due to the surface disorder. In this case the arsenate complexes may well not make much contribution to any of the CTRs, as the range of x, y, z positions would be too large. However, the interatomic As–Fe vector distance would be similar in all cases, and the GIXAFS measurement would still provide useful (though not long-range) results. This difference in sensitivity to disorder is manifested in the type of Debye-Waller (DW) term used in theoretical expressions for the two experiments. In GIXAFS (as in EXAFS), the DW term uses a factor with the interatomic backscatterer vectors contained in the exponent, but no lattice or surface cell vectors. In the CTR intensity expression, the unit cell and/or surface cell periodicity are used in the exponential term, but no interatomic vectors. This means that in particular cases the two methods can be expected to yield different results depending on the precise nature of the surface disorder. Hence this is another reason for utilizing only the highest quality substrates when comparing or combining the methods. A more rigorously quantitative analysis of these issues is beyond the scope of this paper, and readers are referred to the discussions in references [25], [27], [30] and [31] for further perspectives. A more thorough work-up of all considerations mentioned herein specific to the arsenate/hematite system and types of possible surface disorder will be published elsewhere.
Future work: refinement of CTR data with sorbates present
The most direct complementarity between the CTR and GIXAFS techniques is achieved if the CTR analysis can be performed on substrates with the sorbed species. Our group is beginning such measurements, and Fig. 11 shows the effect on two CTRs of arsenate sorption on the hematite (0001) surface. Related work is also in progress to determine the effects of varied pH and ionic strengths on near-surface structures, independent of sorption. It is clear from Fig. 11 that large changes occur on the measured CTRs over a single sorption period. Other work we have done shows that additional sorption periods result in continued changes of the same type, suggesting that the CTR variations are proportional directly to sorption density rather than to other changes in the substrate surface. CTR measurements of hematite surfaces with sorbed selenate, silicate and uranyl have also been performed, and are being analyzed at this time. Competitive sorption studies are also planned. The most important need for CTR-refinement of the surfaces with sorbed complexes is to resolve the effects due to a surface relaxation instigated by binding with the sorbate. That this may occur is obvious from considering the bond-valence sums for surface oxygens that would be involved in complexation. With no attached arsenate, but assuming protonation, a surface oxygen in the partial second FeO6 layer on hematite would have a bond valence of 3/6 (trivalent Fe divided by CN 6)+0.83 (typical contribution from single proton bond)=1.33. The oxygen is thus underbonded and can either form additional hydrogen bonds with nearby water molecules, or will be pulled closer to the Fe atom to gain bond strength. If arsenate is bound to this oxygen in complexation, the bond valence will be 2.58, indicating considerable overbonding. This is likely to extend the Fe–O bond and move the complex further from the hematite surface. Hence CTR refinement of a complexed surface will need to include not only addition of scattering due to surface complexes, but changes in scattering phases due to relocated oxygens (and possibly Fe atoms) in the substrate surface. Molecular dynamics simulations of complexation may be helpful for bootstrapping the fitting of the CTR data in such cases.

CTR data showing the effects of arsenate sorption under reaction conditions similar to those in the GIXAFS experiment. Arrows show regions in diffraction space where the CTR has changed significantly
Conclusions
-
1.
Combined use of CTR and GIXAFS measurements have enabled quantitative identification of two types of dominant arsenate compexes and their orientations on hematite (0001) and (10–12) surfaces. These results could not have been obtained with powder EXAFS analysis, or via indirect structural methods.
-
2.
The main complexation topology is that of an edge-sharing complex, in contrast to most arsenate complexation observed with other iron oxyhydroxides. This appears to be due to the abundance of isolated FeO6 octahedral edges among the surface species, and a relative paucity of adjacent corner sites for bidentate binuclear complexation, especially on the (0001) surface.
-
3.
The particular placement of complexes on the (10–12) surface is largely dictated by rows of FeO6 polyhedra. No depopulation of the Fe surface layers occurs as no surface FeO6 shares a polyhedral face with a deeper FeO6. However, in general we expect depopulation of surface Fe sites with respect to bulk structure wherever polyhedral face sharing would be present.
-
4.
The CTR measurements are unable by themselves to distinguish between differing models for the hematite (0001) surface, such as a single layer of Fe with random occupation, or a single layer with “islands” of full occupation on an otherwise depleted layer. However, the GIXAFS results are consistent with the proposed CTR model, and indicate a high proportion of free edge sites, as would be found with random partial Fe occupation of the surface layer, or perhaps small irregular clustering of the top Fe-octahedra. This finding indicates the complementarity of the two techniques.
References
Davis JA, Kent DB (1990) Surface complexation modeling in aqueous geochemistry. Rev Mineral 23:177–260
Waychunas GA, Brown GE, Ponader CW, Jackson WE (1988) Evidence from X-ray absorption for network-forming Fe2+ in molten alkali silicates. Nature 332:251–253
Farges F, Brown GE, Rehr JJ (1997) Ti K-edge XANES studies of Ti coordination and disorder in oxide compounds: comparison between theory and experiment. Phys Rev B 56:1809–1819
Sposito G (1986) Distinguishing adsorption from surface precipitation. ACS Symp Ser 323:217–229
Arai Y, Sparks DL, Davis JA (2004) Effects of dissolved carbonate on arsenate adsorption and surface speciation at the hematite-water interface. Environ Sci Tech 38:817–824
Hayes KF, Redden G, Ela W, Leckie JO (1991) Surface complexation models: an evaluation of model parameter estimation using FITEGL and oxide mineral titration data. J Colloid Interf Sci 142:448–469
Brown GE, Sturchio NC (2002) Overview of synchrotron radiation applications to low temperature geochemistry and environmental science. Rev Mineral Geochem 49:1–115
Waychunas GA (2002) Grazing-incidence X-ray absorption and emission spectroscopy. Rev Mineral Geochem 49:267–316
Waychunas GA, Brown GE (1994) Fluorescence yield XANES and EXAFS experiments: application to highly dilute and surface samples. Adv X Ray Anal 37:607–617
Stohr J (1992) NEXAFS spectroscopy (Springer Series in Surface Science, vol 25). Springer, Berlin Heidelberg New York
Manceau A, Marcus MA, Tamura N (2002) Quantitative speciation of heavy metals in soils and sediments by synchrotron X-ray techniques. Rev Mineral Geochem 49:341–428
Robinson IK, Tweet DJ (1992) Surface X-ray diffraction. Rep Prog Phys 55:599–651
Eng PJ, Trainor TP, Brown GE, Waychunas GA, Newville M, Sutton SR, Rivers ML (2000) Structure of the hydrated α-Al2O3 (0001) surface. Science 288:1029–1033
Trainor T, Eng P, Brown GE, Waychunas G, Newville M, Sutton S, Rivers M (2004) Structure and reactivity of the hydrated hematite (0001) surface. Surf Sci 573:204–224
Trainor TP, Eng P, Brown GE, Robinson IK, DeSantis M (2002) Crystal truncation rod diffraction study of the clean and hydrated α-Al2O3 (1–102) surface. Surf Sci 496:238–250
Fenter P, Teng H, Geissbuhler P, Hanchar JM, Nagy KL, Sturchio NC (2000) Atomic-scale structure of the orthoclase (001)-water interface measured with high-resolution X-ray reflectivity. Geochem Cosmochim Acta 64:3663–3673
Pauling L (1960) The nature of the chemical bond, 3rd edn. Cornell University Press, Ithaca, NY
Pauling L (1929) The principles determining the structure of complex ionic crystals. J Am Chem Soc 51:1010–1026
Burdett JK, McLarnan TJ (1984) An orbital interpretation of Pauling’s rules. Am Mineral 69:601–621
Rogers JH, Bargar JR, Waychunas GA, Yoon TH, Brown GE (2004) A novel spectrometer system for hard X-ray interfacial environmental chemistry. Am Inst Phys Conf Proc 708:981–984
George GN (2001) EXAFSPAK homepage. See http://www-ssrl.slac.stanford.edu/exafspak.html, last accessed August 2005
Webb S (2004) Sam’s Interface for XAS Package (SixPACK) homepage. See http://www-ssrl.slac.stanford.edu/~swebb/sixpack.htm, last accessed August 2005
Waychunas GA, Rea BA, Fuller CC, Davis JA (1993) Surface chemistry of ferrihydrite: Part 1. EXAFS studies of the geometry of coprecipitated and adsorbed arsenate. Geochim Cosmochim Acta 57:2251–2269
Waychunas GA, Fuller CC, Rea BA, Davis JA (1996) Wide angle X-ray scattering (WAXS) study of “two-line” ferrihydrite structure: Effect of arsenate sorption and counterion variation and comparison with EXAFS results. Geochim Cosmochim Acta 60:1765–1781
Robinson IK (1986) Crystal truncation rods and surface roughness. Phys Rev B 33:3830–3836
Dosch H (1992) Critical phenomena at surfaces and interfaces. Springer Tracts Mod Phys 126:1–145
Sinha SK (1996) Surface roughness by X-ray and neutron scattering methods. Acta Phys Polonica A 89:219–234
Vlieg E (2000) ROD: A program for surface X-ray crystallography. J Appl Cryst 33:401–405
Trainor TP, Eng IK, Robinson IK (2002) Calculation of crystal truncation rod structure factors for arbitrary rational surface terminations. J Appl Cryst 35:696–701
Als-Nielsen J, McMorrow D (2001) Elements of modern X-ray physics. Wiley, New York
Fenter PA (2002) X-ray reflectivity as a probe of mineral-fluid interfaces: a users guide. Rev Mineral Geochem 49:149–220
Acknowledgements
We are grateful to Corwin Booth for helpful discussions regarding GIXAFS and CTR disorder effects. Use of the APS and SSRL was supported by the US Department of Energy, Basic Energy Sciences, Office of Energy Research, under contracts W-31-109-Eng-38 and DE-AC03-76SF00515, respectively. GW is supported by the Office of Basic Energy Sciences through the division of Chemical Sciences, Geosciences and Biosciences through the Lawrence Berkeley National Laboratory. We also acknowledge grants BES-0404400 (TT, PE and GB), and NSF CHE-0431425 (GB and TT).
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00216-006-0922-3
Rights and permissions
About this article
Cite this article
Waychunas, G., Trainor, T., Eng, P. et al. Surface complexation studied via combined grazing-incidence EXAFS and surface diffraction: arsenate on hematite (0001) and (10–12). Anal Bioanal Chem 383, 12–27 (2005). https://doi.org/10.1007/s00216-005-3393-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-005-3393-z