
Abstract
This work describes the development of a method to determine cadmium in coal, in which iridium is used as a permanent chemical modifier and calibration is performed against aqueous standards by high-resolution continuum source atomic absorption spectrometry (HR-CS AAS). This new instrumental concept makes the whole spectral environment in the vicinity of the analytical line accessible, providing a lot more data than just the change in absorbance over time available from conventional instruments. The application of Ir (400 μg) as a permanent chemical modifier, thermally deposited on the pyrolytic graphite platform surface, allowed pyrolysis temperatures of 700 °C to be used, which was sufficiently high to significantly reduce the continuous background that occurred before the analyte signal at pyrolysis temperatures <700 °C. Structured background absorption also occurred after the analyte signal when atomization temperatures of >1600 °C were used, which arose from the electron-excitation spectrum (with rotational fine structure) of a diatomic molecule. Under optimized conditions (pyrolysis at 700 °C and atomization at 1500 °C), interference-free determination of cadmium in seven certified coal reference materials and two real samples was achieved by direct solid sampling and calibrating against aqueous standards, resulting in good agreement with the certified values (where available) at the 95% confidence level. A characteristic mass of 0.4 pg and a detection limit of 2 ng g−1, calculated for a sample mass of 1.0 mg coal, was obtained. A precision (expressed as the relative standard deviation, RSD) of <10% was typically obtained when coal samples in the mass range 0.6–1.2 mg were analyzed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The conventional method of solvating coal includes a high-temperature ashing step in a vented furnace, followed by acid digestion of the resulting ash using a mixture of concentrated hydrofluoric, nitric and sulfuric acids [1]. This procedure is not only time-consuming, but also risks the loss of volatile trace elements during the ashing stage and contamination from reagents and sample handling. Microwave-assisted acid digestion can speed up the second part of the process, but it still requires prior ashing of the coal [2], unless perchloric acid is added [3–5], and/or excessively long heating times are used. Alkaline fusion with lithium metaborate has also been proposed to solvate coal; however, the relatively high dilution involved in this process and the resulting matrix are not really suited for trace element determination [6].
The direct analysis of solid samples using graphite furnace atomic absorption spectrometry (GF AAS) has a number of advantages over solution analysis in cases where the samples are difficult to digest, such as: (i) a significantly reduced risk of contamination and loss of analyte; (ii) an increased sensitivity, as samples are not diluted; (iii) the use of expensive and/or hazardous reagents is not required, resulting in both economic and environmental benefits; (iv) results are obtained more rapidly [7, 8]. Obviously, direct solid sampling also has some limitations, and the most frequently mentioned ones are: (i) sample introduction is more difficult; (ii) calibration is more complex; (iii) the precision is inferior to solution analysis. However, first, with state-of-the-art instruments, equipment is available that greatly facilitates the introduction of solid samples [9]; second, it has been demonstrated that aqueous standards can often be used in solid sample analysis when the conditions are optimized [10–14]; third, a relative standard deviation (RSD) of 5–10%, typical of direct solid sampling, is considered much more acceptable for the analysis of a natural sample with an inherent inhomogeneity, such as coal, than a wrong result due to contamination or analyte loss during sample preparation.
Chemical modifiers are routinely used in GF AAS as part of the stabilized temperature platform furnace (STPF) concept [15]. The platinum group metals, mainly Pt, Rh, Ru and Ir, have been used as modifiers (either alone or mixed) for the determination of several volatile elements, such as As, Bi, Se, Hg, Cd, Ge, Sn, Sb and Pb [16–19]. Belarra et al [20] investigated Pd as a modifier for the determination of cadmium in sewage sludge using solid sampling analysis. Da Silva et al [21] used ruthenium as a permanent modifier for the determination of cadmium in biological samples solubilized with tetramethylammonium hydroxide. Piascik and Bulska [22] demonstrated that all electrodeposited modifiers could stabilize cadmium up to 800 °C in the presence of HNO3 and aqua regia. Vale et al [10] have shown that permanent chemical modifiers can in fact be used for direct solid sampling GF AAS, although the sample is not in intimate contact with the modifier. Obviously, the use of a permanent modifier simplifies the whole procedure of solid sampling significantly, compared to the addition of a modifier in solution to each sample aliquot, in addition to minimizing the risk of introducing high blank values with the modifier [23].
The determination of cadmium in coal using solid sampling GF AAS with conventional equipment turned out to be difficult, particularly as calibration against aqueous standards was not possible, necessitating the use of certified reference materials (CRM) for that purpose [10]. High-resolution continuum-source AAS (HR-CS AAS) has been used successfully in earlier works for the analysis, identification and correction of complex and/or structured molecular background [24–26]. This technique has proven to be a powerful tool for detecting and eliminating spectral interferences, and also for method development in GF AAS. Optimization of instrumental parameters is greatly facilitated by the possibility of altering conditions such as the start and end of signal integration and the pixels used for measurement and background correction, after the measured data have been stored [14].
The aim of this work was to investigate the feasibility of the direct determination of cadmium in solid coal samples using HR-CS AAS and calibration against aqueous standards. It was possible to identify the problems associated with this determination and to establish a reliable method for this kind of analysis.
Experimental
Instrumentation
All method development work and the final determinations were carried out using a prototype high-resolution continuum source atomic absorption spectrometer, built at ISAS, Department Berlin. The prototype is based on a Model AAS 6 Vario (Analytik Jena AG, Jena, Germany), from which the entire optical compartment including detector and associated controls had been removed and replaced by a high-resolution double monochromator (DEMON), similar to the system described by Heitmann et al [27]. The instrument, which utilizes a high-intensity xenon short-arc lamp as a continuum source, and a linear CCD array detector with 512 pixels, among other features, has been described in detail in previous publications [14, 24, 26, 27]. At 228.802 nm, the main Cd absorption line, the resolution per pixel was 1.9 pm, and an intermediate slit width of 428 μm was used, making possible the simultaneous evaluation of 200 pixels, corresponding to about ±0.2 nm around the analytical wavelength. Cadmium absorption was measured using the central pixel (CP) only, and CP±1, in other words over a spectral interval of about 6 pm. However, only the data obtained using CP±1 are presented in this work due to the better sensitivity and signal-to-noise ratio obtained under these conditions; otherwise the difference between the two modes of evaluation was of negligible influence on the analytical results. The system was controlled by a Pentium III, 1000 MHz personal computer, running an in-house-developed data acquisition program.
A conventional transversely-heated graphite tube atomizer system, supplied by Analytik Jena together with the Model AAS 6 Vario, was used throughout. All experiments were carried out using pyrolytic graphite-coated solid sampling graphite tubes without a dosing hole (Analytik Jena AG, Part No. 07-8130325), and the samples were introduced on pyrolytic graphite-coated solid sampling (SS) platforms (Analytik Jena AG, Part No. 407-A81.312), using a pre-adjusted pair of tweezers, which is part of the standard SSA 5 solid sampling accessory. Solid samples were weighed directly onto the platforms using an M2P microbalance (Sartorius, Göttingen, Germany). All calibration or modifier solutions were pipetted manually onto the platform. The sample mass was used to calculate the “normalized integrated absorbance” (the integrated absorbance calculated for 1 mg of sample) for each measurement.
Argon with a purity of 99.996% (White Martins, São Paulo, Brazil) was used as the purge gas. The graphite furnace temperature program used when treating the SS platforms with the Ir permanent modifier is presented in Table 1. A total mass of 400 μg of the Ir modifier was applied using ten injections of 40 μL each of modifier solution (1000 mg L−1), executing the temperature program in Table 1 after each injection. The optimized graphite furnace temperature program used for Cd determination in aqueous solutions and for direct solid sampling is given in Table 2.
Reagents and reference materials
Analytical grade reagents were used unless otherwise specified. Nitric acid (Carlo Erba, Milan, Italy, No. 4080150) was further purified by double sub-boiling distillation using a quartz sub-boiling still (Kürner Analysentechnik, Rosenheim, Germany). Water was purified in a Milli-Q system (Millipore, Bedford, MA, USA), resulting in water with a resistivity of 18 MΩ cm. The cadmium stock standard solution, 1000 mg L−1, was prepared from cadmium nitrate (Merck) in 0.5 mol L−1 HNO3. A 1000 mg L−1 Ir atomic absorption standard solution (Fluka, Buchs, Switzerland) was used to treat the platforms with the permanent modifier. The calibration solutions were prepared by serial dilution of the stock standard solution with 5% (v/v) nitric acid.
The following reference materials were used in this work: BCR No. 40 Trace elements in coal, BCR No. 180 Gas Coal and BCR No. 181 Coking Coal (Community Bureau of Reference, Brussels, Belgium); SARM 20 Coal Sasolburg (South Africa Bureau of Standards, Pretoria, South Africa); NIST SRM 1630a Trace Mercury in Coal, SRM 1632b Trace Elements Coal, and NIST SRM 1635 Trace Elements in Coal (National Institute for Standards and Technology, Gaithersburg, MD, USA). In addition, two real coal samples were investigated, a Brazilian coal from the Candiota mine in Rio Grande do Sul, termed “Candiota”, and a coal from Spitzbergen, Norway, termed “Spitzbergen”. For all experiments, the samples were ground manually in an agate mortar to a particle size ≤50 μm.
Results and discussion
Pyrolysis and atomization temperatures
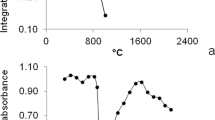
Two coal samples, SARM 20 and NIST 1630a, were used for method development and optimization procedures. Figure 1 shows the pyrolysis curves obtained for the above-mentioned coal samples and for an aqueous solution containing 2 μg L−1 Cd in 0.5% v/v HNO3, with and without the use of thermally deposited Ir as permanent modifier. The thermal stabilization provided by the modifier is obvious for all samples, although for NIST 1630a the pyrolysis curve obtained without modifier is rather similar to the one obtained with the permanent modifier. This suggests that the coal matrix itself, in this specific case, contributes to stabilize cadmium at higher pyrolysis temperatures. The stabilization effect provided by the modifier is noticeably higher for Cd in aqueous solution, in which about 15% of the analyte is lost at a pyrolysis temperature of 400 °C, and much greater losses occur at higher temperatures. The presence of iridium in this case allowed a pyrolysis temperature of 700 °C to be used, which facilitated the use of aqueous standards for calibration.

Pyrolysis temperature curves for Cd in aqueous solution (2 μg L−1) (open diamonds) without modifier and (rotated triangles) with Ir permanent modifier; SARM 20 coal (open squares) without modifier and (filled upside-down triangles) with Ir permanent modifier and NIST 1630a coal (open circles) without modifier and (filled triangles) with Ir permanent modifier. Atomization temperature: 1500°C
An interesting effect that occurred in the case of coal samples is the very strong continuous background absorption that preceded the analyte signal at pyrolysis temperatures ≤600 °C, as shown in Fig. 2 for NIST 1630a, using a pyrolysis temperature of 400 °C. The signal registration in Fig. 2 starts with the Auto Zero step (1 s prior to the atomization stage), and the cleaning stage starts at 9 s. At such low pyrolysis temperatures, the matrix obviously cannot be eliminated efficiently, and the remaining particles are vaporized in the atomization stage, which generates smoke in the graphite tube and, consequently, a great deal of scattering of the radiation emitted by the source. This then results in the pronounced baseline noise early in the atomization stage that can be seen in Fig. 2, as only less than 0.1% of the source radiation reaches the detector under these conditions, a situation that cannot be handled by any optical spectrometer. Although this background absorption and the analyte signal appeared at different time periods during the atomization stage, there was still some overlap between these two signals, which resulted in a slight loss in the measured integrated absorbance when the integration intervals were chosen so as to exclude the extreme baseline noise. This effect was most pronounced in NIST 1630a coal, and is responsible for the lower absorbance values that are measured when using pyrolysis temperatures <600 °C, as can be seen in Fig. 1. A much more efficient matrix elimination was achieved when a pyrolysis temperature of 700 °C was used, resulting in a considerable reduction in the continuous background for most samples, and in a complete elimination of this background absorption for the Candiota coal. The fact that Ir, applied as permanent modifier, allowed a pyrolysis temperature of 700 °C to be used for the coal samples, is yet another example that proves that permanent chemical modifiers can be applied successfully to direct solid sample analysis, although the sample is not in intimate contact with the modifier. The action of the modifier in this case can only be explained by a migration of the analyte from the coal matrix to the modifier-treated platform at relatively low temperatures (during the pyrolysis stage).

Time- and wavelength-resolved absorbance spectrum for cadmium in NIST 1630a coal at 228.802±0.2 nm, using 400 °C pyrolysis temperature and atomization at 1500 °C
The temperature used for atomization is responsible for another spectral event that can also be seen in Fig. 2. At temperatures above 1600 °C, structured background absorption, characteristic of the electron excitation spectra of diatomic molecules with rotational fine structure, appeared after the atomic signal. The identity of the molecule(s) that is/are causing this absorption spectrum is still under investigation, but a sulfur-containing molecule is undoubtedly responsible for at least part of this absorption. Nevertheless, the structured background that is visible in Fig. 2 only appeared during the cleaning stage at 2100 °C, as the atomization temperature in this case was 1500 °C, and so this did not interfere with the determination. A well-defined absorption peak can also be seen during the cleaning stage, which corresponds to a secondary iron line at 228.725 nm [28]; this obviously does not interfere with the determination either, since this peak can easily be separated from the atomic peak both temporally and spectrally, although it might cause spectral interference with deuterium background correction unless the temperature program is optimized carefully.
The absorbance spectrum integrated over time, which is shown in Fig. 3 for SARM 20 coal at an atomization temperature of 1700 °C, exhibits an extremely dense pattern of atomic and molecular absorption lines. The Cd absorption line is barely resolved in wavelength from the molecular absorption “lines”, a situation that would require least-squares background correction using a model spectrum [24, 25] in the case where the atomic and molecular absorption could not be separated in time, although this was possible in the present case. Cadmium is a relatively volatile element, which does not usually require atomization temperatures above 1500 °C. This was, in fact, the optimum temperature finally adopted in this work, since the integrated absorbance signal decreases at higher temperatures due to faster diffusion and hence a shorter residence time of the atoms in the absorption volume.

Wavelength-resolved absorbance spectrum for SARM 20 coal integrated over time in the vicinity of the cadmium resonance line at 228.802±0.2 nm; atomization temperature 1700 °C
Correction for continuous spectral events
The automatic correction for any continuous spectral “event”—any alteration in the radiation flux that affects all of the pixels at the same time and to the same extent—is another feature of the HR-CS AAS system. The fact that only a few pixels, typically three, are used to measure analyte absorption makes it possible to use any set of the remaining 197 pixels for correction purposes. And as all pixels are illuminated and read out simultaneously, this feature results in kind of a simultaneous double beam system and a simultaneous correction for continuous background absorption and/or emission. The effect of this specific type of correction is shown in Fig. 4a and b. In Fig. 4a, at a low pyrolysis temperature (400 °C), the background absorption reaches values well above A=3, meaning that essentially no radiation reaches the detector, as was already discussed in connection with Fig. 2. After proper correction, which occurs automatically, using a set of software-selected pixels, a more effective distinction can be made between the background absorption and the analyte signal, although there is still some overlap between the baseline noise (caused by the background absorption) and the analyte signal. Using the optimum pyrolysis temperature of 700 °C, which results in a much lower background absorption (apparent in Fig. 4b) the correction for continuous events not only eliminates the residual background absorption, but also the lamp flicker noise, resulting in a well-defined absorbance pulse with an extremely low noise level.

Absorbance over time for cadmium in NIST 1630a coal at the center pixel (228.802 nm) with and without the correction for continuous background absorption: a 400 °C pyrolysis temperature and b 700 °C pyrolysis temperature, using iridium as permanent modifier and atomization at 1500 °C
Optimization of sample mass
One of the biggest difficulties involved in direct solid sampling analysis is the micro-inhomogeneity of the samples to be analyzed. To evaluate this specific property, an optimization of the coal mass to be introduced into the graphite furnace was performed using NIST 1630a and SARM 20, with masses ranging from 0.1 to 2.2 mg. The final signal obtained was normalized to 1 mg and the results for each of the two CRM separated into eight groups of three masses each, which were typically within 0.1 mg, except for sample masses >1 mg, for which the mass range was greater. The relative standard deviation of the normalized signal for each mass interval was evaluated, and the results are shown in Fig. 5. For mass aliquots smaller than 0.6 mg, relative standard deviations between 15 and 35% were obtained, which shows that the micro-inhomogeneity of the coal samples could become critical at lower mass aliquots. Although higher masses, like those usually introduced, result in better precision, it should also be considered that adequate matrix elimination during the pyrolysis stage is more difficult under these conditions, necessitating a compromise between these two parameters. In fact, mass aliquots higher then 1 mg for NIST 1630a resulted in such a considerable overlap between the continuous background and the analyte signal that the precision deteriorated severely, resulting in relative standard deviations of around of 35% for mass aliquots above 1.5 mg. Sample aliquots between 0.6 and 1.2 mg were found to be optimum for all coal samples investigated, resulting in good precision (RSD<10%) and adequate matrix elimination at a pyrolysis temperature of 700 °C.

Effect of the coal sample mass introduced into the graphite furnace on the repeatability, evaluated by means of the relative standard deviation, for cadmium in (filled triangles) NIST 1630a, and (filled inverted triangles) SARM 20; pyrolysis temperature 700 °C and atomization temperature 1500 °C
Analytical results
The results obtained for the determination of cadmium in the CRM and in the coal samples using solid sampling and calibration against aqueous standards are shown in Table 3. The determined values are in good agreement, at a 95% confidence level, according to a Student t-test, with the certified values (where available). Since certified values are not available for Cd in SARM 20 and NIST 1630a, the results were compared to those obtained by slurry sampling ETV-ICP-MS, following the procedure described by Maia et al [29]. The determined concentration of Cd in Candiota coal is also in good agreement with the reference value determined by conventional GF AAS after acid digestion [30], which indicates that the method can be efficiently applied to “real” samples. No reference value was available for the Spitzbergen coal sample.
Figures of merit
A slope of 0.005 s pg−1 was obtained, along with a correlation coefficient of 0.9992, for the calibration curve established with aqueous standards. The same characteristic mass of 0.4 pg was found for coal samples and aqueous solutions, which suggests that calibration against aqueous standards is feasible for determining Cd in coal. This value is also considerably lower than the characteristic mass of 1.3 pg usually obtained for Cd in transversely heated graphite tube atomizers with Zeeman-effect background correction [31], the only other configuration that could potentially deal with this kind of analytical task. Largely due to the complete absence of dilution, which is certainly one of the most attractive characteristics of direct solid sampling analysis, a detection limit of 2 ng g−1 was achieved, calculated for a sample mass of 1 mg, using the “zero mass response” [7]: three times the standard deviation of the signal obtained for ten repetitive introductions and “atomizations” of an empty SS platform. This shows that the method developed in this work is extremely sensitive and adequate for the determination of the lowest trace levels of cadmium in coal.
Conclusion
It has been shown in this work that the determination of cadmium, a very volatile element, in coal, a complex matrix, which is extremely difficult to bring into solution, can be successfully carried out using direct solid sample analysis and calibration against aqueous standards. This is one more example that shows that the frequently mentioned problem of calibration in direct solid sample analysis is not as severe as often claimed. Obviously, calibration against aqueous standards requires the complete absence of any matrix effect, which, for a complex matrix such as coal, can only be achieved through careful optimization of the analytical procedure. This optimization has been facilitated dramatically in HR-CS AAS, where the entire spectral environment becomes visible, and optimization can be carried out using a stored set of data. The fact that calibration against aqueous standards in the determination of the same element in the same matrix is often not possible using conventional line source (LS) AAS might be due to the limited optimization possibilities of the latter technique. However, as the same graphite atomizer is used for both techniques, and hence the same non-spectral interferences must be expected, it might be assumed that determinations by LS AAS are in fact limited by spectral interferences, which obviously do not exist, or can be controlled much more easily in HR-CS AAS.
It might be expected that other difficult-to-determine trace elements in coal could be determined in the same way—using direct solid sample analysis and calibration against aqueous standards—after careful selection of a suitable modifier and program optimization using HR-CS AAS.
References
Pollock EN (1975) Trace impurities in coal by wet chemical methods. In: Babu SP (ed) Trace elements in fuel. American Chemical Society, Washington, DC, pp 23–34
Kingston HM, Haswell S (1997) Microwave enhanced chemistry. American Chemical Society Professional Book Series, ACS, Washington, DC
Fadda S, Rivoldini A, Cau I (1995) Geostandards Newslett 19:41–54
Lachas H, Richaud R, Jarvis KE, Herod AA, Dugwell DR, Kandiyoti R (1999) Analyst 124:177–184
Liu J, Sturgeon RE (1995) Ciencia 3:127–138
Figueroa LB, Alvarado JI, Dominguez JR (1997) Atom Spectrosc 18:13–16
Kurfürst U (1998) Solid sample analysis. Springer, Berlin Heidelberg New York
Belarra MA, Resano M, Vanhaecke F, Moens L (2002) Trends Anal Chem 21:828–839
Nowka R, Müller H (1997) Fresen J Anal Chem 359:132–137
Vale MGR, Silva MM, Welz B, Lima EC (2001) Spectrochim Acta Part B 56:1859–1873
Vale MGR, Silva MM, Welz B, Nowka R (2002) J Anal Atom Spectrom 17:38–45
Silva AF, Welz B, Curtius AJ (2002) Spectrochim Acta Part B 57:2031–2045
Silva MM, Vale MGR, Damin ICF, Welz B, Mandaji M, Fett JP (2003) Anal Bioanal Chem 377:165–172
Silva AF, Borges DLG, Welz B, Vale MGR, Silva MM, Klassen A, Heitmann U (2004) Spectrochim Acta Part B 59:841–850
Slavin W, Manning DC, Carnrick GR (1981) Atom Spectrosc 2:137–145
Tsalev DL, Slaveykova VI (1992) Spectrosc Lett 25:221–238
Tsalev DL, D’Ulivo A, Lampugnani A, Di Marco M, Zamboni RJ (1995) J Anal Atom Spectrom 10:1003–1009
Volynski AB, Krivan V (1997) J Anal Atom Spectrom 12:333–340
Volynski AB, Tikhomirov SV (1998) J Anal Chem 53:718–721
Belarra MA, Resano M, Rodríguez S, Urchaga J, Castillo JR (1999) Spectrochim Acta Part B 54:787–795
Da Silva JBB, Borges DLG, da Veiga MAS, Curtius AJ, Welz B (2003) Talanta 60:977–982
Piascik M, Bulska E (2001) Spectrochim Acta Part B 56:1615–1623
Da Silva AF, Welz B, Curtius AJ (2002) Spectrochim Acta Part B 57:2031–2045
Becker-Ross H, Florek S, Heitmann U (2000) J Anal Atom Spectrom 15:137–141
Welz B, Vale MGR, Silva MM, Becker-Ross H, Huang MD, Florek S, Heitmann U (2002) Spectrochim Acta Part B 57:1043–1055
Vale MGR, Damin ICF, Klassen A, Silva MM, Welz B, Silva AF, Lepri FG, Borges DLG, Heitmann U (2004) Microchem J 77:131–140
Heitmann U, Schütz M, Becker-Ross H, Florek S (1996) Spectrochim Acta Part B 51:1095–1105
Welz B, Becker-Ross H, Florek S, Heitmann U (2005) High-resolution continuum source AAS—the better way to do atomic absorption spectrometry. Wiley-VCH, Weinheim
Maia SM, Silva JBB, Curtius AJ, Welz B (2000) J Anal Atom Spectrom 15:1081–1086
Silva MM, Vale MGR, Caramão EB (1999) Talanta 50:1035–1043
Welz B, Sperling M (1999) Atomic absorption spectrometry. Wiley-VCH, Weinheim, p 493
Acknowledgements
The authors are grateful to Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), to Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and to Analytik Jena. D.L.G.B and A.J.C. have scholarships from CNPq and F.G.L has a scholarship from CAPES. The authors are also grateful to Analytik Jena for financial support and donation of the HR-CS atomic absorption spectrometer. We also thank Michael Kriews for the donation of the coal sample “Spitzbergen”.
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to the memory of Wilhelm Fresenius
Rights and permissions
About this article
Cite this article
da Silva, A.F., Borges, D.L.G., Lepri, F.G. et al. Determination of cadmium in coal using solid sampling graphite furnace high-resolution continuum source atomic absorption spectrometry. Anal Bioanal Chem 382, 1835–1841 (2005). https://doi.org/10.1007/s00216-005-3327-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-005-3327-9