
Abstract
This technical note reports on a new procedure to on-column-label organelles sampled from a tissue cross section into a fused silica capillary. These organelles are then analyzed by capillary electrophoresis with postcolumn laser-induced fluorescence detection. In this procedure, the fluorescent label does not come in contact with the tissue, which facilitates visualization of the sampled tissue cross section. In addition, on-column labeling allows for better control of the reaction time and fluorescent label concentrations. As a proof-of-principle, we show results of mitochondria from rat gastrocnemius muscle cross sections that were on-column-labeled with 10-N-nonyl acridine orange (NAO), a mitochondrion-specific probe, and compare them with results for NAO in-tissue labeling of the same tissue. The new organelle labeling procedure reported here may easily be extended to the analysis of individual organelles in other biological samples and may become a valuable tool in studies investigating the role of mitochondria in muscle aging and exercise physiology.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Analysis of mitochondrial properties in highly heterogeneous muscle tissue cross sections usually relies on histological procedures in which micron-thick tissue slices are stained with specific markers for enzymatic activity or biochemical function [1]. These histochemical procedures are fast, report simultaneously on the status of all the fibers in a muscle cross section, aid in fiber typing, and may provide reference maps for other techniques yielding complementary information, e.g., tissue profiling by mass spectrometry [2]. The emergence of other complementary techniques would be highly beneficial to histology.
We have reported the use of capillary electrophoresis (CE) with postcolumn laser-induced fluorescence (LIF) detection for direct tissue analysis [3]. In this technique, the injection end of the capillary is used to directly suction a small sample from a tissue cross section. Because of the capillary dimensions (e.g., 50-μm inner diameter, i.d.), areas usually smaller than the cross section of a single fiber are sampled. After sampling, the injection end of the capillary is placed in a running buffer vial. By applying an electric field, mitochondria electromigrate toward an LIF detector where they are detected individually [3, 4]. The number of mitochondria, their electrophoretic mobility distribution, and their fluorescence intensity distribution are then calculated from the detector’s measurements. The number of mitochondria is relevant in determining the role that these organelles may have in the production of ATP in the muscle [5]. The electrophoretic mobility of individual organelles is related to the electrical charge status of the organelle and its dimensions [3, 4], and is a largely unexplored property in organelles. The fluorescence intensity depends on chemical content or function. For example, 10-N-nonyl acridine orange (NAO) fluorescence intensity is related to the cardiolipin content of mitochondria [6].
Instead of fluorescently labeling mitochondria while they are still in the tissue cross section, a more versatile approach would be to carry out labeling on-column after sampling. On-column labeling has been used to label proteins and peptides [7–9]. After sequentially introducing separate nanoliter volumes of solutions containing the analytes and the labeling reagents into the capillary, a short electric pulse causes electrophoretic mixing, thus allowing for the labeling reaction to occur. The precise timing of these events and the subsequent CE separation provide an excellent control over the mixing of the labeling reagent and the sample. Furthermore, on-column labeling uses nanoliters of sample and labeling reagents, which is possible when using low-volume samples and is fairly attractive when expensive reagents are involved.
In this report, we extend on-column labeling to the CE-LIF analysis of biological particles (i.e., mitochondria). Instead of using electrophoretic mixing, the sample containing mitochondria is sandwiched between two NAO plugs, and then sufficient time is allowed for mixing by diffusion. Besides the advantages of the on-column labeling procedures mentioned before, this labeling procedure prevents the user from treating the entire tissue cross section with the labeling reagents. The CE-LIF results reveal that this procedure effectively labels the sampled mitochondria.
Experimental
Reagents
Sucrose, dimethyl sulfoxide (DMSO) and ethanol were purchased from Fisher Scientific (Pittsburg, PA, USA). Trypsin and N-(2-hydroxyethyl)piperazine-N-ethanesulfonic acid (HEPES) were purchased from Sigma (St. Louis, MO, USA). KOH was purchased from Aldrich (Milwaukee, WI, USA). Fluorescein and NAO were purchased from Molecular Probes (Eugene, OR, USA). The separation buffer contained 10 mM HEPES, 250 mM sucrose and was adjusted to pH 7.4 with KOH (CE buffer). Stock solutions of 1 mM fluorescein and 1 mM NAO were made in ethanol and DMSO, respectively. For tissue treatment prior to on-column NAO labeling, trypsin was diluted to 0.5 g L−1 with CE buffer before the experiment. Fluorescein was diluted to 1 nM and NAO was diluted to 5 μM with CE buffer prior to use. All buffers were made with Milli-Q deionized water and filtered with a 0.22-μm filter before use.
Animal and tissue preparation
The gastrocnemius muscle was dissected from an anesthetized rat according to the protocol approved by the Animal Use Committee of the University of Minnesota. Next, the superficial portion of the muscle (defined as white gastrocnemius) was delineated and quickly frozen in isopentane, cooled in liquid nitrogen, and stored at −80°C. Serial sections (10 μm) were cut using a cryostat microtome maintained at −25°C. Each cross section was mounted onto a silanized-coated glass slide and stored at −20°C. On the day of the analysis, the tissue was brought to room temperature immediately prior to direct sampling.
On-column NAO labeling and direct sampling of mitochondria
Figure 1 shows the steps used for direct sampling and on-column labeling of mitochondria. Using a micropipette, 10 μL of 5 μM NAO was delivered onto the microscope glass slide close to the region of the tissue cross section (Fig. 1a). The injection end of a 45-cm-long CE capillary (50-μm i.d.) was mounted in a single-cell injector that moved in the x–y–z direction with micromanipulators (MX100L and MW1, Soma Scientific, Irvine, CA, USA) as previously described [10]. The capillary was brought into the NAO drop on the microscope glass slide and 3.8 nL of the NAO solution was introduced into the capillary by applying −11 kPa for 1.0 s. An electronic three-way solenoid valve (part no. 4-8-900, VAC-100PSIG, Parker Instrumentation, Fairfield, NJ, USA) that hydrodynamically connects the detector end with a reservoir placed 110 cm below the tissue cross section was used to apply the pulse of negative pressure. The process was monitored with an inverted fluorescence microscope (Eclipse TE300 Nikon, Huntley, IL, USA).

Procedure for direct sampling of mitochondria from a single muscle fiber cross section followed by their on-column labeling with 10-N-nonyl acridine orange (NAO). a The injection end of the capillary is immersed into a drop of NAO solution and approximately 3.8 nL is hydrodynamically introduced into the capillary. b The capillary is brought in contact with a fiber and −11 kPa for 1 s introduces the tissue sample into the capillary. c The capillary is brought back into the NAO solution and a second approximately 3.8-nL plug is introduced into the capillary. d While the capillary is kept in a vial containing capillary electrophoresis run buffer, diffusion causes mixing, making it possible to label mitochondria with NAO. e, f Bright-field images showing a white gastrocnemius muscle fiber cross section; the star marks a fiber before (e) and after (f) sampling, respectively
After injection of the first plug of NAO, the capillary was brought slightly above the fiber to be sampled (marked with a star in Fig. 1e,f) and 1 μL of 0.5 mg mL−1 trypsin was delivered to the same region of the tissue using a micropipette and then allowed to sit for 5 min. For sampling, the capillary was lowered until it established minimal contact with the targeted fiber and −11-kPa pressure was then applied for 1 s. This process resulted in the introduction of mitochondria from the selected fiber cross section and surrounding buffer onto the capillary. Then, the capillary was brought back into the NAO drop to introduce another 3.8 nL of NAO solution into the capillary (Fig. 1c). The second NAO plug prevented the sample from leaking out from the capillary during the on-column reaction that was allowed to occur for 5 min while the capillary was being held in a vial containing CE buffer (Fig. 1d). The second plug of NAO also provided a second source of labeling reagent from which NAO could diffuse toward the sample. In order to minimize perturbations during the labeling reaction, the injection end of the capillary was not removed from the holder, but the capillary holder was moved away from the slide so that the vial containing the CE run buffer could be placed underneath the holder and clamped in place. Upon termination of the reaction, CE analysis was begun by applying −9-kV high voltage on a 45-cm-long capillary (electric field −200 V cm−1). Control experiments were performed under the same conditions using either the tissue without NAO (an equal amount of buffer was sampled instead of NAO) or just two NAO plugs sandwiching a plug of CE buffer.
After sampling, the status of the tissue cross section near the sampled fiber was observed under the microscope. Figure 1f shows a cross section in which a sampled fiber is marked by a star. The curved dark lines on the tissue are the result of the capillary tip deforming the tissue owing to contact during the sampling.
In-tissue NAO labeling and direct sampling of mitochondria
A previously described procedure with minor modifications was used for the in-tissue labeling of mitochondria prior to CE-LIF analysis [3]. Briefly, 1 μL of a solution containing 10 μM NAO and 1 μL of a solution containing 0.5 g L−1 trypsin was delivered on top of the tissue cross section and allowed to incubate for more than 5 min. The exact timing was difficult to determine owing to the extra time spent on positioning and manipulating the tissue prior to sampling. Upon completion of the incubation period, mitochondria were sampled from the targeted fiber at −11-kPa pressure for 1 s and then analyzed by CE-LIF, as described before for the on-column NAO labeling procedure.
Instrumentation
The CE-LIF instrument has been previously described [3, 11]. Mitochondria separations were performed using a 50-μm i.d., 150-μm outer diameter poly(acrylaminopropanol)-coated capillary as previously reported [3, 4]. The electroosmotic flow mobility was (1.1±0.1)×10−5 cm2 V−1 s−1 as determined by the current monitoring method [12]. The excitation source was a 488-nm, 4-mW, Ar-ion laser (JDS Uniphase, San Jose, CA, USA). Emission in the range 522–552 nm was spectrally filtered using a 535DF35 interference filter (Omega Optical, Bratteboro, VT, USA) and detected with a photomultiplier tube (Hamamatsu, Omega Optical). Scattering of the 488-nm excitation line caused by bubbles, particulates, and mitochondria in the sample was reduced using an additional rejection band filter (488-53D, OD4, Omega Optical) that was placed in front of the interference filter. The photomultiplier tube output was electronically filtered (RC=0.01 s) and then digitized using a PCI-MIO-16E-50 I/O board controlled by Labview software (National Instruments, Austin, TX, USA). The data collection rate was 100 Hz and the data were stored in binary files. The detector alignment was optimized in a separate run for a maximum signal to-noise ratio (S/N) (900±20) by using 1 nM fluorescein. The instrument variability for particle detection was determined in a separate run by continuously electromigrating 1-μm-diameter fluorescently labeled polystyrene beads (Fluoresbrite, Polysciences, Warrington, PA, USA) suspended in CE buffer (S/N=400±50, n=3). The capillary was reconditioned between runs by pressure flushing 2 min each time with water, methanol, water, and CE buffer.
The data analysis procedure was done using Igor Pro software (Wavemetrics, Lake Oswego, OR, USA) as previously described with minor modifications [11]. Briefly, the Igor Pro routines measure the peak intensities and migration times for the detected individual events with S/N>3. The electrophoretic mobility for each individual event (μ) is calculated as μ=Lt M −1 E −1, where L is the capillary length, t M is the migration time, and E is the electric field.
Results and discussion
Successful on-column labeling of proteins or peptides as well as on-column enzymatic reactions have been previously reported [7–9, 13, 14]. Although this approach would seem to be applicable to the fluorescent labeling and analysis of sub-cellular-size particles such as mitochondria, no reports have appeared on the subject. Here, we demonstrate the use of on-column reactions to fluorescently label mitochondria directly sampled from the cross section of a single muscle fiber before their analysis by CE-LIF.
As shown in Fig. 1, the sample is sandwiched between two 3.8-nL, 1.9-mm-long NAO plugs from which this reagent diffuses into the sample region and labels mitochondria. Since buffer also enters the capillary during the sample introduction, the maximum sample volume and plug length would be 3.8 nL and 1.9 mm, respectively. Using Fick’s second law of diffusion, after a 5-min incubation time, an estimate of the NAO concentration at the center of the sample plug is 4 μM. This estimate assumes that (1) the NAO diffusion coefficient is approximately 4±10−6 cm2 s−1, (2) sub-micron-size mitochondria do not significantly diffuse during the incubation time, and (3) diffusion of NAO toward the sample occurs from both flanking NAO plugs. Use of only one NAO plug is not adequate for on-column labeling because it would result in a more asymmetric NAO concentration profile across the plug sample. In addition, electrophoretic mixing is not practical owing to the highly heterogeneous electrophoretic mobilities of mitochondria (Fig. 3). The two-NAO-plug on-column labeling strategy was adopted for the rest of the experiments.

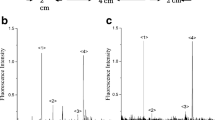
Electropherograms of samples taken directly from single muscle fiber cross sections and labeled on-column. A Muscle sample labeled with NAO. B NAO control without mitochondria. C Control, mitochondria not labeled with NAO. Sampling and NAO labeling conditions were as in Fig. 1. Separation conditions A 50-μm inner diameter, 45-cm-long poly(acrylaminopropanol)-coated capillary; 10 mM N-(2-hydroxyethyl)piperazine-N-ethanesulfonic acid and 250 mM sucrose run buffer, pH 7.4; and −200 V cm−1 applied voltage. Detection conditions as described in the Experimental section
Figure 2, trace A shows the electropherogram for the sample taken from a single fiber cross section and then on-column-labeled with the mitochondrion-selective NAO. Control electropherograms, NAO without mitochondria (Fig. 2, trace B) and mitochondria without NAO (Fig. 2, trace C), confirm that most of the individual events in Fig. 2, trace A result from labeling mitochondria with NAO. While there were no events in the NAO control (Fig. 2, trace B), the events in the mitochondria control electropherogram (Fig. 2, trace C) are only a small fraction (5%) of the total number of detected events for labeled mitochondria (19±6, n=3) and have low intensities (Fig. 2, trace C, Fig. 3b, trace 3). These low-intensity events may result from particulate autofluorescence or scattering that cannot be completely rejected, given the optical configuration of the detector. Figure 2 and the selectivity of NAO toward mitochondria both support the observation that individually detected events are indeed caused by mitochondria [6].

Electrophoretic mobility and fluorescence intensity distributions for on-column and in-tissue NAO labeling of mitochondria. Average electrophoretic mobility distributions (a) and sorted signal intensities (b) of individual mitochondrial events for on-column labeling (1) and in-tissue labeling (2). After calculating the electrophoretic mobility for each event, the data were binned into a separate histogram distribution for each sample, normalized by the total number of events in the sample, and then averaged out (n=3). The standard deviation for each bin is shown as a thinner line. In a, the on-column labeling distribution is offset on the y-axis for clarity. The sorted fluorescence intensities show the combined data of the distributions shown in a and also include a control experiment (3) in which the samples were not labeled with NAO. Other conditions are as in Fig. 2
Trypsin treatment prior to sampling was necessary in order to detect mitochondrial events. In the absence of trypsin treatment, a very low number of events were detected (6±3, n=3; data not shown). Trypsin has been shown to cause partial digestion of the cytoskeleton network and the subsequent redistribution of mitochondria in muscle fibers [15]. This enzyme appears to digest the contact points between the cytoskeleton and mitochondria, making it possible to sample these organelles into the capillary.
The results for on-column labeling and in-tissue labeling of mitochondria from white gastrocnemius muscle cross section and their corresponding CE-LIF analysis are shown in (Table 1 and Fig. 3). Table 1 shows that there are fewer mitochondrial events for on-column than for in-tissue NAO labeling (i.e., 36±10, n=3, and 19±6, n=3, respectively). This table also shows average and median measurements that would be typical of bulk measurements. For electrophoretic mobility, the averages are (−2.4±0.6)×10−4 and (−2.6±0.4)×10−4 cm2 V−1 s−1, respectively; the medians are (−2.3±0.2)×10−4 and (−2.5±0.2)×10−4 cm2 V−1 s−1, respectively. Both parameters are statistically undistinguishable at 95% confidence level between labeling methods. Moreover, the fluorescence intensity averages, 1.54±0.30 and 0.63±0.15, respectively, and medians, 1.42±0.34 and 0.70±0.21, respectively, are statistically different at the 95% confidence level.
As already discussed and previously reported [3], lengthening of the trypsin treatment appears to result in the release of more mitochondrial events and the disengagement of organelles that may have been tethered to each other through cytoskeletal proteins. Since the length of the trypsin treatment for the in-tissue NAO labeling procedure was not rigorously timed (e.g. more than 5 min), it is not surprising to detect more events for the in-tissue labeling procedure. Similarly, the decrease in fluorescence intensity for the in-tissue labeling procedure (Table 1), even when the effective NAO concentration (i.e. 5 μM NAO) is higher than the maximum NAO concentration predicted for the on-column labeling procedure (i.e. 4 μM NAO), is consistent with more extensive trypsin treatment resulting in greater breakage between tethered organelles.
One of the advantages of individual organelle analysis by CE-LIF is that the results can be represented as distributions of individual electrophoretic mobility and intensity values, which can indicate the extent of heterogeneity of such properties within a given sample. Qualitatively, as shown in (Fig. 3a), the mobility distributions appear different for the on-column NAO labeling and in-tissue labeling procedures, and this may be caused by their different trypsin treatments, as previously reported [3]. The results of the shorter trypsin treatment of the on-column labeling show a subgroup of mitochondrial events with electrophoretic mobilities less than −3×10−4 cm2 V−1 s−1 that is not present for the longer trypsin treatment of the in-tissue labeling. In contrast, an additional subgroup of mitochondrial events with electrophoretic mobilities greater than −2×10−4 cm2 V−1 s−1 is present for the in-tissue labeling results. Owing to the differences in trypsin treatments, the present data cannot assess the contribution of histological heterogeneity within the tissue samples to the observed variations in electrophoretic mobilities.
A more comprehensive comparison between the electrophoretic mobility data reported here and those in the previous report [3] could not be pursued because the muscle types are histologically different: here we used a white gastrocnemius muscle, while a soleus muscle was used in the previous report. The former is a fast-twitch glycolytic muscle, while the latter is a slow-twitch oxidative muscle which is expected to have more, highly interconnected interfibrillar mitochondria. Morphological differences in mitochondria may be related to the variations in electrophoretic mobilities of these organelles, a hypothesis based on studies carried out on particles of similar dimensions [16] that has never been directly tested on organelles.
As suggested by the sorted fluorescence intensities of the individual events (Fig. 3b), the relative NAO content in the individual mitochondrial events varies over a wide range. Control experiments in which no NAO was used indicate that the fluorescence intensity for these events is low (Fig. 3b, trace 3); therefore, on-column and in-tissue NAO labeling truly results in the detection of fluorescence associated with the NAO content of each event, which in turn is related to the relative abundance of cardiolipin in each detected event [6].
Conclusions
The hyphenation of solid tissue sampling with separation techniques (e.g., CE) is in its infancy. The on-column labeling of mitochondria sampled from single muscle fiber cross sections is a new analytical CE-LIF procedure compatible with the analysis of highly variable organelle properties in highly heterogeneous tissue. Since only the fiber cross section that comes in contact with the capillary is perturbed, neighboring fibers can be subsequently sampled, allowing for comparisons among single fibers. Furthermore, on-column labeling strategies may be easily adapted to other labeling reagents and other organelles sampled from muscle or other tissue cross sections. The on-column procedure reported here could become an important resource for investigating changes in tissues as they develop, age, or are affected by disease.
References
Dubowitz V (1985) Histological and histochemical stains and reactions, Bailliere Tindall, London
Chaurand P, Sanders ME, Jensen RA, Caprioli RM (2004) Am J Pathol 165:1057–1068
Ahmadzadeh H, Johnson RD, Thompson L, Arriaga EA (2004) Anal Chem 76:315–321
Duffy CF, Fuller KM, Malvey MW, O’Kennedy R, Arriaga EA (2002) Anal Chem 74:171–176
Drew B, Leeuwenburgh C (2004) Acta Physiol Scand 182:333–341
Petit JM, Maftah A, Ratinaud M–H, Julien R (1992) Eur J Biochem 209:267–273
Harmon BJ, Patterson DH, Regnier FE (1993) Anal Chem 65 2655–2662
Miller KJ, Leesong I, Bao J, Regnier FE, Lytle FE (1993) Anal Chem 65:3267–3270
Taga A, Nishino A, Honda S (1998) J Chromatogr A 822:271–279
Krylov SN, Starke DA, Arriaga EA, Zhang Z, Chan NWC, Palcic MM, Dovichi NJ (2000) Anal Chem 72:872–877
Duffy CF, Gafoor S, Richards DP, Ahmadzadeh H, O’Kennedy R, Arriaga EA (2001) Anal Chem 73:1855–1861
Ahmadzadeh H (2000) PhD dissertation, University of Alberta, Edmonton
Burke BJ, Regnier FE (2001) Electrophoresis 22:3744–3751
Lee IH, Pinto D, Arriaga EA, Zhang Z, Dovichi NJ (1998) Anal Chem 70:4546–4548
Vendelin M, Beraud N, Guerrero K, Andrienko T, Kuznetsov AV, Olivares J, Kay L, Saks VA (2005) Am J Physiol Cell Physiol 288:C757–C767
Radko SP, Chrambach A (2002) Electrophoresis 23:1957–1972
Acknowledgements
This work is supported by NIH R01-AG20866. Janice Shoeman prepared the muscle cross sections. Dmitry Andreyev designed Igor Pro routines that automate data analysis. E.A.A. is supported by 1K02-AG21453. L.V.T. is supported by AG17768 and AG21626.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ahmadzadeh, H., Thompson, L.V. & Arriaga, E.A. On-column labeling for capillary electrophoretic analysis of individual mitochondria directly sampled from tissue cross sections. Anal Bioanal Chem 384, 169–174 (2006). https://doi.org/10.1007/s00216-005-0171-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-005-0171-x