
Abstract
To obtain quantitative information on human metabolism of selenium, we have performed selenium speciation analysis by HPLC/ICPMS on samples of human urine from one volunteer over a 48-hour period after ingestion of selenium (1.0 mg) as sodium selenite, L-selenomethionine, or DL-selenomethionine. The three separate experiments were performed in duplicate. Normal background urine from the volunteer contained total selenium concentrations of 8–30 μg Se/L (n=22) but, depending on the chromatographic conditions, only about 30–70% could be quantified by HPLC/ICPMS. The major species in background urine were two selenosugars, namely methyl-2-acetamido-2-deoxy-1-seleno-β-D-galactopyranoside (selenosugar 1) and its deacylated analog methyl-2-amino-2-deoxy-1-seleno-β-D-galactopyranoside (selenosugar 3). Selenium was rapidly excreted after ingestion of the selenium compounds: the peak concentrations (∼250–400 μg Se/L, normalized concentrations) were recorded within 5–9 hours, and concentrations had returned to close to background levels within 48 hours, by which time 25–40% of the ingested selenium, depending on the species ingested, had been accounted for in the urine. In all experiments, the major metabolite was selenosugar 1, constituting either ∼80% of the total selenium excreted over the first 24 hours after ingestion of selenite or L-selenomethionine or ∼65% after ingestion of DL-selenomethionine. Selenite was not present at significant levels (<1 μg Se/L) in any of the samples; selenomethionine was present in only trace amounts (∼1 μg/L, equivalent to less than 0.5% of the total Se) following ingestion of L-selenomethionine, but it constituted about 20% of the excreted selenium (first 24 hours) after ingestion of DL-selenomethionine, presumably because the D form was not efficiently metabolized. Trimethylselenonium ion, a commonly reported urine metabolite, could not be detected (<1 μg/L) in the urine samples after ingestion of selenite or selenomethionine. Cytotoxicity studies on selenosugar 1 and its glucosamine isomer (selenosugar 2, methyl-2-acetamido-2-deoxy-1-seleno-β-D-glucosopyranoside) were performed with HepG2 cells derived from human hepatocarcinoma, and these showed that both compounds had low toxicity (about 1000-fold less toxic than sodium selenite). The results support earlier studies showing that selenosugar 1 is the major urinary metabolite after increased selenium intake, and they suggest that previously accepted pathways for human metabolism of selenium involving trimethylselenonium ion as the excretionary end product may need to be re-evaluated.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The study of selenium metabolism has considerable relevance to human health because of selenium's dual role as a toxicant and an essential trace element. The physiological requirements for selenium, an integral part of several important enzyme systems [1], have been estimated to be about 70 μg/day for an adult man [2], and there are several studies linking specific diseases such as Keshan’s disease to diets low in selenium [3]. Selenium is also a purported anticancer agent, and a large 12-year study is currently investigating the effects of long-term selenium supplementation as a protective measure against prostate cancer [4]. Collectively, these perceived health benefits attributed to selenium have lead to a growing market for selenium supplements.
But selenium can also have toxic effects at relatively modest intakes [2]. Because urine is a major excretory route, investigations into the selenium species present in urine can provide information on the metabolic processes, perhaps delineating those pathways leading to beneficial and toxic effects. The first identified selenium urinary metabolite was trimethylselenonium ion, isolated in 1969 from the urine of rats after high exposure to selenite [5]. This species was subsequently claimed to be in human urine [6] and was generally thought to be produced as a way of excreting excess, potentially toxic selenium. More recent studies, however, with greatly improved analytical methods such as HPLC/ICPMS (high performance liquid chromatography/inductively coupled plasma mass spectrometry) have failed to demonstrate that trimethylselenonium ion is a significant constituent of human urine [7].
An important advance in the field came with the identification of a selenium-containing carbohydrate (selenosugar 1, Fig. 1) as a significant metabolite in urine from rats administered selenite [8]. This work was quickly followed by the report by Gammelgaard and coworkers [9] that human volunteers given selenized yeast (containing largely L-selenomethionine bound in proteins) also produced selenosugar 1 as a significant urinary metabolite. Further work by this research group demonstrated the presence of a second selenosugar, selenosugar 2, [7, 10] and then a third, selenosugar 3, [10] as trace urinary metabolites. In contrast to these results, Cao et al. [11] claimed that selenocystamine was the major selenium metabolite in human urine after ingesting DL-selenomethionine, although some concerns regarding the rigour of this assignment have been expressed [12].

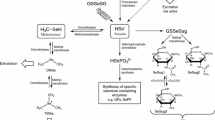
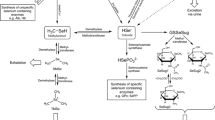
Structures of selenium species relevant to this study
The studies describing selenosugars have opened up a new phase in research into selenium metabolism. In addition to providing the unambiguous assignment of a significant selenium metabolite, they have flagged the existence of an alternative metabolic pathway that does not end with trimethylselenonium ion. Although a recent study [13] measured concentrations of selenosugars in urine after selenium supplementation, the work reported so far has been essentially of a qualitative nature, and has provided no data on excretion rates of selenium and the absolute quantities of the various selenium metabolites. We report results from a quantitative case study of selenium excretion and metabolites in human urine from one individual after the administration of selenium as sodium selenite, DL-selenomethionine, or L-selenomethionine. In addition, results from cytotoxicity testing of the major selenium urinary metabolite, selenosugar 1, and its isomer selenosugar 2, are reported.
Experimental
Chemicals and reagents
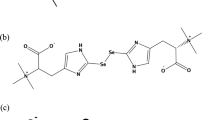
The selenium compounds used in the ingestion experiments were: sodium selenite (Na2SeO3·5 H2O, >99%) obtained from Merck (Darmstadt, Germany), DL-selenomethionine (CH3SeCH2CH2CH(NH2)COOH, >99%) from Fluka (Buchs, Switzerland), and L-selenomethionine (>98%) from Sigma-Aldrich (Vienna, Austria). Selenium species standards used for HPLC/ICPMS, in addition to selenite and selenomethionine, were: selenate (as sodium selenate, >99% from Fluka); trimethylselenonium iodide (synthesized in-house); selenosugars 1 and 2 synthesized as previously reported [14]; and selenosugar 3, prepared from selenosugar 1 by a modification of the analytical scale synthesis reported by Bendahl and Gammelgaard [10]. Thus, selenosugar 1 (20 mg, 67 μmol) was dissolved in a mixture of 98% hydrazine/water (0.5 mL, 9+1 v/v) and heated in a sealed tube at 110°C for 24 h. The solvent was evaporated, and the residue taken up in water and applied to a small column containing cation exchange resin (DOWEX 50). The column was washed with water (100 mL), and the desired selenogalactosamine, selenosugar 3, was stripped from the column with 1 M NH3, and obtained as a white solid (10 mg, 58% yield). m/z (ESMS, 100 V): 162 ([M-CH3Se]+ 100%), [M+H]+ 255 (13%), 256 (35%), 258 (71%), 260 (13%). 1H-NMR (500 MHz, D2O) δ 4.44 (1H, d, H-1, J 10 Hz), 3.78 (1H, d, H-4, J 3 Hz), 3.65-3.56 (3H, m, H-5, H-6), 3.39 (1H, dd, H-3, J 10, 3 Hz), 2.89 (1H, t, H-2, J 10 Hz), 1.99 (3H, s, SeMe). 13C-NMR (100 MHz, D2O) δ 81.7, 80.2, 73.7, 68.2, 61.2, 51.9, 1.8.
For total selenium analyses by ICPMS, stock standard solutions of 1000 mg Se/L were purchased from CPI (Santa Rosa, USA). Nitric acid (p.a.), aqueous ammonia solution (25%, suprapur) and pyridine (p.a.) were obtained from Merck; formic acid (p.a.), ammonium formate (p.a.) and citric acid (p.a.) were purchased from Fluka; and methanol (p.a.) was obtained from Carl Roth GmbH (Karlsruhe, Germany). Dilutions of standard solutions and digest solutions were performed with Milli-Q water (18.2 MΩ cm). Three certified reference materials were used: NIES CRM No. 18 (Human Urine, National Institute for Environmental Studies, Ibaraki, Japan); Lyophilized Seronorm Trace Elements in Urine Lot FE1113 (Sero A/S, Billingstad, Norway); and NIST SRM 1640 Trace Elements in Natural Water (National Institute of Standards and Technology, Gaithersburg, MD, USA).
Background samples
A minimum of three background samples were collected before each experiment, including a morning urine sample (about t=−12 h), and a sample (t=0) collected immediately before ingestion of the selenium dose.
Ingestion of selenium and collection and storage of urine
A total of six ingestion experiments were performed in two sets, each comprising three forms of selenium. In each duplicate set of experiments, a known quantity of selenium (1.0 mg Se) as pure sodium selenite, DL-selenomethionine, or L-selenomethionine was ingested by one male volunteer (51 years of age) in about 100 cm3 of water on a designated day at 18:00 (t 0). The volunteer gave informed consent and was aware of the experimental details and possible effects of ingesting the selenium; the quantity of selenium ingested was about fivefold that taken daily by people on selenium supplements.
The urine samples were individually collected in 500 mL polyethylene bottles over the following 48 hours (24 h in the case of the second DL-selenomethionine experiment). The masses of the urine samples were recorded and specific gravity was measured with a Leica TS 400 total solids refractometer (Leica Microsystems, Buffalo, NY, United States); urine volumes were then calculated from the masses of the samples. The samples were stored at 4°C until analysis for total selenium and for selenium species (completed within ten days). The total selenium concentrations were normalized to the mean specific gravity using the equation:
Determination of total selenium
Digestion
A portion (1.00 mL) of urine or solution of reconstituted certified reference material was added to a quartz digestion vessel (12-mL capacity) of an ultraCLAVE 2 microwave autoclave (EMLS, Leutkirch, Germany). HNO3 (4.00 mL) was added to the quartz vessels. The system was closed, loaded with argon to 4×106 Pa, and the mixture was heated at 250°C for 30 min. The digested urine samples were then diluted with Milli-Q water (to 10–50 mL depending on the Se content) before analysis for selenium by ICPMS. Matrix effects, investigated by the method of standard additions, were shown to cause about a 10% increase in the selenium signal when urine digests were diluted to 10 mL, but these effects were acceptably small (<5%) when samples were diluted to 20 mL or more. Although occasional checks for matrix effects were carried out, our standard method was performed with external calibration on digest solutions diluted to 20 mL.
Inductively coupled plasma mass spectrometry for selenium-selective detection
An Agilent 7500c or 7500ce inductively coupled plasma mass spectrometer (Agilent, Waldbronn, Germany) equipped with a PFA microconcentric nebulizer, a Scott double pass spray chamber and an octopole reaction cell served as the selenium-selective detector. The instrumental and acquisition parameters were: rf power (1500 W); carrier gas (1 L/min); reaction cell gas (H2 at 3.0 mL/min); monitored masses (m/z 77, 78).
Standard solutions of selenium (0.1–50 μg Se/L) were used for calibration; quantification was performed with selenium isotope m/z 78. As a source of carbon to enhance Se ionisation, a solution of isopropanol/water (1+1) at a flow rate of 0.02 mL/min was mixed with the sample/standard solution just prior to nebulization. The accuracy of the method was checked by analysis of the three standard reference materials: NIES No. 18 Human urine (certified [Se]=59±5 μg/L) returned 61±2 μg/L, n=3; Seronorm Trace Elements in Urine (certified [Se] range 14–26 μg/L) returned 22±1 μg/L, n=3; and NIST SRM 1640 Trace Elements in Natural Water (certified [Se]=21.96±0.51 μg/L) returned 22.2±0.2 μg/L, n=3.
Determination of selenium species by HPLC/ICPMS and HPLC/ESMS
Urine samples were filtered through 0.22 μm polyamide filters (Macherey-Nagel, Dueren, Germany) prior to injection into the HPLC/ICPMS system. Separation of selenium species was performed using ion-exchange chromatography (duplicate set 1) and ion-exchange and reversed-phase chromatography (duplicate set 2). Anion-exchange chromatography was performed on a PRP-X100 anion-exchange column (100 mm×4.1 mm) from Hamilton Company (Reno, NV, USA), and a mobile phase of an aqueous solution of citric acid (10 mM) at pH 4.8, adjusted with aqueous ammonia, at 40°C and a flow rate of 1.5 mL/min. Cation-exchange chromatography was performed on a Zorbax 300-SCX cation-exchange column (150 mm×4.6 mm) from Agilent, or on a Hamilton PRP-X200 column (250×4.1 mm). Conditions applied to the Zorbax 300-SCX column were: aqueous solutions of pyridine (7 or 20 mM) at pH 2.1, adjusted with formic acid, as mobile phase, at 30°C and a flow rate of 1.5 mL/min. Conditions applied to the Hamilton PRP-X200 column were: aqueous solution of pyridine (10 mM) at pH 1.6, adjusted with formic acid, as mobile phase, at 30°C and a flow rate of 1.0 mL/min. Reversed-phase chromatography was performed at 30°C on a Walters Atlantis C18 column (150 mm×4.6 mm; Waters Corporation, Milford, MA, USA) and a mobile phase of 20 mM ammonium formate at pH 3.0, adjusted with formic acid, containing 3% methanol at a flow rate of 1.0 mL/min. Injection volume was always 20 μL. ICPMS selenium-selective detection was performed with an Agilent 7500c or 7500ce equipped with a Babington nebulizer under the conditions described above. Selenomethionine and selenosugars 1, 2, and 3 were quantified in the urine samples by external calibration with standard solutions of the respective pure compounds; unknown selenium compounds were quantified by external calibration with standard solutions of selenosugar 1 or selenosugar 3. Under our measurement conditions, the signal response for selenium was not greatly influenced by the type of selenium compound (slopes of calibration curves were all within 10% of the mean slope for the various species).
HPLC/Electrospray mass spectrometry was performed on one of the crude urine samples (second L-selenomethionine experiment, sample t=4.9 h) with an Agilent 1100 LCMSD system (SL type) under reversed-phase conditions with a Walters Atlantis C18 column, 150 mm×4.6 mm at 30°C, and a mobile phase of 20 mM ammonium formate at pH 3.0, adjusted with formic acid, containing 3% methanol at a flow rate of 1.0 mL/min. MSD conditions were: nebulizer pressure: 55 psig, drying gas flow: 12 L/min, drying gas temp. 350°C, capillary voltage 4,000 V, single ion monitoring positive polarity.
Cytotoxicity of selenosugars and selenite
Details of the general procedure employed for our cytotoxicity tests have been previously reported [15]. In brief, HepG2 cells derived from human hepatocarcinoma were grown in a monolayer in Dulbecco’s modified Eagle’s medium and supplemented with 10% fetal bovine serum. The cells were cultured in an incubator in an atmosphere of 5% CO2 in humidified air. HepG2 cells, seeded at a density of 1×104 cells/well in 96-well plates (IWAKI, Japan) and incubated for 24 h, were placed in fresh medium that contained test chemicals and incubated for 48 h. Cytotoxicity was assayed by the method of WST-8 (Cell Counting Kit-8, Wako, Japan), in which WST-8 was converted to soluble formazan by the action of mitochondrial dehydrogenase in viable cells. The cultures in 96-well plates were placed in 100 mm3 of medium that contained WST-8 and incubated for 1 h at 37°C. The absorbance at 450 nm was determined by a multiplate reader.
Results and discussion
The experiments aimed to compare the selenium metabolism in a single volunteer after ingestion of the same quantity of selenium in different chemical forms. The first two experiments investigated selenite and an equal mixture of D- and L-selenomethionine, and a third experiment was performed with pure L-selenomethionine. The three experiments showed clear differences which required confirmation. For this reason, a second set of experiments duplicating the first set was performed with the same volunteer. The results from the duplicates agreed well, both in terms of the total and rate of selenium excretion, and the selenium metabolites produced. In the second set of experiments (data set 2), however, an extra chromatographic system (reversed-phase on Atlantis C18) was employed in addition to the ion-exchange systems used in the first set of experiments (data set 1). Reversed-phase separations were generally superior to those obtained from ion exchange, giving sharper peaks and better detection limits, and hence a clearer data set. Consequently, the following discussion will focus on the second set of experiments.
Background levels and time courses of selenium elimination
Background levels were determined for 12 hours before the ingestion of selenium, and found to range from 11–18 μg Se/L (normalized data). This is within the range for urine from a healthy person consuming natural selenium at adequate nutritional levels [2]. The total selenium excretion data, displayed in Tables 1–3, show interesting differences. After ingestion of either L-selenomethionine (Table 2) or DL-selenomethionine (Table 3), selenium was rapidly excreted: the first urine sample collected (∼t=2.2 h) contained greatly elevated selenium concentrations (∼250 μg Se/L, normalized data), and the peak samples were obtained within about six hours, after which time the concentrations quickly decreased (<50 μg Se/L after 22 hours). The excretion for selenite (Table 1) was markedly slower; the peak sample was obtained at nine hours and the concentration was still ∼160 μg/L after 18 hours.
Excretion profiles of selenium, based on normalized selenium concentrations in urine, for the selenite, L- and DL-selenomethionine experiments are presented in Fig. 2. Pseudo first-order elimination rate constants (K e) for selenium were obtained by fitting the data to a one-compartment toxicological model: the pseudo K e values were about 0.07 h−1 (t 1/2 ∼10 h), 0.13 h−1 (t 1/2 ∼5 h) and 0.18 h−1 (t 1/2 ∼4 h) after selenite, DL-selenomethionine and L-selenomethionine ingestion, respectively. For comparison, data from the first set of experiments returned half-lives of 9 hours, 6 hours and 4.5 hours for selenite, DL-selenomethionine and L-selenomethionine, respectively. This difference in half-lives presumably reflects different rates of formation of selenosugar 1, the major metabolite (see below), from the precursors. The mixture of DL-selenomethionine showed a more complicated excretion profile for both data sets, which was explainable when the selenium species were determined later (see below). These preliminary data suggest that selenosugar 1 may be biosynthesized more slowly when selenite is the source of selenium, but clearly more experiments involving more volunteers are needed to test this hypothesis. We note that the study of Thomson et al. [16] also reported that peak excretion of selenium occurred sooner after ingestion of selenomethionine than with selenite ingestion.

Time courses of selenium excretion after ingestion of 1.0 mg selenium as selenite, DL-selenomethionine, or L-selenomethionine. Data have been normalized to average specific gravity
The total amounts of selenium excreted over 24 and 48 hours respectively, were 27% and 32% (selenite), 29% and 32% (L-selenomethionine) and 25% (DL-selenomethionine, 24 hours only). For comparison, the first set of experiments showed total excretion after 48 hours of 40% (selenite), 30% (L-selenomethionine) and 25% (DL-selenomethionine). Presumably the “missing” selenium is explainable by its incorporation into essential proteins; excretion in the feces and as the respiratory product dimethylselenide [17] may also account for some of the selenium. For comparison, the study of Thomson et al. [16] reported that 22% (one volunteer) of the 1 mg of selenium ingested as selenomethionine was excreted in the urine in 24 hours, whereas 72% and 77% (two volunteers) was excreted when 1 mg of selenium was ingested as selenite.
Determination of selenium species: analytical aspects
The most common method currently used for the determination of urinary selenium species is HPLC with selenium-selective detection by ICPMS [12]. Although a total of 16 selenium compounds have been reported in human urine, only four—selenite and selenosugars 1 to 3—appear to have been identified based on sound data; another three species—selenomethionine, trimethylselenonium ion and selenate—have also been reported in normal human urine although the data appear less rigorous [12]. There is no compelling evidence for the presence of the other nine purported selenium urinary metabolites. Accordingly, we applied chromatographic systems to determine the above-named seven selenium species. These systems were based on anion and cation exchange, and reversed-phase separations.
The anionic selenium species and the selenosugars 1 and 2 were resolved with the PRP-X100 anion-exchange column, whereas trimethylselenonium ion and selenosugar 3, which eluted at the solvent front on anion exchange, chromatographed well under cation-exchange conditions. The fact that the selenosugars 1 and 2, which are neutral molecules at the pHs employed here, were retarded under anion-exchange conditions suggested that they were experiencing hydrophobic interactions with the polymer backbone of the column. Selenosugars 1 and 2 were similarly retarded on PRP-X200 (a cation exchanger), which also has a polymer backbone, but they eluted at the void volume from the Zorbax 300-SCX cation exchanger, which has a silica backbone. These presumed hydrophobic interactions suggested that reversed-phase chromatography might be suitable for the selenosugars, and subsequently good retention and separation of the selenosugars 1 and 2 were achieved on an Atlantis C18 reversed-phase column; selenomethionine was also adequately retained on this column, whereas selenosugar 3 and trimethylselenonium ion eluted close to the solvent front. The improved resolution obtained with the Atlantis reversed-phase column, in comparison to our ion-exchange separations, resulted in considerably lower detection limits (two- to tenfold depending on the species and sample composition).
When quantifying selenium species (selenium speciation analysis) by HPLC/ICPMS, several factors must be considered, such as possible chromatographic losses and variable matrix interferences in the ICPMS. In this regard, mass balance calculations on the total selenium applied to the HPLC column and the sum of selenium species eluting from the column can provide useful information. Following HPLC analysis of the urine samples, the sum of selenium species was generally 5–20 μg/L lower than the total selenium concentration obtained following acid digestion. This “recovery” was acceptable (≥90%) at the higher selenium concentrations, but was poor (30–70%) for the samples with low selenium concentrations. The apparent poor column recovery for background samples is probably, at least in part, a direct consequence of low total selenium concentrations and the presence of several selenium species; when this small quantity of selenium is distributed among several species, some of them are present at levels below the limit of quantification and hence do not contribute to the recorded sum of species. In support of this was the observation that HPLC recoveries for background samples were higher when using the Atlantis column because the lower detection limits enabled more (small) peaks to be quantified. It is also possible that some selenium species do not elute from the columns.
Another factor impacting on the quantitative aspects of this study is the lability of the selenium metabolites. Gammelgaard and Bendahl [7] reported that selenosugars 1 and 2 and selenomethionine when stored in urine at 25°C degraded over a period of days. We carried out a preliminary stability study which indicated that losses of selenosugar 1 were small (∼5%) when urine samples were stored for two weeks at 4°C. In our current experiments, all samples were analyzed for selenium species within ten days of collection (stored at 4°C), and thus significant losses of selenosugar 1 were unlikely.
Selenium metabolites in urine
Speciation analysis of background urine samples revealed the presence of two significant and several minor selenium species. The two major species were identified as selenosugars 1 and 3 (each between ∼1–4 μg/L for the various background samples) by co-chromatography with authentic specimens under reversed phase and cation exchange chromatographic conditions (Fig. 3). A recent study [13] demonstrated that selenosugars 1 and 3 occurred as normal constituents of human urine at about 1–8 μg Se/L (eight subjects). We could not find evidence for the presence of trimethylselenonium ion (<1 μg Se/L) in the background urine samples. In recent papers Gammelgaard and Bendahl [7, 13] reported that trimethylselenonium ion was not detectable in their samples of human urine. Similarly, we could not detect other selenium species sometimes reported in urine such as selenite, selenate (both <1 μg Se/L) and selenomethionine (<0.5 μg/L). We note here that our stated detection limits are based on actual urine samples containing mixtures of selenium compounds.

Chromatograms (HPLC/ICPMS) of a background urine sample (solid line) and the same background urine sample spiked with 3 μg Se/L of selenosugars 1 and 3 (dashed line): a Reversed phase chromatography (Atlantis C18, 150 mm×4.6 mm; 20 mM ammonium formate pH 3.0 containing 3% methanol at a flow rate of 1.0 mL/min); b cation exchange chromatography (Hamilton PRP-X200, 250 mm×4.1 mm; 10 mM pyridine pH 1.6 at a flow rate of 1.0 mL/min)
Analysis of the urine samples after ingestion of selenite showed a large increase in the peak assigned to selenosugar 1, which accounted for about 80% of the total selenium (Table 1). A smaller but clear increase was also observed for selenosugar 3, and traces of selenosugar 2 were also detected in the more concentrated samples. Assignments were made on the basis of spiking (addition) experiments performed under three sets of chromatographic conditions with ICPMS detection (Fig. 4).


Chromatograms (HPLC/ICPMS) of the urine sample 18.5 hours after selenite ingestion (solid line), and the same urine sample spiked with selenosugar 1 (140 μg Se/L), selenosugar 2 (1.5 μg Se/L), selenosugar 3 (20 μg Se/L), and selenomethionine (2 μg Se/L) (dashed/dotted lines): a Reversed phase chromatography (Atlantis C18, 150 mm×4.6 mm; 20 mM ammonium formate pH 3.0 containing 3% methanol at a flow rate of 1.0 mL/min); b anion exchange chromatography (Hamilton PRP-X100, 100 mm×4.1 mm; 10 mM citric acid pH 4.8 at a flow rate of 1.5 mL/min); c cation exchange chromatography (Hamilton PRP-X200, 250 mm×4.1 mm; 10 mM pyridine pH 1.6 at a flow rate of 1.0 mL/min)
In addition to the main selenium metabolites, several minor selenium species (collectively <10% of sum of species) were also present in the urine samples; although these species were not identified, spiking experiments clearly showed that they were not trimethylselenonium ion, selenomethionine, selenite, or selenate.
After ingestion of L-selenomethionine, the pattern of selenium species in urine was very similar to that obtained for the selenite experiment (Fig. 5a, Table 2). The major species (∼80% of total selenium) was again selenosugar 1, and selenosugars 2 and 3 were also formed. These assignments were supported by spiking experiments (Fig. 5a). Further support for the structural assignment of selenosugar 1 was provided by HPLC/electrospray MS with detection at m/z 302, 300, 298, 297 (representing the [M+H]+ pseudo-molecular ion for four Se isotopes) and m/z 204 ([M-CH3Se]+) (Fig. 6). In addition, there was a trace of selenomethionine (<0.5%) in the most concentrated of the urine samples, presumably representing unmetabolized ingested compound. These results showing the presence of selenosugars 1, 2, and 3 after ingestion of pure L-selenomethionine agree well with the qualitative data obtained by Gammelgaard and coworkers [7, 10] from similar experiments with selenized yeast which contains largely L-selenomethionine bound in proteins. They do, however, differ in regard to the formation of selenosugar 3. Gammelgaard and coworkers [10] report the presence of selenosugar 3 in normal (unsupplemented) urine, but thought that its concentration did not increase after selenium supplementation despite the formation of large quantities of selenosugar 1. We can confirm that selenosugar 3 is present in normal urine, but quantitative results from six separate experiments clearly show that its concentration increases (∼tenfold) after selenium supplementation. These increases, however, were much less than those shown by selenosugar 1 (∼100–200-fold), and, hence, might have been overlooked in the earlier qualitative study [10].

Chromatograms (HPLC/ICPMS) of urine samples after ingestion of L-selenomethionine (t=8.7 h) or DL-selenomethionine (t=6.2 h). Reversed phase chromatography (Atlantis C18, 150 mm×4.6 mm; 20 mM ammonium formate pH 3.0 containing 3% methanol at a flow rate of 1.0 mL/min). a Urine sample after L-selenomethionine ingestion (solid line) and the same urine sample spiked with selenosugar 1 (200 μg Se/L), selenosugar 2 (5 μg Se/L), selenosugar 3 (20 μg Se/L), and selenomethionine (1 μg Se/L) (dashed/dotted lines); b urine sample after DL-selenomethionine ingestion (solid line) and the same urine sample spiked with selenosugar 1 (250 μg Se/L), selenosugar 2 (3 μg Se/L), selenosugar 3 (15 μg Se/L), and selenomethionine (65 μg Se/L) (dashed/dotted lines)

Chromatograms (HPLC/ESMS) of a urine sample after L-selenomethionine ingestion (t=4.9 h) (solid line), and the same urine sample spiked with selenosugar 1 (200 μg Se/L) (dashed line). Reversed phase chromatography (Atlantis C18, 150 mm×4.6 mm; 20 mM ammonium formate pH 3.0 containing 3% methanol at a flow rate of 1.0 mL/min); positive ion mode with variable fragmentor voltage (m/z 204 at 125 V; m/z 297, 298, 300, and 302 at 75 V). Signals for m/z 297 and 302 not shown
The anomaly of the earlier reported results of Gammelgaard and coworkers [10] has been discussed previously [12], and this may have been the stimulus for these researchers, in a subsequent study [13], to measure the concentrations of the selenium metabolites before and after selenium supplementation. Their results more or less matched their earlier report in that, except for one of eight subjects, no clear increase in concentration of selenosugar 3 was observed after selenium supplementation, whereas selenosugar 1 concentrations measurably increased in most cases. These data were interpreted [13] as resulting from individual variability in the way humans metabolize selenium to the various selenosugars, with the suggestion that most humans do not produce any selenosugar 3 after selenium supplementation. We consider this unlikely and suggest that the quantities of selenium in the urine samples after supplementation were still too small to detect significant increases in selenosugar 3.
We believe, however, that the relative quantities of selenosugars 1 and 3 could be dose-dependent, and thus might be relevant to a possible detoxification pathway for selenium. The pathway may involve transformation of excess, potentially toxic, selenium to selenosugar 1, which is then deacylated to selenosugar 3. At normal exposure, both selenosugars are present at low levels (∼1–4 μg/L), but at high exposure the deacylation step is overloaded leading to a large excess of selenosugar 1.
The first experiment (data set 1) with the mixture of DL-selenomethionine gave clearly different results from those obtained with L-selenomethionine; selenosugar 1 represented less of the total selenium excreted (∼65%) and unchanged selenomethionine was a significant species (∼20%). The repeat experiment duplicated this result exactly (Fig. 5b, Table 3). Probably, this unchanged selenomethionine corresponds to D-selenomethionine in the ingested mixture, reflecting the selectivity of enzymes for the natural L-selenomethionine. Unfortunately, D-selenomethionine is not a commercially available product, which deterred us from performing a separate experiment with this compound. Although chiral separations of D and L-selenomethionine are possible [18], the methods we employed in this study were unable to distinguish these two compounds.
It is interesting to compare our results from ingestion of DL-selenomethionine with a previous similar study reported by Cao et al. [11]. In that study, one volunteer ingested 400 μg of DL-selenomethionine, and the two major “metabolites” in urine were identified as selenocystamine and selenomethionine at a ratio of about 4:1 (total quantities excreted were not reported). Presumably, the selenium “metabolite” identified as selenomethionine in that study was unmetabolized D-selenomethionine, and thus our data are in agreement with that result. The assignment of the major metabolite to selenocystamine, however, could not be confirmed in our two experiments with DL-selenomethionine; in both cases the major metabolite was selenosugar 1. The discrepancy between these two data sets remains to be explained; we consider it unlikely, however, that it is due to individual variability between the two human subjects. We also note that Gammelgaard et al. [9] have stated that they have never detected selenocystamine in any urine sample.
Cytotoxicity of selenosugars 1 and 2 compared with selenite
When the HepG2 cells were incubated for 48 h with sodium selenite, toxicity was observed at concentrations over 0.001 mM (Fig. 7). By contrast, both selenosugars 1 and 2 were about 1000-fold less toxic than selenite, and cytotoxicity was not observed until concentrations were over 1 mM (∼80,000 μg Se/L). These data indicating low toxicity for selenosugars are consistent with the likely role of these compounds as end products in a process, invoked when selenium intake is in excess of physiologic requirements, to biotransform (detoxify) potentially toxic selenium species. It is of interest, and perhaps reassuring for people on selenium supplements, that excess ingested selenium is excreted as nontoxic metabolites.

Cytotoxicity of selenosugars compared with selenite
General discussion
Previous qualitative experiments with rats and selenite [8], and humans and selenized yeast [7, 10], which contains largely L-selenomethionine in proteins, have demonstrated that one significant metabolite in both cases was the novel selenium-containing carbohydrate, selenosugar 1. These data suggested that the form of selenium ingested may have little effect on the final metabolic product excreted in the urine. Our results from the experiments with selenite and L-selenomethionine support that view, because the different selenium species were metabolized primarily to the same metabolite, which accounted for about 80% of the excreted selenium in both cases.
On the other hand, the results from the experiments with DL-selenomethionine suggest that the form of selenium ingested can be important at times; it appears as though D-selenomethionine was poorly metabolized and that large quantities were excreted unchanged. It is interesting that two similar compounds such as D- and L-selenomethionine should be handled so differently, and yet selenite and L-selenomethionine appear to converge to the same metabolic pathway with comparable efficiency. Collectively, these data are consistent with the accepted metabolic pathway for selenium, which has various selenium sources first converting to selenide which is then transformed into selenocysteine and incorporated into selenoproteins [19].
There may be implications for the metabolism of various selenium compounds in regard to the way that selenium is administered for health promoting purposes (selenium supplementation). The difference between the metabolism of D- and L-selenomethionine may be seen as providing support for the contention that “natural L-selenomethionine”, for example from selenised yeast, is a preferred source of this essential trace element. Our contrary results showing similarities in excretion products after ingestion of either L-selenomethionine or selenite should not, however, be seen as mitigating this claim, because the biosynthesis of selenosugar 1 may only be a mechanism to remove excess, possibly toxic, selenium, and this could be incidental to the manner in which essential selenoproteins are formed from the two selenium sources. Further studies with various selenium sources involving analysis of blood in addition to urine may help clarify this distinction.
It has been noted recently [12] that despite apparently solid early data showing that trimethylselenonium ion was a major urinary selenium metabolite in rats, its presence in human urine has never been conclusively proven. We have specifically looked for trimethylselenonium ion in normal (unsupplemented) human urine and in human urine collected after supplementation with selenite or selenomethionine. In no case could we identify this species in the samples; we estimate that if it were present it was at a level of <1 μg/L, equivalent to <0.5% of the total selenium in those urine samples containing the highest selenium concentrations. Recent studies [7, 13] indicate that trimethylselenonium ion is not a significant metabolite in normal human urine, and our results certainly support that view; whether it is produced at all by humans, even at elevated selenium intake, is now also open to question.
There are several interesting unresolved aspects concerning the formation of selenosugars. Presumably H2Se is the common intermediate in the conversion of either selenite or selenomethionine into selenosugars; this species is considered central to human metabolism of selenium [19] and serves as the precursor to selenoproteins in addition to the excretion products dimethylselenide and trimethylselenonium ion. The existence of these other methylated excretion products (dimethylselenide at least) suggests that methylation of H2Se precedes the addition of the sugar moiety in the biosynthesis of selenosugars. The preferred use of a galactose sugar over a glucose sugar as the “selenium scavenger” is also of interest, and a clear biochemical basis for this selectivity is not readily apparent. A study of selenium metabolites from other animal species may provide helpful data in this regard.
In summary, our work on the biotransformation of selenium has confirmed the earlier studies of Kobayashi et al. [8] and Gammelgaard and co-workers [7, 9, 10, 13], and provided quantitative data for the formation and excretion of three selenosugars (one major, one minor, and one in trace amounts) after ingestion of selenite, L-selenomethionine, or DL-selenomethionine. Cytotoxicity studies have shown low toxicities for the two selenosugars tested so far (1000-fold lower than selenite). Our future work will look at the effect of dose and type of ingested selenium on the formation of selenosugars and other, currently unknown, selenium species, and investigate individual variability in the way humans handle exposure to selenium.
References
Birringer M, Pilawa S, Flohe L (2002) Nat Prod Rep 19:693–718
Alaejos MS, Romero CD (1993) Clin Chem 39:2040–2052
Yang G, Ge K, Chen J, Chen X (1988) World Rev Nutr Diet 55:98–152
Klein EA, Lippman SM, Thompson IM, Goodman PJ, Albanes D, Taylor PR, Coltman C (2003) World J Urol 21:21–27
Byard JL (1969) Arch Biochem Biophys 130:556–560
Burk RF (1976) Trace elements in human health and diseases, vol 2. Academic, New York, pp 105–133
Gammelgaard B, Bendahl L (2004) J Anal Atom Spectrom 19:135–142
Kobayashi Y, Ogra Y, Ishiwata K, Takayama H, Aimi N, Suzuki KT (2002) Proc Natl Acad Sci U S A 99:15932–15936
Gammelgaard B, Madsen KG, Bjerrum J, Bendahl L, Jons O, Olsen J, Sidenius U (2003) J Anal Atom Spectrom 18:65–70
Bendahl L, Gammelgaard B (2004) J Anal Atom Spectrom 19:950–957
Cao TH, Cooney RA, Woznichak MM, May SW, Browner RF (2001) Anal Chem 73:2898–2902
Francesconi KA, Pannier F (2004) Clin Chem 50:2240–2253
Gammelgaard B, Bendahl L, Wessel Jacobsen N, Stürup S (2005) J Anal Atom Spectrom (in press)
Traar P, Belaj F, Francesconi KA (2004) Aust J Chem 57:1051–1053
Ochi T, Suzuki T, Isono H, Kaise T (2004) Toxicol Appl Pharmacol 200:64–72
Thomson CD, Burton CE, Robinson MF (1978) Br J Nutr 39:579–587
Ganther HE (1986) J Am Coll Toxicol 5:1–5
Sutton KL, de Leon CAP, Ackley KL, Sutton RMC, Stalcup AM, Caruso JA (2000) Analyst 125:281–286
Rayman MP (2004) Br J Nutr 92:557–573
Acknowledgements
We thank the Austrian Science Fund (FWF project number P 16816) for financial support, and Matthias Uray for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kuehnelt, D., Kienzl, N., Traar, P. et al. Selenium metabolites in human urine after ingestion of selenite, L-selenomethionine, or DL-selenomethionine: a quantitative case study by HPLC/ICPMS. Anal Bioanal Chem 383, 235–246 (2005). https://doi.org/10.1007/s00216-005-0007-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-005-0007-8