
Abstract
The recombinant photoprotein aequorin was used as a reporter in highly sensitive and automatable hybridization assays for the analysis of transgenic sequences in genetically modified organisms (GMO). The terminator of the nopaline synthase gene (NOS) from Agrobacterium tumefaciens and the 35S promoter sequence were detected in genetically modified soybean. The endogenous, soybean-specific, lectin gene was also detected for confirmation of the integrity of extracted DNA. A universal detection reagent was produced through conjugation of aequorin to the oligonucleotide (dA)30. Biotinylated (through PCR) products for the three target sequences were captured onto streptavidin-coated wells, and one strand was removed by NaOH treatment. The immobilized single-stranded DNAs were then hybridized with oligonucleotide probes consisting of a target-specific segment and a poly(dT) tail. This allowed the subsequent determination of all hybrids through the use of the (dA)30-aequorin conjugate as a universal reagent. The bound aequorin was measured by adding Ca2+ and integrating the light emission for 3 s. As low as 2 pM (100 amol per well) of amplified DNA was detectable for all three targets, with a signal-to-background ratio of about 2. The analytical range extended up to 2000 pM. As low as 0.05% GMO content in soybean can be detected with a signal-to-background ratio of 8.2. The overall repeatability of the proposed assay, including DNA extraction, PCR, and hybridization assay, ranged from 7.5–19.8%. The use of a (dA)30-aequorin conjugate renders the assay configuration general for any target DNA, provided that the specific probe carries a poly(dT) tail.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genetically modified organisms (GMOs) are products of gene technology characterized by improved functional properties. This is achieved by the introduction of a gene that expresses a novel protein conferring new characteristics, such as herbicide tolerance and resistance to viruses and insects. A promoter, a structural gene (encoding the novel protein) and a terminator are the main constituents of the inserted foreign DNA.
In many countries, labeling of grains and foodstuffs is mandatory if the GMO content exceeds a certain level. For instance, the European Union and Japan have set threshold values of 1 and 5%, respectively, of genetically modified (GM) material in a non-GM background as the basis for food labeling [1, 2]. The enforcement of these threshold values has created a demand for the development of reliable, highly sensitive and automatable methods for GMO analysis. The analytical procedure for GMO testing of food samples involves a screening step followed by quantitative analysis of those samples that were found positive.
Detection of GMOs and derived materials can be achieved by identifying either the expressed proteins encoded by the transgenic sequences [3, 4] or the introduced DNA [5, 6]. DNA is the preferred analyte in GMO testing due to its superior stability compared to proteins, especially in processed foods. Target DNA sequences used frequently for GMO screening include genetic control elements of the constructs, as well as genes from cloning vectors coding for antibiotic resistance. The 35S promoter of the cauliflower mosaic virus and the nopaline synthase (NOS) terminator from Agrobacterium tumefaciens are the most commonly used regulatory elements for the production of transgenic plants [7]. It is estimated that 100 ng of DNA extracted from soybean contains 80000 copies of genomic DNA [8]. If only 1% of this amount is of GMO origin, then the challenge is to detect 800 genome copies of genetically modified soybean in a background of about 80000 copies of unaltered genome. Hence, DNA amplification by polymerase chain reaction (PCR) constitutes an essential step of the methods used for GMO detection. Usually a large number of amplification cycles (40 to 50) are performed [9, 10].
PCR methods based on common DNA sequences with potential for the detection of GMOs have been developed [11, 12]. Amplification products are usually analyzed by agarose gel electrophoresis and ethidium bromide staining of separated DNA fragments. Other techniques such as capillary electrophoresis [13] have also been used for separation and detection of PCR products. Electrophoretic methods, however, do not give any sequence information. To verify the amplified sequences, methods such as specific cleavage of the amplicons by restriction endonucleases, Southern blot followed by hybridization with a DNA probe specific to the target sequence, direct sequencing, or nested-PCR assays are used [12]. Research is also carried out on real-time PCR [14] and biosensors [15, 16].
Bio- and chemiluminescence, the emission of light from chemically generated excited states, was introduced in nucleic acid analysis in response to the need for alternatives to radioactive labels. Bio(chemi)luminometric methods, despite the relatively low quantum yields, offer higher detectability and dynamic range than spectrophotometric and fluorometric ones, because problems arising from scattering of excitation radiation and fluorescence of sample components are avoided. In spite of this distinct advantage, bio(chemi)luminescence has not been exploited as a detection technique in the analysis of GMO.
Aequorin is a photoprotein composed of a 189-amino acid polypeptide chain (apoaequorin), coelenterazine, and oxygen, that is attached to coelenterazine as peroxide [17, 18, 19]. Upon Ca2+ binding, aequorin undergoes a conformational change that triggers the oxidative decarboxylation of coelenterazine to produce coelenteramide and light at 470 nm. Aequorin can be detected at the attomole level in the presence of excess Ca2+ [20]. Moreover, the detection of aequorin is complete within 3 s following the simple addition of Ca2+, which is a significant advantage over enzyme reactions that require much longer incubation times. Because of these unique characteristics, aequorin is an excellent reporter molecule for the development of binding assays [21, 22, 23]. Recently, we reported simple methods for the purification of hexahistidine-tagged recombinant aequorin from bacterial cultures, and direct conjugation of the photoprotein to oligonucleotide probes [24, 25].
In the present work, we demonstrate the potential of aequorin-oligonucleotide conjugates for the development of simple and highly sensitive bioluminometric hybridization assays of 35S promoter, NOS terminator and lectin DNA sequences in transgenic soybean in a high-throughput format.
Experimental
Apparatus
Luminescence measurements were carried out using the PhL microplate Luminometer/Photometer from Mediators (Vienna, Austria). PCR amplification reactions were performed in a Hybaid Omn-E thermal cycler (Middlesex, UK). A digital camera, Kodak DC 120, and the Gel Analyzer software for DNA and protein documentation were purchased from Kodak (New York). The microcentrifuge Mikro 20 was from Hettich Gmbh (Tuttlingen, Germany).
Reagents
Sulfo-succinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxylate (Sulfo-SMCC) and N-succinimidyl S-acetylthioacetate (SATA) were obtained from Pierce Chemical Co (Rockford, USA). Bovine serum albumin (BSA) and cetyltrimethylammonium bromide (CTAB) were from Serva (Heidelberg, Germany) and blocking reagent was from Roche (Mannheim, Germany). Sephadex G-25 Spin Pure columns were purchased from CPG (New Jersey, USA). Ni-NTA agarose was obtained from Qiagen (Hilden, Germany). Microcon YM-10 centrifugal filter devices were from Millipore (Bedford, USA). Ultrapure 2′-deoxyribonucleoside 5′ triphosphates (dNTPs) were purchased from HT Biotechnology (Cambridge, UK). Tth DNA polymerase was from Biotools (Madrid, Spain). Terminal deoxynucleotidyl transferase (TdT) and ΦX174 DNA/BsuRI marker were from MBI Fermentas (Vilnius, Lithuania). The NucleoSpin Plant kit was from Macherey-Nagel (Duren, Germany). Soybean powder reference material with various GMO contents, that was certified by the Institute for Reference Materials and Measurement (IRMM) [26] was purchased from Fluka Biochemica, (Geel, Belgium). (His)6-tagged recombinant aequorin was expressed in an E. coli JM109 bacterial strain and purified as described previously [24].
All oligonucleotides used as primers and probes throughout this work were synthesized by MWG-Biotech AG (Ebersberg, Germany). A summary of their sequences and characteristics is given in Table 1. The upstream and downstream primers for the amplification of 35S promoter and NOS terminator were created according to [9]. The upstream and downstream primers employed for amplification of the lectin (le1) gene were as in [27]. All the upstream primers were biotinylated at the 5′ end. Oligonucleotide probes specific for each amplification product (lectin, 35S and NOS) were designed using Primer Premier 5 software.
The phosphate-buffered saline (PBS) contained 0.14 M NaCl, 2.7 mM KCl, 10 mM sodium phosphate and 1.7 mM potassium phosphate, pH 7.4. The wash solution consisted of 50 mM Tris, pH 7.5, 0.15 M NaCl, 2 mM EGTA, and 1 mL L−1 Tween-20. The blocking solution contained 10 g L−1 blocking reagent, 0.1 M maleic acid, 0.15 M NaCl, and 2 mM EGTA (final pH 7.5).
DNA extraction
DNA extraction from soybean powder was performed using the NucleoSpin Plant kit, following the manufacturer’s instructions with minor modifications. 40–100 mg of soybean powder was transferred to a sterile reaction tube, followed by addition of 500 μL of lysis buffer C1 and 10 μL of Rnase A. The mixture was vortexed thoroughly and incubated at 60 °C for 30 min. The mixture was then centrifuged at 11000 g in a microcentrifuge for 5 min. 300 μL of the supernatant was transferred to a NucleoSpin filter and centrifuged for 5 min at 11000 g. To the clarified solution, 300 μL of binding buffer C4 and 200 μL of ethanol were added. The solution was mixed, transferred to a NucleoSpin column, and centrifuged at 11000 g for 1 min. The column was washed three times with 400 μL of wash buffer CW and centrifuged at 11000 g for 1 min. Then, 100 μL of elution buffer CE, preheated at 70 °C, was added to the column followed by incubation at 70 °C for 5 min. DNA was eluted by spinning the column at 11000 g for 1 min. The integrity, as well as the quantity of the extracted DNA, was determined using 1% agarose gel electrophoresis and DNA markers (Lamda DNA/EcoRI, Hind III).
Polymerase chain reaction
All PCR reactions were carried out in a final volume of 50 μL using 50 mM Tris-HCl, pH 9.0, 15 mM (NH4)2SO4, 0.1% Triton X-100, 2 mM MgCl2, 0.2 μM of each of the primers, 0.2 mM of each of the dNTPs, 1.5 U Tth DNA polymerase and 20–25 ng of DNA extract (except for lectin, where 2–2.5 ng of sample DNA was used). The cycling conditions for each target DNA were as follows.
Le1 gene: 95 °C for 3 min followed by 35 cycles at 94 °C for 25 s and 62 °C for 30 s. At the end of the cycling, the mixture was held at 72 °C for 3 min.
35S promoter and NOS terminator: 95 °C for 3 min followed by 35 cycles at 95 °C for 25 s and 57 °C for 30 s. At the end of the cycling, the mixture was held at 72 °C for 3 min.
Labeling of probes with dTTP
Oligonucleotide probes, p 1 , p 2 and p 3 , specific for le1, 35S promoter and NOS terminator sequences, respectively, were tailed at the 3′ end with multiple dTTPs by using terminal deoxynucleotidyl transferase. The tailing reactions were carried out in a final volume of 20 μL consisting of 0.2 M potassium cacodylate, pH 7.2, 0.1 mM DTT, 0.1 mL L−1 Triton 100, 1 mM CoCl2, 2 mM dTTP, 30 units of TdT and 400 pmol of oligonucleotide. The reaction mixture was incubated at 37 °C for 1 h, and 50 mM EDTA was added to terminate the reaction. The labeled probes were used without purification.
Preparation of (dA)30-aequorin conjugate
Protected sulfhydryl groups were introduced to aequorin by reacting with SATA at a 1:1 molar ratio. A solution (61 μL) containing 10 nmol in 50 mM MOPS, pH 7.2, 1 M KCl, 5 mM EGTA and 0.3 M glucose was mixed with 1.8 μL of 1.25 g L−1 SATA (dissolved in DMSO) and 6.9 μL of 0.2 M sodium phosphate, pH 8.0, 5 mM EDTA and 2 M KCl. Following a 90 min incubation period at ambient temperature, the unreacted SATA was removed by ultrafiltration on the microcon filters. The volume was brought to 0.5 mL with 20 mM phosphate, pH 7.5, 0.5 mM EDTA, and 0.2 M KCl, and the solution was centrifuged at 10000 rpm for 50 min at 4 °C. This step was repeated once. The 5′ amino-modified oligonucleotide (dA)30 was derivatized with a 100-fold molar excess of sulfo-SMCC. The reaction was carried out in a final volume of 12.4 μL containing 3.4 nmol oligo and 340 nmol of sulfo-SMCC in 0.16 M bicarbonate buffer, pH 8. Following 30 min incubation, the excess of the linker was removed by gel filtration on G-25 columns. Subsequently, the maleimide-activated (dA)30 was mixed, at 1:1 molar ratio, with the SATA-derivatized aequorin in 20 mM phosphate, pH 7.5, 0.5 mM EDTA, and 2 M KCl. The reaction was initiated by the addition of 0.1 M NH2OH to deprotect the sulfhydryl group and the mixture was incubated for 12–16 h at 4 °C. Purification of the conjugate from the excess oligo was carried out by diluting the mixture ten times with water and loading onto a Ni-NTA agarose column. Prior to the application of the conjugate, Ni-NTA agarose was treated with 20 mM phosphate, 0.5 mM EDTA, 0.2 M KCl, pH 7.5, 10 g L−1 BSA and 0.15% Tween-20 to prevent nonspecific binding of the conjugate to the matrix. Following loading, the column was washed with 100 μL of 20 mM phosphate, 0.5 mM EDTA, 0.2 M KCl, pH 7.5. The conjugate was then eluted with 3×100 μL of 20 mM phosphate, 0.1 M EDTA, and 0.2 M KCl, pH 7.5.
Bioluminometric hybridization assays of PCR-amplified 35S, NOS and lectin DNA sequences
All incubation steps were carried out at ambient temperature unless otherwise stated. Opaque polystyrene wells were coated overnight with 50 μL of 1.4 mg L−1 streptavidin diluted in phosphate-buffered saline. Before use, the wells were washed three times with wash solution. Then, 50 μL of 5′ biotinylated PCR product diluted ten times in blocking solution was added into the wells and incubated with shaking for 30 min. The wells were then washed as above and 50 μL of 0.2 M NaOH was added. After incubating for 20 min, the non-biotinylated DNA strand was removed by washing the wells as above. Then, 50 μL of 14 nM poly(dT)-tailed target-specific probe, diluted in blocking solution containing 10% DMSO, was added in the wells and incubated for 30 min at 37 °C, to allow hybridization with the immobilized single-stranded amplification product. The wells were washed three times to remove the unbound probe, and then 50 μL of a solution containing 11 nM (with respect to aequorin) of (dA)30-aequorin conjugate, diluted in blocking solution containing 10% DMSO, was added into the wells. The hybridization was allowed to proceed for 15 min, and then the excess of reagent was removed by washing. The wells were then placed in the luminometer and the activity of aequorin was measured by dispensing 50 μL of light-triggering solution (20 mM Tris-HCl, 25 mM CaCl2, pH 7.5) and integrating for 3 s.
Results and discussion
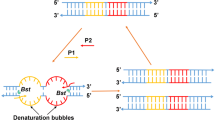
The principle of the bioluminometric assay for the determination of amplified 35S, NOS and lectin DNA is presented in Fig. 1. Biotinylated amplification fragments with sizes of 195, 180 and 181 bp were produced by PCR of the 35S, NOS and lectin DNA sequences, respectively. The extension step of PCR could be omitted without compromising the amplification efficiency. The hybridization assays were performed in microtiter wells coated with streptavidin. The PCR products for 35S, NOS and lectin were captured on separate wells through biotin/streptavidin interaction, and then one strand was removed by NaOH treatment. The immobilized single-stranded target DNAs were hybridized with oligonucleotide probes comprising a target-specific segment and a poly(dT) tail. This allowed the determination of all the hybrids by using the (dA)30-aequorin conjugate as a universal reagent.

Schematic presentation of the hybridization assay configuration. The biotinylated target DNA is captured on streptavidin-coated wells. One strand is dissociated with NaOH treatment and washed away. Immobilized single-stranded DNA is then hybridized with a target-specific probe labeled at the 3′ end with a poly(dT) tail. Then hybrids are allowed to hybridize with the universal aequorin-labeled (dA)30 oligonucleotide probe and the activity of aequorin is measured after the addition of Ca2+. SA=streptavidin, B=biotin, AEQ=aequorin
DNA from soybean powder certified reference material with known GMO content (0, 0.1, 0.5, 1, 2, 5%) was extracted by using the NucleoSpin plant DNA extraction kit, and served as a template for PCR. The extraction method was based on cell lysis, followed by binding of the DNA to a silica spin column at a high ionic strength and elution at a low ionic strength. Two alternative buffers (included in the kit) were tested for cell lysis. Buffer C0 contained a proprietary formulation of detergents optimized for a wide range of species. Buffer C1 was based on the established CTAB lysis procedure for plant material [9]. We found that buffer C0 gave a higher yield of DNA. The quantity and quality of extracted DNA were assessed by electrophoresis on a 1% agarose gel using DNA markers (lambda DNA/EcoRI, Hind III) as standards. DNA fragments of 23 Kbp were obtained, and the yield of the DNA from 40 mg of soybean powder ranged between 0.7 and 1.5 μg (100 μL solution). Removal of cell debris before binding of the DNA on the silica resulted in increased extraction yields. A “negative control for extraction”, which contained water instead of soybean powder and was subjected to all extraction steps, was carried out with each series of samples to confirm the absence of contamination in the reagents used for DNA extraction. “Negative controls for PCR”, containing water instead of extracted DNA, were also carried out for both 35S and NOS, to ensure the absence of contamination in the PCR reagents. The optimum amount of DNA template for the amplification of 35S and NOS was found to be 25 ng. Higher amounts of extracted DNA did not improve the yield amplification. This was attributed to the increased concentration of PCR inhibitors associated with the use of higher volumes of extracted DNA in the PCR mixture. It is known that the yield of PCR can be affected by the presence of substances originating from the sample, such as polysaccharides, lipids and phenols [8]. The amplification of the lectin gene was performed in order to assess the integrity of the extracted DNA.
The effect of the concentration of (dA)30-aequorin conjugate on the signal of the hyridization assay was studied in the range 4.4–17.6 nM while keeping the target DNA (NOS terminator amplicon) concentration constant at 0.4 nM. The signal increased continuously with increasing (dA)30-aequorin conjugate concentration. However, at conjugate concentrations higher than 11 nM, the signal-to-background ratio decreased, due to a higher nonspecific binding of the conjugate to the solid phase. The background was defined as the luminescence obtained in the absence of target DNA. Therefore, a conjugate concentration of 11 nM was used for all subsequent studies.
The detectability and analytical range of the hybridization assay for each target DNA (35S, NOS and lectin) were established as follows. Stock solutions of biotinylated amplification products were prepared by pooling several PCRs and determining the DNA concentration of each pool via picture densitometry (taken with a Kodak DC120 digital camera) of ethidium bromide-stained agarose gels. The ΦX174 DNA markers were used as calibrators. Then, serial dilutions of the three stock solutions were prepared and analyzed by hybridization to specific probes. The results are presented in Fig. 2. As low as 2 pM (100 amol per well) of the amplified 35S promoter, NOS terminator and lectin gene can be detected with signal-to-background ratios of 1.8, 2.4, and 2.2, respectively. The analytical range of the assays extended up to 2000 pM.

Luminescence versus the concentration of amplification product. Calibration graphs for the hybridization assays of amplification products from lectin (triangles), 35S promoter (circles) and NOS terminator (squares) sequences. All signals are corrected for background. The background is defined as the luminescence obtained from a solution containing no amplified DNA sequences
The repeatability of the hybridization assays was assessed by analyzing samples containing 2, 125 and 2000 pM of amplified DNA for 35S promoter and lectin, and 4, 125 and 1000 pM for NOS terminator. The CVs were 2.8, 4.2 and 2.6% for lectin, 9.9, 11.3 and 1.9% for 35S promoter, and 8%, 5.3% and 6% for NOS terminator, respectively (n=3).
Next, the proposed method for GMO detection was applied to DNA extracted from soybean powder certified reference material with known GMO contents of 0, 0.1, 0.5, 1, 2 and 5%. An additional sample containing 0.05% of GMO was included in the assay. This sample was prepared by mixing equal amounts of DNA extracted from samples containing 0.1% and 0% GMO. In Figs. 3 and 4, the luminescence signals obtained for NOS terminator and 35S promoter amplification products, respectively, are plotted versus the GMO content of the samples. It can be observed that the signals increase with the GMO content of the sample. The signal-to-background ratios for samples containing 0.05, 0.1 and 1% GMO were found to be 8.2, 13.2 and 42.2, as determined from the assay of the NOS terminator. The signal-to-background ratio for 0.1% GMO containing material was 9.2, as determined from the 35S promoter assay. The background is defined as the luminescence obtained when the extraction negative control, containing no sample DNA, was subjected to PCR followed by the hybridization assay. It should be noted that the 0.05% GMO content corresponds to about 10 genome copies [8] of transgenic material in the 25 ng of extracted DNA that served as template in the PCR. Also, only a tenth of the PCR mixture was used for the hybridization assay that contains amplification product from a single copy of transgenic material. Furthermore, due to the high detectability of the proposed system, we detected NOS terminator sequences in the 0% (nominal value) reference material with a signal-to-background ratio of 2.3. This is in accordance with the certificate of reference materials, where a non-GMO reference material (IRMM-410S-0) is referred to as containing less than 0.03% transgenic soybean powder. The absence of any contamination during the extraction procedure and PCR amplification was confirmed by comparing analytical signals obtained for extraction negative control and PCR negative control.

Application of the proposed method to the detection of transgenes in soybean certified reference material containing various amounts of Round Ready soybean. The method is based on the detection of NOS terminator. All signals are corrected for background

Application of the proposed method to the detection of transgenes in soybean certified reference material containing various amounts of Round Ready soybean. The method is based on the detection of the 35S promoter. All signals are corrected for background
In order to estimate the overall repeatability of the proposed method, including DNA extraction, PCR, and bioluminometric hybridization assay, DNA was extracted three times from soybean powder certified reference materials containing 0, 0.05, 0.1, 0.5, 1, 2 and 5%. PCR for the NOS terminator was performed with each extract, and the amplification products were analyzed four times by the hybridization assay. The CVs ranged from 7.5–19.8%.
Conclusions
Agarose gel electrophoresis and ethidium bromide staining of PCR-amplified DNA is the most widely used method for detection of GMO. The proposed bioluminometric method, however, offers significantly higher (about 80 times) detectability, and allows the confirmation of the amplified sequences by a rapid post-PCR hybridization assay. Furthermore, microtiter wells allow many samples to be analyzed in parallel in a single plate. As a consequence, microtiter well-based assay formats are highly automatable and suitable for high throughput screening. GMO analysis (detection and quantification) by real-time PCR has also been reported [14] but the cost (labeled probes and instrument) is much higher than the proposed system. Biosensors have also been proposed [15,16] for GMO analysis, but the detectabilities achieved are similar to those obtained by gel elctrophoresis. Since the luminescence signal increases with the GMO content, the present method may be used for semiquantitative assessment of the GM content of the sample. However, in order to obtain accurate quantitative information, a suitable internal standard should be used that contains the same primer binding sites and is distinguishable from the target by hybridization. Although the proposed method was developed for the detection of transgenes in soybean, it can be easily extended to the detection of other genetically modified organisms in a variety of samples. Indeed, the method presented here is general because (a) it detects the 35S promoter and NOS terminator sequences which are the most commonly-used regulatory elements for the production of genetically modified plants, and (b) the same universal detection reagent can be used with other target sequences in combination with suitable poly(dT)-tailed specific probes.
References
Erickson BE (2000) Anal Chem 72:454A-459A
Anklam E, Neumann DA (2002) J AOAC Int 85:754–756
Lipp M, Anklam E, Stave JW (2000) J AOAC Int 83:919–927
Stave JW (2002) J AOAC Int 85:780–786
Ahmed FE (2002) Trends Biotechnol 20:215–223
Holst-Jensen A, Ronning SB, Lovseth A, Berdal KG (2003) Anal Bioanal Chem 375:985–993
Anklam E, Gadani F, Heinze P, Pijnenburg H, Van Den Eede G (2002) Eur Food Res Technol 214:3–26
Bonfini L, Heinz P, Kay S, Van den Eede G (2002) Review of GMO detection and quantification techniques. European Commission Joint Research Center, Institute for Health and Consumer Protection, Food Products and Consumer Goods Unit, Ispra, Italy, pp 1–67
Lipp M, Brodmann P, Pietsch K, Pauwels J, Anklam E (1999) J AOAC Int 82:923–928
Lipp M, Bluth A, Eyquem F, Kruse L, Schimmel H, Van den Eede G, Anklam E (2001) Eur Food Res Technol 212:497–504
MacCormick CA, Griffin HG, Underwood HM, Gasson MJ (1998) J Appl Microbiol 84:969–980
Meyer R (1999) Food Control 10:391–399
Garcia-Canas V, Gonzalez R, Cifuentes A (2002) J Agr Food Chem 50:1016–1021
Permingeat HR, Reggiardo MI, Vallejos RH (2002) J Agr Food Chem 50:4431–4436
Feriotto G, Borgatti M, Mischiati C, Bianchi N, Gambari R (2002) J Agr Food Chem 50:955–962
Mariotti E, Minunni M, Mascini M (2002) Anal Chim Acta 453:165–172
Jones K, Keenan M, Keenan M (1999) Trends Biotechnol 17:477–481
Shimomura O, Johnson FH (1978) P Natl Acad Sci USA 75:2611–2615
Head JF, Inouye S, Teranishi K, Shimomura O (2000) Nature 405:372–376
Kendall JM, Badminton MN (1998) Trends Biotechnol 16:216–224
Lewis LC, Daunert S (2000) Fresen J Anal Chem 366:760–768
Verhaegen M, Christopoulos TK (1998) Anal Chem 70:4120–4125
Laios E, Ioannou PC, Christopoulos TK (2001) Anal Chem 73:689–692
Glynou K, Ioannou PC, Christopoulos TK (2003) Protein Expres Purif 27:384–390
Glynou K, Ioannou PC, Christopoulos TK (2003) Bioconjugate Chem 14:1024–1029.
Trapmann S, Catalani P, Conneely P, Corbisier P, Gancberg D, Hannes E, Le Guern L, Kramer GN, Prokisch J, Robouch P, Schimmel H, Zeleny R, Pauwels J, van den Eede G, Weighardt F, Mazzara M, Anklam E (2002) Certified Reference Material IRMM-410S. European Commission Joint Research Center, Institute for Reference Materials and Measurements, Geel, Belgium, pp 1–20
Wurz A, Bluth A, Zeltz P, Pfeifer C, Willmund R (1999) Food Control 10:385–389
Acknowledgements
The financial support by the Secretariat of the Research Committee of the University of Athens and a Karatheodory research grant from the University of Patras are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Glynou, K., Ioannou, P.C. & Christopoulos, T.K. Detection of transgenes in soybean via a polymerase chain reaction and a simple bioluminometric assay based on a universal aequorin-labeled oligonucleotide probe. Anal Bioanal Chem 378, 1748–1753 (2004). https://doi.org/10.1007/s00216-004-2507-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-004-2507-3