
Abstract
The degradation of Methyl Orange (C14H14N3SO3Na), chosen as a model sulfonated azo dye, was investigated in aqueous solutions containing suspended polycrystalline TiO2 particles under irradiation with simulated sunlight. The dye disappearance and the formation of the mineralization end products were monitored; the formation of the main transient intermediates was also examined in detail. Particular attention was devoted to the identification and to the evolution of fragments retaining the chromophoric group. The comparison of data coming from various analytical techniques led to a possible reaction mechanism for the degradation process, giving insight into an aspect of the treatment which has not been considered in previous studies.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Photocatalysis is one of the most promising advanced oxidation technologies (AOT) [1], based on the generation of active radical species which can lead to the complete destruction of a large number of organic and inorganic pollutants. This treatment, which has great potential for environmental remediation purposes and in particular for water and wastewater purification [2, 3, 4, 5, 6] has been developed over the past 15 years. It is based on a series of processes taking place at the semiconductor–solution interface which can be summarized as follows: irradiation with light of proper wavelength causes electron excitation from the semiconductor valence band to the conduction band, generating electron–hole pairs. Both electron and holes migrate to the surface of the semiconductor particles where they can react with water and dissolved oxygen giving rise to the formation of various oxidising species, including the highly reactive hydroxyl radicals [7, 8], whereas superoxide and perhydroxyl radicals are formed from the reaction of excited electrons with oxygen. The highly reactive oxygen-containing species are, in turn, able to attack and oxidise the organic molecules, generally leading to a complete mineralization of the substrate after a proper irradiation period.
The photocatalytic treatment of aqueous wastes containing azo dyes has been reported in the literature [9, 10, 11, 12, 13, 14, 15, 16, 17, 18]. These studies mainly focused on the discolouration monitoring and on the study of the substrate abatement (primary process), whereas less attention has been devoted to the investigation of reaction mechanisms. As a consequence, the identification and determination of intermediates and end products have often been disregarded.
The characterisation and the fate of transient species is however of great concern, since these intermediates can in some cases be even more dangerous than the initial substrate. Moreover, the analysis of their evolution with time can give insight into the reaction mechanism.
Taking into account the poor thermal stability and low volatility of most sulfonated azo dyes, the HPLC-MS technique can represent a suitable tool to analyse the reaction products as we demonstrated in a recent work concerning the characterisation of various Methyl Orange (MO) degradation products [18].
In the present article we discuss the results obtained from a detailed study of the photocatalytic degradation of Methyl Orange, chosen as a model azo dye because of its relatively simple structure. The effect of initial pH on the kinetics of MO abatement was investigated, together with the mass balance and evolution of the end products coming from the substrate mineralization, thus allowing us to obtain a general picture of the overall process. We then focused our attention on the structures of the transient molecules retaining the chromophore, looking at their evolution and trying to assess the sequence of reactions occurring among them.
Experimental
Reagents
The following analytical-grade reagents (all from Merck) were used as received: Methyl Orange (MO), ammonium acetate, acetonitrile (Lichrosolv), NaOH, HNO3, H2SO4, Na2CO3, NaHCO3, and KNO3. Ultrapure water was provided by a Milli-Q System (Millipore). TiO2 P25 from Degussa (ca. 80% anatase form) with a surface area of approximately 55 m2 g−1 was used in all the photocatalytic experiments. The semiconductor was preliminarily irradiated overnight to eliminate adsorbed organics and successively washed repeatedly with water. After centrifugation, the solid material was dried in an oven (ca. 12 h at 80°C) and a stock suspension containing 2 g L−1 of the dry powder in water was prepared by sonication. Stock solutions of the dye in water were prepared, protected from light and stored at 5°C.
Degradation experiments
Most degradation experiments were performed under aerobic conditions in stirred cylindrical closed Pyrex cells (40-mm i.d.×25-mm high), on 5 mL of aqueous TiO2 dispersions containing 20 mg L−1 of Methyl Orange and 200–600 mg L−1 of semiconductor. In some experiments the pH of the suspension was modified by adding either H2SO4 or NaOH. A 1,500-W xenon lamp, equipped with a 340-nm cut-off filter was used (Solarbox, from Cofomegra, Milan) to produce a simulated sunlight to irradiate the stirred dispersions. The temperature measured within the cell was approximately 55°C.
Analytical determinations
Samples were taken from the reaction vessels and filtered through 0.45-μm cellulose membranes (Millex, Millipore). After a defined irradiation time, the residual dye, its degradation products and the transient intermediates were determined according to the procedures detailed below.
The amount of residual dye was determined, after each irradiation cycle, by HPLC. Separations were obtained under isocratic conditions by using an RP-C18 column (Lichrospher RP-18 5 μm, 250-mm long and 4.6-mm i.d., from Merck) and a mobile phase composed of acetonitrile–ammonium acetate 10 mM (24/76 v/v). The eluent pH was 6.8; flow rate 0.8 mL min−1. Detector UV-Vis, wavelengths 472 nm, 270 nm and 220 nm.
The formation of CO2 was followed by head-space gas chromatography, after acidification of the reacted mixture with 5 mL of H2SO4 5 M and further thermal equilibration at 25°C. Samples of the gas phase (typically 200 μL) were taken through the cell Teflon stopcock by using a gas syringe and successively injected on a Carlo Erba 4600 gas chromatograph equipped with a Hayesep 80/100-mesh column (2-m long, 6-mm i.d.). Helium was used as the carrier gas at a flow rate of 30 mL min−1. The column and injector temperatures were 110°C and 130°C, respectively. A TCD detector was used (block temperature 150°C, filament temperature 250°C). Calibration curves were obtained by analysing standard solutions of Na2CO3, placed in the cells and treated as described above. Blanks obtained after long-term irradiation experiments indicated a negligible contribution from the reagents to the measured CO2.
Analysis of inorganic anions formed upon the treatment (NO3 − and SO4 2−) was performed by using a Dionex DX 500 apparatus equipped with a 200-mm-long×4-mm-i.d. AS9-HC column (Dionex) and electrochemical detector ED 40 (Dionex). Elution of the analytes was performed by using a solution composed of Na2CO3 12 mM and NaHCO3 5 mM.
Ammonium was determined by using the Berthelot colourimetric method. Absorbances were measured at 690 nm with a Uvikon 930 spectrophotometer (Kontron).
The analysis of organic transients was accomplished by HPLC-MS. The apparatus consisted of a pump Thermoquest TP 4000, a Thermoquest AS300 auto sampler and a Thermoquest UV 6000 diode array detector. The HPLC separation conditions were the same used for the MO determination, as described above. The eluent from the chromatographic column successively enter the UV-Vis diode array detector, the ESI interface and the quadrupole ion trap mass analyser. The detailed operative conditions can be found elsewhere [18].
Results and discussion
It is known that upon irradiation of TiO2 particles with light of wavelength < 400 nm, the formation of e- CB–h+ VB pairs occurs, leading to the formation of reactive hydroxyl radicals and superoxide radicals from water and dissolved oxygen according to the following reactions:
When dyes are present they can be excited by the incident radiation and a successive electron injection from the excited dye to the conduction band of the semiconductor occurs, leaving the corresponding cationic dye radical [20, 21]:
Both the dye molecules and the dye radicals can further react with the species generated at the semiconductor–solution interface and the degradation proceeds [22]. The above-mentioned degradation mechanisms can be present simultaneously and are indistinguishable due to the nature of the light source employed.
Decomposition of MO (primary process)
Preliminary blank experiments performed in a Solarbox without addition of TiO2 showed no appreciable decolourisation of the irradiated solution, thus confirming a good photostability of the dye under simulated solar light irradiation conditions.
In the presence of irradiated TiO2 the dye degradation occurs following a pseudo-first-order kinetic law, as previously reported in the literature for most organic substrates. Experiments performed at initial pH 5.6 at different TiO2/substrate ratios (10:1 and 30:1) showed, as expected, faster degradation rates in the presence of higher semiconductor concentrations (k obs=0.08 min−1 and 0.17 min−1, respectively). A further increase of semiconductor concentration is generally useless, since it is known that above approximately 2 g L−1 the increased light scattering drastically reduces the photonic flux entering the irradiated solution.
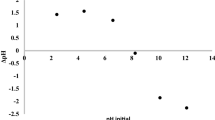
In order to investigate the pH effects on MO degradation, experiments were performed at a fixed substrate/TiO2 ratio (1:10) at three different initial pH values (2.2, 5.6 and 7.6) which were below, around and above the isoelectric point of the semiconductor oxide, respectively. A value of pI of approximately 6.2 was reported in the literature for the employed TiO2 [23].
The degradation curves (Fig. 1) indicate that the process is slower at pH 5.6 and is markedly faster at both pH 2.2 and 7.6. The observed results can be explained by taking into account the effect of pH upon the OH radical production and on dye adsorption onto the semiconductor particles, respectively. At higher pH values the formation of active .OH species is favoured, due to an improved transfer of holes to the adsorbed hydroxyls [24], but repulsive effects between the negatively charged TiO2 particles and the anionic dye start to be present. In contrast, at pH 2.2, electrostatic attraction between positively charged catalyst particles and the anionic dye operates, thus favouring the process at the semiconductor–solution interface. This hypothesis, sustained by literature data concerning the adsorption of similar sulfonated dyes [12, 17], is confirmed by the observed yellowish colour exhibited by the filtered TiO2.

Effect of pH on the MO degradation. MO 20 ppm; TiO2 200 ppm
The obtained results indicate that adsorption effects seem to predominate at pH 2.2, at which the TiO2 particles are neatly positively charged, whereas the increased radical formation reasonably predominates over repulsion at pH 7.6. At pH 5.6 there is a negligible dye adsorption and a moderate production of active surface radicals, thus justifying the observed slower dye abatement. Nevertheless most of the degradation experiments were run at an initial pH value of 5.6, as it corresponds to the unmodified pH of the aqueous Methyl Orange solution. By working at these conditions with a 30:1 semiconductor/substrate ratio, the complete degradation of the dye was observed, within the sensitivity limits of the applied technique, after approximately 45 min irradiation.
Evolution of the degradation products
Formation of CO2
The complete mineralization of one mole of the dye molecule implies the formation of the equivalent amount (14 moles) of CO2. However, the formation profile of CO2 in the presence of a semiconductor/dye ratio=10 (see Fig. 2a) clearly indicates that the mineralization is incomplete. In fact, after 150 min irradiation only about 70% of the initial organic carbon is transformed into CO2, thus implying that other organic compounds are still present in the solution at that time. In contrast, quantitative formation of CO2 was observed after about 1 h in experiments performed with a semiconductor/dye ratio=30 (see Fig. 2b). These last conditions were chosen for most of the successive experiments.

CO2 evolution. a MO 20 ppm, TiO2 200 ppm, b MO 20 ppm, TiO2 600 ppm; pH 5.6 in both cases
Formation of nitrogen-containing products
Previous studies on photocatalytic degradation of nitrogen-containing aromatics demonstrated that the relative abundance of the main mineralization end products (NH4 + and NO3 −) largely depends on the initial oxidation state of nitrogen in the substituent group and on substrate structure [25, 26].
Figure 3 shows that ammonium reaches about 61% of the stoichiometric nitrogen content after 3 h irradiation in closed cells, whereas after this time approximately 15% of the total nitrogen is found as NO3 −. The relatively high ammonium/nitrate ratio observed is not surprising, since similar ratios were usually found starting from organic substrates containing organic nitrogen in low oxidation states. Since the sum of both products gives about 76% of the stoichiometric nitrogen, only a partial mineralization of organic nitrogen is inferred.

Nitrate and ammonium evolution. MO 20 ppm, TiO2 600 ppm, pH 5.6
By increasing the oxygen excess in the reaction medium, for example by fluxing air or simply by opening the cells for some minutes during the treatment, the formation of NH4 + and NO3 − increases. In fact, after 4 h irradiation the sum of nitrate and ammonium contributions arising from the dye degradation in opened cells corresponded to more than 90% of the expected stoichiometric value. Taking into account that the formation of N2 from compounds containing azo groups has been recently proven [17] and that the formation of nitrogen oxides cannot be completely excluded during the photocatalytic treatments [27], a partial formation of such products could justify the observed lack of nitrogen. The absence of residual organic carbon after the above reported irradiation times suggests that nitrogen should be effectively mineralised.
Formation of SO4 2−
The evolution of the total concentration of this anion, the typical transformation product of sulfonic groups, is shown in Fig. 4. It can be seen that a plateau is reached after about 1 h irradiation, which corresponds to approximately 98% of the amount of sulfate expected assuming complete mineralization of the dye. The difference between the stoichiometric and found values is negligible taking into account that SO4 2− can adsorb onto the TiO2 particles [28].

Sulfate evolution. MO 20 ppm, TiO2 600 ppm, pH 5.6
Analysis of organic transients
We focused our attention on the identification of transient species still containing the chromophoric group and on the detection of typical hydrophilic aromatic products (polyhydroxybenzenes) which precede the aromatic ring opening. The analysis of compounds coming from the ring breaking, already described in detail in the literature, has not been undertaken in our study.
Figure 5 shows the HPLC profile recorded at 470 nm for a sample irradiated for 15 min. At this time nearly 50% of the initial carbon amount has been mineralised, but many peaks of coloured intermediates are still present. By analysing the same sample by means of HPLC-MS (under the same chromatographic conditions) a very similar pattern of peaks was observed. Methyl Orange (MO, t R=13.4 min) exhibits a clear MS signal corresponding to a negative ion of m/z 304. An intermediate (A) with m/z=320 (value higher than that of the parent molecule) exhibits a longer retention time. The chromatographic peaks C and D (m/z=290 and 276, respectively) correspond to fragments of the starting molecule, whereas the peak B (m/z=306) should originate from MO through the simultaneous introduction and leaving of groups having a mass difference of 2.

HPLC profile of a sample irradiated for 15 min. MO 20 ppm, TiO2 600 ppm, pH 5.6, detection at 470 nm
Chromatographic profiles similar to that shown in Fig. 5 were also obtained after 10, 22, 27 and 35 min irradiation. The disappearance of some components (or neat reduction of their concentrations) and/or the formation of new derivatives have been observed, as expected, depending on the sampling time.
A detailed MS analysis of the HPLC peaks following the previously described procedure [18] allowed us to suggest the formation of derivatives coming from the introduction of an hydroxyl group on MO (peak A, m/z=320), on compound C (peak B, m/z=306) and on derivative D (peak G, m/z=292). Further hydroxylation of compound B can lead to peak E (m/z 322), whereas hydroxylation of G can produce peak F (m/z=308). Compounds originating from MO through the loss of one methyl group (C, m/z=290) or two methyl groups (D, m/z=276) are also evidenced in the reaction system.
The above observations suggest that the presence of coloured intermediates is related to the presence of the sulfonic group; in fact, all the identified compounds containing the conjugated azo group retain the sulfonic moiety.
Plots of the peak areas of each of the detected intermediates against the irradiation time show bell-shaped profiles, thus indicating that photocatalytic degradation of such transient species occurs. Two sets of components have been grouped, one of them (Fig. 6a) representing the more abundant intermediates, whereas the second one (Fig. 6b) shows the behaviour of components detected at low concentration levels. None of these intermediates appear to be more persistent than MO in the reaction system. After few minutes of irradiation, HPLC and HPLC-MS analysis confirm the presence of compounds A–C as main degradation products; these intermediates can thus be considered as possible precursors of compounds observed at longer irradiation times.

Evolution profile of the intermediates identified by means of HPLC-MS analysis
Taking into account the above considerations and data, the reaction mechanism shown in Scheme 1 can be suggested.

Oxidation of methyl radicals can give rise to the formation of formic acid, in turn oxidised to CO2 through the photo-Kolbe reaction.
Compounds A–F can further degrade through different reactions paths including: sulfonic group removal (c), with successive formation of sulfate; removal of amino groups (d) and formation of ammonia; breaking of the azo group (e) with possible N2 loss. Some of these reactions can be summarized as follows:
in which Ar and Ar′ represent the (C6H4) and (C6H3) groups, respectively.
Coloured compounds originating through sulfonic group removal should be more hydrophobic than the starting molecules. However, we have not observed the appearance of chromatographic peaks absorbing at 472 nm at longer retention times. Moreover, the gradient elution up to 90% acetonitrile did not show the presence of any peak, thus suggesting that the sulfonic group removal and the chromophore breaking could proceed simultaneously.
The chromatographic analysis with UV detection at 220 nm of solutions sampled after 37 min irradiation still showed the presence of residual organic products. However, the HPLC-MS analysis of the same samples did not give any results, both in negative and in positive ion detection modes, thus allowing us to exclude the presence of molecules containing sulfonate groups or other groups (e.g. amino) able to take part in an acid–base equilibrium. After that time, only small amounts of very hydrophilic compounds having retention times slightly higher than t 0 are detected. The presence of catechol, hydroquinone, and 1,3,5-trihydroxybenzene (verified by using authentic standards) is consistent with the chromatographic pattern observed at lower retention times and confirms the typical results obtained during the photocatalytic treatment of most aromatic substrates.
The identification of residual compounds present at this reaction stage was not undertaken; this task is difficult due to the complex nature of the mixture (various ring-opening aliphatic products are usually simultaneously present together with aromatic by-products) and to the low concentration levels of these components.
Conclusions
The photocatalytic degradation of Methyl Orange can be readily performed under the investigated experimental conditions, leading to the complete mineralization of the dye. The structures of the reaction transient products formed during the treatment are consistent with a degradation route based mainly on hydroxylation and demethylation processes. This implies a considerable persistence of a yellowish colour in the solution, which remains even in the presence of very low concentrations of Methyl Orange. The observed intermediates are themselves degraded within about 45 min through further reaction steps including the crucial C–N bonds breaking and sulfonic group removal, mainly responsible for the observed waste discoloration. The evolution of both the end products and the organic transients during the time interval between the complete removal of MO and the CO2 formation plateau suggests that a further fast oxidation of the residual organics occurs, with a complete dye mineralization achievable after about 1 h irradiation.
References
Jackman AP, Powell RL (eds) (1991) Hazardous waste treatment technologies. Noyes Publications, Park Ridge (NJ)
Serpone N, Pelizzetti E (eds) (1989) Photocatalysis. Fundamentals and applications. Wiley, New York
Ollis DF, Al-Ekabi H (eds) (1993) Photocatalytic purification and treatment of water and air. Elsevier, Amsterdam
Legrini O, Oliveros E, Braun AM (1993) Chem Rev 93:671–698
Hoffmann MR, Martin ST, Choi W, Bahnemann DW (1995) Chem Rev 95:69–96
Brillas E, Mur E, Sauleda R, Sanchez L, Peral J, Domenech X, Casado J (1998) J Appl Catal B 16:31–42
Matthews RW (1984) Nature 11:23–28
Turchi CS, Ollis DF (1990) J Catal 122:178–192
Chen LC, Chou TC (1993) J Mol Catal 85:201–214
Vinodgopal K, Bedja I, Hotchandani S, Kamat PV (1994) Langmuir 10:1767–1771
Nasr C, Vinodgopal K, Hotchandani S, Chattopadhyay AK, Kamat PV (1997) Res Chem Intermed 23:219–231
Wang Y (2000) Water Res 34:990–994
Tanaka K, Padermpole K, Hisanaga T (2000) Water Res 34:327–333
Zhu C, Wang L, Kong L, Yang X, Wang L, Zheng S, Chen F, Mai Zhi F, Zong H (2000) Chemosphere 41:303–309
Grzechulska J, Morawski AW (2002) Appl Catalysis B Environ 36:45–51
Lachheb H, Puzenat E, Houas A, Ksibi M, Elaloui E, Guillard C, Herrmann (2002) J M Appl Catalysis B Environ 39:75–90
Augugliaro V, Baiocchi C, Bianco Prevot A, Garcia Lopez E, Loddo V, Malato S, Marcì G, Palmisano L, Pazzi M, Pramauro E (2002) Chemosphere 49:1223–1230
Baiocchi C, Brussino MC, Pramauro E, Bianco Prevot A, Palmisano L, Marcì G (2002) Int J Mass Spectrom 214:247–256
Kamat PV, (1990) Langmuir 6:512–513
Kamat PV, Das S, Thomas KG, George MV (1991) Chem Phys Lett 178:75–79
Wu T, Liu G, Zhao J, Hidaka H, Serpone N (1999) J Phys Chem 103:4862–4867
Kohrmann C, Bahnemann DW, Hoffmann MR (1988) J Phys Chem 92:5196–5201
Mills A, Davies RH, Worsley D (1993) Chem Soc Revs 22:417–425
Low G KC, McEvoy SR, Matthews RW (1991) Environ Sci Technol 25:460–467
Nohara K, Hidaka H, Pelizzetti E, Serpone N (1996) Catal Lett 36:115–118
Nohara K, Hidaka H, Pelizzetti E, Serpone N (1997) J Photochem Photobiol A 102:265–272
Vauthier M, Guillard C, Herrmann JM (2001) J Catal 201:46–59
Abdullah M, Low GKC, Matthews RW (1990) J Phys Chem 94:6820–6825
Acknowledgements
The authors wish to thank the MIUR (Rome) for financial support. Funding from COFIN 2000 is also gratefully acknowledged by E.P. and A.B.P.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bianco Prevot, A., Basso, A., Baiocchi, C. et al. Analytical control of photocatalytic treatments: degradation of a sulfonated azo dye. Anal Bioanal Chem 378, 214–220 (2004). https://doi.org/10.1007/s00216-003-2286-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-003-2286-2