
Abstract
Chlorpromazine, belonging to the first-generation antipsychotics, is known to cause some side effects, such as hepatotoxicity and agranulocytosis. The metabolic mechanisms of chlorpromazine remain elusive up to now, but are thought to result in the formation of some reactive metabolites having side effects on the parent drug. The goal of this work was to explore the metabolic mechanisms of chlorpromazine catalyzed by cytochrome P450 isoenzyme 1A2, a highly important activating enzyme of cytochrome P450 family, using DFT calculation. Three types of metabolic mechanisms were characterized, including S-oxidation, aromatic hydroxylation and N-dealkylation. The calculated results demonstrate that N 14-demethylation is the most thermodynamically and kinetically favorable metabolic pathway of chlorpromazine, followed by S5-oxidation. Then, mono-N-desmethylchlorpromazine is the most feasible chlorpromazine metabolite, which can occur further demethylation to form di-N-desmethylchlorpromazine. Besides, chlorpromazine 5-sulfoxide and 7-hydroxychlorpromazine are both the possible metabolites of chlorpromazine. Interestingly, N-methyl hydroxylation, the rate-limiting step of N-demethylation, proceeds predominantly via a single-electron-transfer mechanism. All the proton transfer processes involved in the aromatic hydroxylation and N-dealkylation prefer to occurrence in a water-assisted enzymatic process. Each metabolic pathway proceeds in the spin-selective manner via the low-spin state of Cpd I. Our results are in good accordance with the experimental observations, which can provide some essential implications for the metabolic mechanisms of chlorpromazine-like drugs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Chlorpromazine, a member of phenothiazine neuroleptics of an aliphatic type, belongs to the first generation of antipsychotics. Acting as a strong antagonist of the dopaminergic D2 receptor, chlorpromazine exhibits the antipsychotic effect and wide application in the treatment of schizophrenia, psychotic disorders and manic phase of bipolar disorders [1–4], which, however, is associated with the extrapyramidal side effect of chlorpromazine [5, 6]. Besides, the prolonged periods or high dosages (>2 g/day) of chlorpromazine may also cause skin disorders, ocular effects and rare, but severe irreversible corneal edema if not promptly identified [7–11].
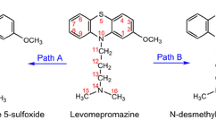
Chlorpromazine clearance is an enzymatic process and occurs extensively in the liver. Cytochrome P450 (CYP), an important heme-containing enzyme superfamily existing in multiple and distinct isoenzymes, is potentially responsible for the metabolism of chlorpromazine [12]. The biotransformation of chlorpromazine mainly proceeds via N-dealkylation, S-oxidation and aromatic hydroxylation, which result in the formation of different metabolites, including mono-N-desmethylchlorpromazine, di-N-desmethylchlorpromazine, chlorpromazine 5-sulfoxide, 7-hydroxychlorpromazine and 8-hydroxychlorpromazine (Scheme 1). Binding studies have shown that 7-hydroxychlorpromazine and mono-N-desmethylchlorpromazine are the most active metabolites of chlorpromazine [13], which own approximately 50 % of the parent compound activity in blocking D2 and a1 receptors. Besides, high levels of chlorpromazine 5-sulfoxide seem to have a side effect on therapeutic response in schizophrenic patients. All these metabolites are weak antagonists of the M1 receptor [14, 15]. Hence, most of the metabolites exhibit biological activity and may contribute to the side effects of the parent drug [16, 17].

Possible metabolic pathways of chlorpromazine catalyzed by CYP1A2
To date, many studies have devoted to find out the particular metabolic processes of chlorpromazine for which different cytochrome P450 isoenzymes are responsible. A recent study on cDNA-expressed human CYP isoenzymes showed that hepatic CYP1A2 is the only CYP isoenzyme that catalyzes the mono-N-demethylation and di-N-demethylation of chlorpromazine and is the main isoenzyme responsible for chlorpromazine 5-sulfoxidation at a therapeutic concentrations of the drug. CYP3A4 contributes to chlorpromazine 5-sulfoxidation to a lesser extent [12]. The catalysis of chlorpromazine N-demethylation and 5-sulfoxidation in humans exhibits a stricter CYP1A2 preference compared with the previously tested perazine, promazine and thioridazine [18–20]. On the other hand, cigarette smoking has been reported to increase the clearance of chlorpromazine [21]. Cigarette smoking is well known to cause marked induction of CYP1A2 and accelerate the metabolism of drugs catalyzed by CYP1A2, such as theobromine and caffeine [22]. Consequently, CYP1A2 is significantly involved in the metabolism of chlorpromazine.
The complete depiction of the metabolic mechanisms of chlorpromazine by CYP1A2 is presented in Scheme 2. The active species of CYP1A2 in catalysis is believed to be a FeIV = O complex with a porphyrin radical, referred to as compound I (Cpd I in brief) [23]. Originating from the high-spin (HS) quartet and low-spin (LS) doublet states of Cpd I, theoretical studies have suggested two possible mechanisms: the spin-selective manner (SSM) scenario and the two-state reactivity (TSR) [24]. For N-methyl hydroxylation, the first step of N-demethylation, two controversial mechanisms, single electron transfer (SET) [25, 26] versus hydrogen atom transfer (HAT) (Scheme 2) [27, 28], have existed for decades. There are not any standard rules to generalize their application, since the preferred mechanism is dependent on the detailed characteristics of the amine [29]. Our previous study on the caffeine metabolism catalyzed by CYP1A2 revealed a HAT mechanism with the TSR mechanism [30]. As of yet, the mechanic details of chlorpromazine metabolism catalyzed by CYP1A2 are still unclear. Some intriguing puzzles still remain, on which theory can provide the undiscovered insight and reveal some new features, such as, which mechanism does chlorpromazine metabolism favor: TSR or SSM? Which kind of mechanism does chlorpromazine N-demethylation proceed by: HAT or SET? Why chlorpromazine 8-hydroxylation is not mentioned in the previous experiments though the C7 and C8 atoms of chlorpromazine have similar characteristic? Which metabolic pathway predominates?

Metabolic mechanisms of chlorpromazine catalyzed by CYP1A2. a S-oxidation, b aromatic hydroxylation and c N-demethylation
The present work aimed to clarify the aforementioned puzzles to resolve the mechanistic details of chlorpromazine by CYP1A2 using density functional theoretical (DFT) calculations. This observation can provide some essential clues for the pharmacological study.
2 Methodology
With the purpose of gaining a theoretical insight into the vital metabolic mechanism of chlorpromazine catalyzed by CYP1A2, a popular six-coordinate oxo-ferryl species model, Fe4+O2−(C20N4H12)−(SH)− [31], was employed as the reactive Cpd I of CYP1A2, which contains a truncated heme and a thiolate axial ligand (SH–) built from the crystal structure of CYP1A2 (PDB:2HI4) [32]. The spin-unrestricted B3LYP was adopted with two basis sets: (a) LACVP(Fe)/6-31G (H, C, N, O, S, Cl) (B1 in brief) for the optimization of all the stationary points without symmetry constraints and (b) LACV3P+*(Fe)/6-311++G(d,p) (the rest) (B2 in brief) for single-point energy calculation both in the gas phase and in the protein environment to obtain reliable energetics. These model and computational methods chosen have been tested extensively and proven to be reliable in solving cytochrome P450 enzymes problems [23, 30, 33–37]. Transition states were affirmed by harmonic frequency analysis to possess only one imaginary frequency, and the stationary points were confirmed as minima with all positive frequencies. The validity of the TS geometry was verified by intrinsic reaction coordinate (IRC) calculations [38]. Natural population atomic (NPA) charges were determined using Reed and Weinhold’s natural bond orbital (NBO) analysis [39]. The binding energies of substrates to heme were corrected using basis set superposition error.
The weak polarization effect of the protein environment was modeled using PCM solvation method [40] with the dielectric constant of ε = 5.62 (chlorobenzene), which was taken as a reasonable compromise for the enzyme active site [41]. All of the single-point energies in chlorobenzene were corrected by the gas-phase thermodynamic quantities. The thermodynamic data reported in this paper are at 298.15 K and 1 atm. Cartesian coordinates for all the optimized geometries are presented in Supporting Information (SI) section. All DFT calculations were done using Gaussian 09 suite of programs [42].
3 Results and discussion
During phase I of metabolism, five possible catalytic pathways of CYP1A2 to chlorpromazine were investigated (Scheme 1), including oxidation at the S5 site of the thiazine ring (S5-oxidation, path A), aromatic hydroxylations at C7 (C7-hydroxylation, path B) and C8 (C8-hydroxylation, path C) sites as well as mono-N-demethylation (path D1) and di-N-demethylation (path D2) at N14 site. The complete reaction mechanisms of S-oxidation, C-hydroxylation and N-demethylation are depicted in Scheme 2. Each pathway is discussed in detail below.
3.1 S5-oxidation (path A)
Path A is a one-step reaction, which involves the direct oxidation of S5 atom by Cpd I (Scheme 2a). The optimized geometries of all the stationary points are presented in Fig. 1. The located transition-state A-TS1 species, A-2TS1 and A-4TS1, is characterized by its single imaginary frequency of 219.7i cm−1 LS and 601.9i cm−1 HS. It implies that heavy oxygen atom motion is comprised in the reaction vector, since, in principle, the magnitude of the imaginary frequency is quite high to exceed 1000 i cm−1 if atom motion in the transition state is largely comprised of light hydrogen atom migration [43]. Animation of the single imaginary frequency of A-TS1 species shows the motion of oxygen atom from Fe to S5 atoms, leading to Fe–O breakage and S–O bond formation. The Fe–O bond in transition-state A-TS1 species is elongated by 0.083 Å LS and 0.179 Å HS with respect to their corresponding reactant complex A-RC species, A-2RC and A-4RC. This is companied with the remarkable curtailment of the S–O distance. In A-TS1 species, it can be readily seen that oxygen atom is closer to iron atom (rFe-O=1.734 Å LS and 1.832 Å HS) than to sulfur atom (r S–O = 2.278 Å LS and 2.044 Å HS). Therefore, A-TS1 species is more reactant like in character. Particularly, this reactant-like character of A-2TS1 on LS state is more obvious than A-4TS1 on HS state. In the product complex A-PC species, A-2PC and A-4PC, a double bond is formed between oxygen and S5 atoms (r S–O = 1.728 Å LS and 1.700 Å HS). Concomitantly, the Fe–O bond is elongated to be 2.029 Å LS and 2.370 Å HS.

Optimized structures (in Å) for S5-oxidation (path A) of chlorpromazine by Cpd I at the B1 level (values out of parentheses, data for LS state; values in parentheses, data for HS state. Definitions are the same in the following structure figures)
The spin density distribution for the stationary points involved in S5-oxidation is listed in Table S1. In the A-RC species, spin density is localized on Cpd I. Then, it shifts from the oxygen, –SH ligand and porphyrin to the iron and sulfur atom of chlorpromazine. In A-TS1 species, the accumulation of spin density on sulfur atom indicates the partial formation of S–O and Fe–O single bonds, resulting in the occurrence of an unpaired electron on sulfur atom. As the reaction proceeds, the unpaired electron on sulfur further pairs with the electron of oxygen atom residing on Fe–O single bond to produce the S=O double bond in A-PC species. Simultaneously, Fe–O single bond is cleaved. There is no spin density on sulfur atom any more. Spin density is mainly localized on iron on both LS and HS states. All the results clearly indicate a stepwise transfer of two electrons.
The calculated energy profiles for S5-oxidation (path A) are shown in Fig. 2. The LS/HS energy barrier of path A is 9.6/14.1 kcal/mol in the gas phase, which corresponds to 8.7/12.9 kcal/mol when ZPE correction and the bulk polarity effect are incorporated. The oxidation process is endothermic with the reaction energy of 21.6 kcal/mol LS and 7.9 kcal/mol HS in chlorobenzene. The lower LS energy barrier agrees well with the more reactant-like character of A-2TS1 than of A-4TS1 as discussed above. It can be readily seen that the solvation of chlorobenzene contributes to the barrier decrease in path A. The energy gap between A-2TS1 and A-4TS1 is large to be 4.2 kcal/mol in chlorobenzene. The ratio of the reaction rate on the LS/HS route is 1198:1. As a consequence, S5-oxidation of chlorpromazine by Cpd I proceeds mainly in a SSM mechanism. The LS state of Cpd I is more feasible than the HS state.

Energy profiles (in kcal/mol) for S5-oxidation (path A) of chlorpromazine by Cpd I (values out of parentheses, relative energies in the gas phase; values in parentheses, relative energies in chlorobenzene. Definitions are the same in the following energy profile figures)
3.2 Aromatic hydroxylation (paths B and C)
Owning to the similar properties of C7 and C8 sites, the hydroxylations of chlorpromazine at C7 (path B) and C8 (path C) sites have been explored for comparison. Aromatic hydroxylation process begins with the nucleophilic addition of Cpd I’s oxygen atom at the carbon atom to form the oxidized chlorpromazine intermediate, which then converts into the hydroxylated chlorpromazine metabolite via intramolecular 1,2-proton transfer (Scheme 2b).
3.2.1 Nucleophilic addition of Cpd I’s oxygen atom
The optimized geometries of all the stationary points for the initial nucleophilic addition of Cpd I’s oxygen atom along paths B and C are shown in Fig. 3. For C7-hydroxylation (path B), the located transition-state B-TS1 species, B-2TS1 and B-4TS1, is characterized by its single imaginary frequency of 269.0i cm−1 LS and 440.1i cm−1 HS (Fig. 3a). Animation of the single imaginary frequency of B-TS1 species indicates the migration of heavy oxygen atom from Fe atom to C7 atom, leading to the C7–O bond formation. Compared with reactant complex B-RC species, B-2RC and B-4RC, the Fe–O bond in the transition-state B-TS1 species is elongated by 0.077 Å LS and 0.094 Å HS, which is concurrent with the curtailment of the C7–O distance by 1.382 Å LS and 1.438 Å HS. In B-TS1 species, the distance of Fe–O bond is elongated to be 1.727 Å LS and 1.747 Å HS, while the C7–O distance is shorten to be 1.948 Å LS and 1.851 Å HS. It can be readily seen that oxygen atom is closer to iron atom than to C7 atom. So B-TS1 species is still more reactant-like in character. Particularly, the reactant-like characters of B-2TS1 are more obvious than HS transition-state B-4TS1. In the intermediate B-IM species, B-2IM and B-4IM, a single bond is formed between oxygen and C7 atom with the C7–O bond length of 1.469 Å LS and 1.488 Å HS. Simultaneously, the Fe–O bond is further elongated to be 1.920 Å LS and 1.823 Å HS.

Optimized structures (in Å) for the nucleophilic attack of Cpd I’s oxygen atom at C7 (a, path B) and C8 (b, path C) sites of chlorpromazine at the B1 level
Considering C8-hydroxylation (path C), as shown in Fig. 3b, the transition-state C-TS1 species, C-2TS1 and C-4TS1, located for the nucleophilic attack of Cpd I’s oxygen atom is characterized by its single imaginary frequency of 314.6i cm−1 LS and 465.5i cm−1 HS. Animation of the single imaginary frequency of C-TS1 species also concerns the motion of heavy oxygen atom from Fe atom to C8 atom, leading to the C8–O bond formation. The geometrical feature tendencies for the nucleophilic addition of Cpd I’s oxygen atom along path C are similar to path B on either the LS or the HS state: The Fe–O distance in C-TS1 species is elongated by 0.088 Å LS and 0.095 Å HS with respect to its corresponding reactant complex C-RC species, C-2RC and C-4RC, and the C8–O distance is dramatically shorted by 1.437 Å LS and 2.546 Å HS. In C-TS1 species, the distance of Fe–O bond is 1.737 Å LS and 1.749 Å HS and the C8–O distance is 1.889 Å LS and 1.833 Å HS, indicating the partial formation of C8–O bond. It can be readily seen that oxygen atom is also closer to iron atom than to C8 atom. Thus, C-TS1 species is still more reactant-like in character and the reactant-like character of C-2TS1 is more obvious than C-4TS1. In the intermediate C-IM species, C-2IM and C-4IM, a single bond is formed between oxygen and C8 atom. The C8–O bond length is 1.504 Å LS and 1.489 Å HS, and the Fe–O bond is elongated to be 1.805 Å LS and 1.812 Å HS.
The spin density distribution of the stationary points involved in the nucleophilic addition processes of paths B and C is listed in Table S2. In the reactant complexes, spin density mainly delocalizes on Cpd I. As the nucleophilic addition proceeds, spin density migrates from the oxygen, –SH ligand and porphyrin to the iron atom and chlorpromazine. In B-TS1 and C-TS1 species, high spin density resides on iron and chlorpromazine. The accumulation of spin density on the beta carbon atom (C8 atom in path B and C7 atom in path C) implies the emergence of an unpaired electron. Further delocalization appears in the B-4IM and C-4IM, with a significant amount of the spin density on chlorpromazine delocalized in the direction of beta carbon. After the nucleophilic addition, spin density is localized on Cpd I on LS state and delocalized between Cpd I and chlorpromazine on HS state.
The calculated energy profiles for the nucleophilic addition of Cpd I’s oxygen atom along paths B and C are shown in Fig. 4. As shown in Fig. 4, the gas-phase LS/HS energy barrier is 15.4/16.7 kcal/mol for B-TS1 species and 16.6/19.5 kcal/mol for C-TS1 species, which correspond to 13.9/16.2 and 16.7/18.9 kcal/mol when ZPE correction and the bulk polarity effect are incorporated. The energy gap between LS and HS is 2.3 kcal/mol for B-TS1 species and 2.2 kcal/mol for C-TS1 species in chlorobenzene. The lower LS energy barriers of B-2TS1 and C-2TS1 with respect to B-4TS1 and C-4TS1 arise from their more reactant-like characters. The ratio of the reaction rate on the LS/HS route is 48.5:1 for B-TS1 species and 34.6:1 for C-TS1 species. Consequently, the nucleophilic addition processes along paths B and C proceed predominantly via the LS state in the SSM mechanism.

Energy profiles (in kcal/mol) for the nucleophilic attack of Cpd I’s oxygen atom at C7 (a, path B) and C8 (b, path C) sites of chlorpromazine
3.2.2 Intramolecular proton transfer
In this step, the formed intermediate B-IM species (B-2IM and B-4IM) and C-IM species (C-2IM and C-4IM) can convert to its enol form product complexes B-PC species (B-2PC and B-4PC) and C-PC species (C-2PC and C-4PC) via intramolecular proton transfer. Owning to the strong interaction between Fe and oxygen atoms in B-IM and C-IM species (r Fe–O = 1.805–1.920 Å), this proton transfer should be an enzymatic process. Considering the presence of one explicit water molecule in the active site of the human P450 1A2 [25], both the direct and one-water-assisted enzymatic proton transfer mechanisms were taken into account for comparison. The optimized structures of transition states and calculated energy profiles for the intramolecular proton transfer processes of paths B and C are shown in Figs. 5 and 6, respectively.

Optimized transition state structures (in Å) for the intramolecular proton transfer along paths B (a, b) and C (c, d) at the B1 level

Energy profiles (in kcal/mol) for the intramolecular proton transfer for the intramolecular proton transfer along paths B (a, b) and C (c, d)
For the direct reaction mechanism, the located transition state B-TS2 species (B-2TS2 and B-4TS2) and C-TS2 species (C-2TS2 and C-4TS2) are all triangle structures. The single imaginary frequencies of B-TS2 and C-TS2 species are greater than 1200i cm−1, which mainly comprises the motion of the light hydrogen atom from the alpha carbon (C7 atom in path B and C8 atom in path C) to the bonding oxygen atom. The gas-phase LS/HS energy barrier is 12.5/17.1 kcal/mol for B-TS2 species and 9.8/11.4 kcal/mol for C-TS2 species, which corresponds to 16.0/12.7 and 12.3/8.9 kcal/mol when ZPE correction and the bulk polarity effect are incorporated. Both proton transfer processes are exothermic with the reaction energies of 47–60 kcal/mol in chlorobenzene.
With the presence of one explicit water molecule, a water bridge is formed, receiving a proton and donating one in turn. The B-TS2w and C-TS2w species involved, B-2,4TS2w and C-2,4TS2w, is expanded to be pentagon structure, which is characterized by the single imaginary frequency of 49–184 i cm−1. Animation of the single imaginary frequency of B-TS2w and C-TS2w species shows the synchronous transfers of two hydrogen atoms, namely H atom from alpha C atom to Ow atom of the water molecule and the Hw atom of water molecule from Ow to O atoms. The gas-phase LS/HS activation energy is 2.7/7.6 kcal/mol for B-TS2w species and −0.7/0.4 kcal/mol for C-TS2w species, which corresponds to 4.8/6.8 and −0.4/0.6 kcal/mol when ZPE correction and the bulk polarity effect are incorporated. The water-assisted proton transfer processes are still exothermic with the reaction energies of 43–55 kcal/mol in chlorobenzene. As expected, the water-assisted energy barriers are dramatically lower than those of the direct reaction processes by 6–12 kcal/mol, attributed to the lower constraint of the pentagon transition state structures, where the energies required for the bond dissociation and formation are smaller than the triangle so much that a large amount of deformation energy is saved. It suggests that the presence of the explicit water is thermodynamically and kinetically beneficial to this proton transfer process. Additionally, the reactant complexes on LS state, B-2IM and C-2IM, are much more stable than those on HS state, B-4IM and C-4IM, indicating clearly that this proton transfer process still proceeds in a SSM mechanism, mainly via the LS state of Cpd I.
As a result, the rate-determining steps along the potential energy surfaces of paths B and C both involve the nucleophilic attack of Cpd I’s oxygen atom, each of which proceeds predominantly via the LS state in the SSM mechanism. Path B is thermodynamically and kinetically more favorable than path C due to its lower activation barrier, and 7-hydroxychlorpromazine therefore is the optimum aromatic hydroxylation product of chlorpromazine by Cpd I.
3.3 N-demethylation (paths D1 and D2)
As depicted in Scheme 2c, the overall reaction of CYP-catalyzed N-dealkylation of chlorpromazine proceeds through two stepwise stages, N-methyl hydroxylation and C–N bond fission to release formaldehyde.
3.3.1 N-methyl hydroxylation
For N-methyl hydroxylation, the reaction pattern starts with proton abstraction from methyl group to the Cpd I’s oxygen atom, forming a N-methylene intermediate, which then acts as a receptor of hydroxyl from the active iron species by oxygen-rebound process. The reaction is concerted on the LS state, forming carbinolamine–heme complex without a distinct oxygen-rebound step. The high-spin proton transfer intermediate, 4INT, just serves as a shoulder but not a real minimum and fall to the corresponding carbinolamine–heme complex with a barrier-free oxygen rebound. Therefore, the N-methyl hydroxylation is an effective concerted process on the two spin states. These results are in agreement with our recent work [30] and follow earlier observation on P450 reactions [35, 44].
The optimized geometries of all the stationary points for the N-methyl hydroxylations of paths D1 and D2 are shown in Fig. 7. As for path D1, the mono-N-demethylation, the transition state D1-TS1 species, D1-2TS1 and D1-4TS1, located for N-methyl hydroxylation is characterized by its single imaginary frequency of 604.1i cm−1 LS and 1032.0i cm−1 HS. Animation of the single imaginary frequency of D1-TS1 species comprises the motion of hydrogen from methyl carbon atom to the oxygen atom of Cpd I atom. Compared with the reactant complex D1-RC species, D1-2RC and D1-4RC, it can be seen that the Fe–O bond is elongated by 0.049 Å LS and 0.082 Å HS in the transition state D1-TS1 species, whereas the O–H distance is considerably reduced by 0.817 Å LS and 0.856 HS. This is accompanied by the elongation of the C–H bond by 0.082 Å LS and 0.174 Å HS. Transition-state D1-TS1 species is characterized by an almost collinear arrangement of the C–H–O atoms with the bond angles of 170.9° LS and 171.3° HS. The geometric parameters in Fig. 7 also indicate the degrees of C–H bond cleavage in the D1-TS1 species. In the D1-TS1 species, hydrogen atom is closer to carbon atom (r C–H = 1.186 Å LS and 1.279 Å HS) than to oxygen atom (r O–H = 1.477 Å LS and 1.301 Å HS). Therefore, they are more reactant like in character. Furthermore, the reactant-like character of D1-2TS1 on the LS state is more obvious than of D1-4TS1 on the HS state. As the reaction progresses from D1-RC species to the intermediate D1-IM1 species, D1-2IM1 and D1-4IM1, the distances between the SH ligand and Fe are shortened by 0.286 Å LS and 0.055 Å HS.

Optimized structures (in Å) for the N-methyl hydroxylation in mono-N-demethylation (a, path D1) and di-N-demethylation (b, path D2) of chlorpromazine by Cpd I at the B1 level
As for the di-N-demethylation path D2, the located transition-state D2-TS1 species, D2-2TS1 and D2-4TS1, is characterized by its single imaginary frequency of 795.7 i cm−1 LS and 1065.7 i cm−1 HS. Animation of the single imaginary frequency of C-TS1 species also concerns the motion of hydrogen from methyl carbon atom to the oxygen atom of Cpd I atom. The geometrical feature tendencies for the N-methyl hydroxylation along path D2 are similar to path D1. The Fe–O bonds in the transition-state D2-TS1 species are elongated by 0.051–0.088 Å, while the O–H distances are reduced by 0.817–0.856 Å. This is accompanied by the elongation of the C–H bonds by 0.084–0.172 Å. Transition-state D2-TS1 species also exhibits an almost collinear arrangement of the C–H–O atoms with the bond angles of 174.5–174.8° and the reactant-like character. In the D2-TS1 species, hydrogen atom is closer to carbon atom (r C–H = 1.186 Å LS and 1.274 Å HS) than to oxygen atom (r O–H = 1.473 Å LS and 1.305 Å HS). Clearly, the reactant-like character of D1-2TS1 on the LS state is still more obvious than of D1-4TS1 on the HS state. In the intermediate D2-IM1 species, D2-2IM1 and D2-4IM1, the distances between the SH ligand and Fe are shortened by 0.074–0.289 Å.
The spin density distribution for the stationary points involved in the N-methyl hydroxylations of paths D1 and D2 is reported in Table S3. Initially, spin density is localized on Cpd I. Then, during the C–H bond activation step it shifts from the oxygen, –SH ligand and porphyrin to the Fe and chlorpromazine. When reaching D1-TS1 species and D2-TS1 species, high spin density resides on the nitrogen atom of chlorpromazine and the iron. The accumulation of spin density on nitrogen in D1-2TS1 and D2-2TS1 suggests that it may be involved in the stabilization of carbon during leaving of the hydrogen atom. In D1-4TS1 and D2-4TS1, there is some spin density locating on the carbon of chlorpromazine. On the porphyrin, on the other hand, excess unpaired spin is no longer observed and spin density is delocalized between Fe-oxo and –SH. Further delocalization appears in the D1-4IM and D2-4IM, with a significant amount of the spin density on chlorpromazine delocalized in the direction of carbon. There is also accumulation of the spin density on iron by a shift from –SH and oxygen. After the hydroxyl rebound step, spin density is localized on iron on the LS and delocalized between iron and –SH on the HS. All the observations indicate that the N-methyl hydroxylations of paths D1 and D2 proceed via the SET mechanism.
The calculated energy profiles for the N-methyl hydroxylations of paths D1 and D2 are shown in Fig. 8. The gas-phase LS/HS energy barrier is 4.7/3.3 kcal/mol for D1-TS1 species and 6.4/5.1 kcal/mol for D2-TS1 species, which decrease to 1.9/1.9 and 4.5/4.3 kcal/mol when ZPE correction and the bulk polarity effect are incorporated. The energy gaps between LS and HS of D1-2TS1 and D2-2TS1 species are approximately zero in chlorobenzene, indicating a TSR mechanism. The N-methyl hydroxylation is greatly exothermic with the reaction energies of 44–61 kcal/mol in chlorobenzene.

Energy profiles (in kcal/mol) for the N-methyl hydroxylation in mono-N-demethylation (a, path D1) and di-N-demethylation (b, path D2) of chlorpromazine by Cpd I
3.3.2 C–N bond fission
As the second step of the N-demethylation, C–N bond fission requires a proton transfer from the hydroxyl oxygen to nitrogen atom, while the C–N bond between the alpha carbon and nitrogen gradually breaks. We examined two different processes for the second step: the direct proton transfer and the water-assisted proton transfer. Both the enzymatic and nonenzymatic environments were taken into account.
The optimized transition state structures and energy profiles for the C–N bond fission along path D1 are depicted in Figs. 9 and 10, respectively. For the direct proton transfer process, the enzymatic and nonenzymatic transition states (D1-2,4TS2 and D1-TS2) are all rhomboid structures, whose single imaginary frequencies are greater than 1473i cm−1. Animation of the single imaginary frequency shows the direct motion of hydroxyl hydrogen to nitrogen atom. For the water-assisted proton transfer process, the involved enzymatic and nonenzymatic transition states (D1-2,4TS2w and D1-TS2w) are expanded to be hexagon structures. Their single imaginary frequencies range from 811 to 1105i cm−1, wherein the water molecule hydrogen bonding with the nitrogen gradually transfers a proton to the nitrogen, while the hydroxyl proton gradually transfers to the water oxygen. Figure 10 shows that the gas-phase energy barriers of the direct proton transfer are 36.1 and 27.8/21.1 kcal/mol for the nonenzymatic and enzymatic LS/HS environments, respectively, which decrease to 30.3 and 23.5/20.1 kcal/mol when ZPE correction and the bulk polarity effect are incorporated. For the water-assisted proton transfer process, the nonenzymatic and enzymatic LS/HS energy barriers are 17.0 and 11.4/11.8 kcal/mol, respectively, in the gas-phase, which decrease to 13.8 and 9.5/10.1 kcal/mol when ZPE correction and the bulk polarity effect are incorporated. Clearly, with the assistance of one explicit water molecule, the energy barrier required for C–N fission is dramatically lowered by 10–17 kcal/mol in chlorobenzene. The large barrier decrease in the water-assisted proton transfer process can be derived from the smaller ring tension of the hexagon compared with rhomboid. Furthermore, the water-assisted enzymatic LS/HS energy barrier is 4.3/3.7 kcal/mol lower than the nonenzymatic energy barrier in chlorobenzene. The binding energy of the carbinolamine to heme in path D1 is 4.2 kcal/mol LS and 1.0 kcal/mol HS (Table S4). The lower binding energy on HS state results in the higher energy of the carbinolamine on HS state (15.5 kcal/mol higher than that on LS state in chlorobenzene). Comparison of the above barrier data with the binding energies of carbinolamine to heme shows that C–N fission proceeds in an enzymatic route, predominately via the LS state.

Optimized transition-state structures (in Å) for the C–N fission along the mono-N-demethylation (path D1) of chlorpromazine by Cpd I at the B1 level

Energy profiles (in kcal/mol) for the C–N fission along the mono-N-demethylation (path D1) of chlorpromazine by Cpd I at the B1 level
Figures 11 and 12 show the optimized transition state structures and energy profiles for the C–N bond fission along path D2, respectively. Analog to path D1, the enzymatic and nonenzymatic transition states (D2-2,4TS2 and D2-TS2) involved in the direct proton transfer process of path D2 are also rhomboid structures, which expand to be the hexagon structures of D2-2,4TS2w and D2-TS2w in the water-assisted proton transfer process. For the direct proton transfer process, the gas-phase nonenzymatic and enzymatic LS/HS energy barriers are 37.6 and 28.0/30.8 kcal/mol, respectively, which decrease to 31.4 and 24.3/27.7 kcal/mol when ZPE correction and the bulk polarity effect are incorporated. With the assistance of one explicit water molecule, the gas-phase nonenzymatic and enzymatic LS/HS energy barriers are 18.5 and 11.6/12.4 kcal/mol, respectively, which decrease to 14.7 and 9.7/10.6 kcal/mol when ZPE correction and the bulk polarity effect are incorporated. By comparison, it can be readily seen that the energy barriers involved in the water-assisted proton transfer processes are 14–17 kcal/mol lower than those of the direct proton transfer in chlorobenzene. Comparison of the above barrier data with the binding energies of carbinolamine to heme (LS: 5.0 kcal/mol; HS: 1.5 kcal/mol, Table S4) indicates that C–N fission should happen in a water-assisted enzymatic environment, mainly via the LS state.

Optimized transition state structures (in Å) for the C–N fission along the di-N-demethylation (path D2) of chlorpromazine by Cpd I at the B1 level

Energy profiles (in kcal/mol) for the C–N fission along the di-N-demethylation (path D2) of chlorpromazine by Cpd I at the B1 level
Taken together, the C–N bond fission proceeds mainly via the water-assisted enzymatic proton transfer process on LS state. N-methyl hydroxylation is the rate-determining step along the potential energy surface of either mono-N-demethylation (path D1) or di-N-demethylation (path D2).
In summary, the LS/HS activation barriers for the rate-determining steps of S5-oxidation (path A), aromatic hydroxylations at C7 (C7-hydroxylation, path B) and C8 (C8-hydroxylation, path C) sites as well as mono-N-demethylation (path D1) and di-N-demethylation (path D2) are 8.7/12.9, 13.9/16.2, 16.7/18.9, 1.9/1.9 and 4.5/4.3 kcal/mol, respectively, in chlorobenzene. It can be concluded that N-demethylation, paths D1 and/or D2, is the most plausible metabolic pathway of chlorpromazine catalyzed by CYP1A2 on either LS or HS state, followed by paths A, B and C in turn. So mono-N-desmethylchlorpromazine (D1-PC) is the optimum metabolite of chlorpromazine, which can undergo the second N-demethylation to yield di-N-desmethylchlorpromazine (D2-PC). All the observations agree well with the experimental results [12]. Generally, all the metabolic pathways proceed in a SSM mechanism, mainly through the LS state.
4 Conclusions
The metabolic mechanisms of chlorpromazine catalyzed by CYP1A2 have been systematically addressed in the present work based on DFT calculation. The mechanistic conclusions have been revealed as follows. S5-oxidation (path A) is a one-step reaction, which involves a stepwise transfer of two electrons. The rate-determining step of aromatic hydroxylation (paths B and C) involves the nucleophilic addition of Cpd I’s oxygen atom. The subsequent intramolecular proton transfer is prone to the water-assisted enzymatic proton transfer mechanism. N-methyl hydroxylation is the rate-limiting step of N-demethylation, which proceeds predominantly via a SET mechanism. The generated carbinolamines then prefer to decomposition in a water-assisted enzymatic process on LS state. N-demethylation is the most thermodynamically and kinetically feasible metabolic pathway of chlorpromazine due to its lowest activation barrier, followed by path A. Mono-N-desmethylchlorpromazine therefore is the most feasible chlorpromazine metabolite catalyzed by CYP1A2, followed by di-N-desmethylchlorpromazine, chlorpromazine 5-sulfoxide, 7-hydroxychlorpromazine and 8-hydroxychlorpromazine. Each metabolic pathway proceeds in a SSM mechanism, predominately via the LS state of Cpd I. Our results keep in good accordance with the experimental observations, which can provide some complementary insights into the N-dealkylation mechanism by CYP and offer general implications for the metabolic mechanism of chlorpromazine-like drugs.
5 Supporting information
Spin densities for the species involved in the metabolic processes of chlorpromazine, binding energies of carbinolamine to heme in the N-demethylation of chlorpromazine and cartesian coordinates of all stationary points along the potential energy profiles.
References
Liu X, De Haan S (2009) Cochrane Database Syst Rev 2:CD007778
Anthérieu S, Bachour-El Azzi P, Dumont J, Abdel-Razzak Z, Guguen-Guillouzo C, Fromenty B, Robin MA, Guillouzo A (2013) Hepatology 57:1518–1529
Tohen M, Vieta E (2009) Bipolar Disord 2:45–54
Morak-Młodawska B, Jeleń M (2007) Pol Merkur Lek 23:459–461
Shin SY, Kim CG, Kim SH, Kim YS, Lim Y, Lee YH (2010) Exp Mol Med 42:395–405
Liperoti R, Pedone C, Corsonello A (2008) Curr Neuropharmacol 6:117–124
Drucker AM, Rosen CF (2011) Drug Saf 34:821–837
Lasic D, Cvitanovic MZ, Uglešic B, Višic V, Hlevnjak I (2011) Psychiatr Danub 23:194–197
Shahzad S, Suleman MI, Shahab H, Mazour I, Kaur A, Rudzinskiy P, Lippmann S (2002) Psychosomatics 43:354–359
Subashini K, Rao VA (2004) Indian J Pharmacol 36:323–324
Toler SM (2004) Exp Biol Med (Maywood) 229:607–615
Wójcikowski J, Boksa J, Daniel WA (2010) Biochem Pharmacol 80:1252–1259
Daniel W (1995) Pol J Pharmacol 47:367–379
Chetty M, Pillay VL, Moodley SV, Miller R (1996) Eur Neuropsychopharmacol 2:85–91
Chetty M, Gouws E, Miller R, Moodley SV (1999) Eur Neuropsychopharmacol 9(1–2):77–82
Abernathy CO, Lukacs L, Zimmerman HJ (1977) Proc Soc Exp Biol Med 155:474–478
Tavoloni N, Boyer JL (1980) J Pharmacol Exp Ther 214:269–274
Wójcikowski J, Pichard-Garcia L, Maurel P, Daniel WA (2003) Br J Pharmacol 138:1465–1474
Wójcikowski J, Pichard-Garcia L, Maurel P, Daniel WA (2004) Eur Neuropsychopharmacol 14:199–208
Wójcikowski J, Maurel P, Daniel WA (2006) Drug Metab Dispos 34:471–476
Chetty M, Miller R, Moodley SV (1994) Eur J Clin Pharmacol 46:523–526
Kot M, Daniel WA (2008) Biochem Pharmacol 76:543–551
Blomberg MRA, Borowski T, Himo F, Liao RZ, Siegbahn PEM (2014) Chem Rev 114:3601–3658
Schrǒder D, Shaik S, Schwarz H (2000) Acc Chem Res 33:139–145
Baciocchi EBM, Gerini MF, Lanzalunga O (2005) J Org Chem 70:5144
Guengerich FP, Yun CH, Macdonald TL (1996) J Biol Chem 271:27321–27329
Jurva U, Bissel P, Isin EM, Igarashi K, Kuttab S, Castagnoli N (2005) J Am Chem Soc 127:12368–12377
Li CS, Wu W, Kumar D, Shaik S (2006) J Am Chem Soc 128:394–395
Chen H, de Groot MJ, Vermeulen NPE, Hanzlik RP (1997) J Org Chem 62:8227–8230
Chen ZQ, Kang Y, Zhang CH, Tao J, Xue Y (2015) Theor Chem Acc 134:110
Shaik S, Cohen S, Wang Y, Chen H, Kumar D, Thiel W (2010) Chem Rev 110:949–1017
Sansen S, Yano JK, Reynald RL, Schoch GA, Griffin KJ, Stout CD, Johnson EF (2007) J Biol Chem 282:14348–14355
Tao J, Kang Y, Xue ZY, Wang YT, Zhang Y, Chen Q, Chen ZQ, Xue Y (2015) J Mol Graph Model 61:123–132
Kwiecien RA, Molinié R, Paneth P, Silvestre V, Lebreton J, Robins RJ (2011) Arch Biochem Biophys 510:35–41
Li DM, Wang Y, Yang CL, Han KL (2009) Dalton Trans 14:291–297
Schyman PUD, Wang Y, Shaik S (2010) J Phys Chem B 114:7078–7089
Kang Y, Tao J, Xue ZY, Zhang Y, Chen ZQ, Xue Y (2016) Tetrahedron 72:2858–2867
Zhang Q, Bell R, Truong TN (1995) J Phys Chem 99:592–599
Reed AE, Schleyer PR (1990) J Am Chem Soc 112:1434–1445
Mennucci B (2012) WIREs Comput Mol Sci 2:386–404
Schutz CN, Warshel A (2001) Proteins 44:400–417
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JJA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2013) Gaussian 09 revision D01. Gaussian Inc, Wallingford
Bach RD, Dmitrenko O (2003) J Phys Chem B 107:12851–12861
Wang Y, Kumar D, Yang CL, Han KL, Shaik S (2007) J Phys Chem B 111:7700–7710
Acknowledgments
This work was supported by grants from National Natural Science Foundation of China (Grant No. 21203153), Science and Technology Department of Sichuan Province (Grant No. 2011JY0136) and Department of Education of Sichuan Province (Grant No. 12ZA174) and China West Normal University (Grant No. 11B002).
Author information
Authors and Affiliations
Corresponding author
Additional information
Zhiyu Xue and Yan Zhang authors have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xue, Z., Zhang, Y., Tao, J. et al. Theoretical elucidation of the metabolic mechanisms of phenothiazine neuroleptic chlorpromazine catalyzed by cytochrome P450 isoenzyme 1A2. Theor Chem Acc 135, 218 (2016). https://doi.org/10.1007/s00214-016-1943-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-016-1943-4