
Abstract
The host–guest inclusion mechanism formed between β-cyclodextrin and those poorly water-soluble drug molecules has important applications in supramolecular chemistry, biology and pharmacy. In this work, the chiral recognition ability of β-cyclodextrin to one of nonsteroidal anti-inflammatory drugs, ketoprofen, has been systematically investigated using molecular dynamics and free energy simulation methods. The R- and S-enantiomers of ketoprofen were explicitly bound within the cyclodextrin cavity in our simulations, respectively. In consistent with experimental observations, tiny structural difference between two isomers could be observed. Calculated absolute binding free energies using adapted biasing force (ABF) method and MM/GBSA approach for both isomers are comparable to experimental values. Significant binding fluctuations along the MD trajectory have been observed. The free energy profiles calculated using two different approaches reveal that the ketoprofen prefers binding in the cavity with the carboxylate group facing the wider edge of β-cyclodextrin. Similar free energy profiles for two enantiomers obtained using ABF calculations indicate that it is very hard to separate and identify the chiral conjugates within the framework of the natural β-cyclodextrin.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The interaction or recognition mechanism of host–guest systems constitutes the core part of supramolecular chemistry, for which the main driving force of the formation of the systems is considered to be the hydrophobic interactions. Correct understanding or quantification of the intermolecular interaction would be particularly important for the “rational planning of new supramolecular systems, including intelligent materials, as well as for developing new biologically active agents” [1]. Among host–guest systems, the inclusion complexes formed between natural cyclodextrins (CDs) with small organic molecules, esp. those hydrophobic molecules, represents a typical example in this field and thus attracts a lot of attentions [2–9].
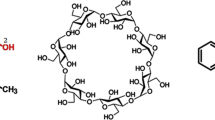
CDs are cyclic oligomers of glucose units. The most common natural CDs are composed of 6 (α), 7 (β) or 8 (γ) pyranose units connected by α-1,4 glycosidic linkage. Due to 4C1 chair conformation of the pyranose units, the overall topology of these CDs can be described as a toroid/hollow truncated cone shape. The significant different hydrophilicity of their exterior environment and interior cavity makes the CDs very unique in the supramolecular chemistry. They can easily bind lipophilic molecules to form a host–guest inclusion complex and thus change the chemical, physical or even biological properties of these ligands. Such ability makes them widely applied in pharmaceutical development, drug delivery or food industry. People have pursued their usage in the synthesis of some certain mechanically interlocked molecular architecture, such as rotaxanes [10] and catenanes [11]. The photochemistry of molecules hosted by CDs is also of particular interests, e.g., arenes [12]. In this work, we will focus on the detailed chiral recognition mechanism of β-CD complexed with a widely used anti-inflammatory drug, ketoprofen. The schematic representations of β-CD and ketoprofen are given in Scheme 1.

Schematic representations and definition of atoms of β-cyclodextrin and (S-) and (R-)ketoprofen molecules
Ketoprofen (IUPACE name of (R/S)2-(3-benzoylphenyl) propionic acid) has long been considered to be the nonsteroidal anti-inflammatory drug (NSAID) with analgesic and antipyretic effects [13]. It has also served as one of pain relief drugs. Ketoprofen is usually marketed as 50:50 racemic mixtures of (R-) and (S-)enantiomers, but with poor solubility. It was demonstrated that only S-enantiomer has anti-inflammatory activity, while same functions can be found for other profen drugs such as ibuprofen and flurbiprofen. However, the chiral inversion of the R- to S-enantiomers of profen drugs has been observed in vivo, e.g., in pigs [14]. Based on this, the physiological functions of (R-)ketoprofen should not be totally neglected and might deserve some more researches. More importantly, some experimental studies have proved that (R-)flurbiprofen can be involved in the antitumor activity in mouse. Clearly, we cannot simply neglect either enantiomer. Due to the importance of the enantiomer drugs in the drug delivery, it might be interesting to pursue the synthesis, separation or chemical properties for different enantiomers. The recognition mechanism and binding structures of β-CD with ketoprofen have been reported in some detail from both experimental and theoretical sides [15–19]. However, little was known about the difference of the binding features, such as binding free energies or structures, for both enantiomers of ketoprofen. Recently, to tackle the photochemistry characteristics of (R-) and (S-)ketoprofen and their chiral recognition in β-CD, it was investigated by Marconi et al. [17] using various experimental techniques such as circular dichroism, isothermal titration calorimetry and NMR. For the association constants for both enantiomers, the experiments gave almost identical values considering experimental uncertainty. Based on circular dichroism and 2D ROESY spectra, tiny structural difference can be probed for both enantiomers. Therefore, it should be pointed out that it is very difficult to use experimental tools to get stereoselective recognition ability of β-CD versus (R-) and (S-)ketoprofen. Given the practically enantio-differentiated anti-inflammatory and analgesic activities of ketoprofen, it is then necessary to understand the detailed recognition mechanism for both chiral isomers from theoretical view.
Besides the stereoselective recognition mechanism of β-CD with ketoprofen, there is another interesting issue not quite well understood. For the profen drugs, there are two sides which bear different polarizability as shown in Scheme 1, with polar carboxylate group at one side and nonpolar aromatic group at the other side. So far there is no definite study to say which rim of the β-CD the carboxylate group is bound with. Indeed, some controversies were reported for other profen drugs, such as ibuprofen. The X-ray structure of the inclusion complex formed between β-CD and S-ibuprofen reported by Braga et al. [20] clearly indicates that the carboxylate group of the ligand faces the smaller rim of the β-CD. On the contrary, another crystallographic structure of S-ibuprofen complexed by a heptakis-substituted β-CD shows that the carboxylate group is facing the bigger rim [21]. In addition, theoretical simulations reported by Cai et al. [22] on steroid drugs recognized by β-CD indicate that the different binding orientations could lead to closed binding free energy profiles along the binding coordinates as well as the association constants. Such observations could further suggest that the formation of inclusion complex has some complicated processes. Therefore, to obtain a complete understanding of the β-CD bound with ketoprofen molecule or other small ligands, it is highly desired to perform extensive simulations on all of possible binding orientations for different enantiomers.
In this work, to obtain a complete understanding of the binding characteristics of ketoprofen/β-CD inclusion complex, we then build four possible binding structures for two ketoprofen enantiomers as shown in Scheme 2. The absolute binding free energies will then be calculated based on molecular mechanics-generalized born surface area (MM-GBSA) method. To understand the detailed formation processes of each inclusion complex, the potential of mean force (PMF) profiles along the inclusion coordinates will be computed using adapted biasing forcing (ABF) method.

Schematic representations of two different inclusion directions of ketoprofen threading through the cavity of β-CD. The origin is set at the center of mass of seven glycosidic oxygen atoms of β-CD. The narrower edge is usually called as primary rim, while the wider edge as secondary rim
2 Computational details
2.1 Models of β-CD/ketoprofen inclusion complex
There is no published X-ray structure for β-CD/ketoprofen inclusion complex. To investigate the dynamics properties of this inclusion complex, it would be necessary to construct the model of inclusion complex firstly. It should be pointed out that since there are two isomers for ketoprofen molecule, we then docked R- and S-ketoprofen into β-CD cavity using AutoDock 4.2 program, respectively [23]. In addition, since there are also two possible binding orientations for each enantiomer as shown in Schemes 1 and 2, the structures for each orientation of different enantiomers with the lowest binding energy were chosen for the following extensive molecular dynamics simulation. For convenience, the glucose units of cyclodextrin are labeled from G1 to G7. We simply give the name for each binding orientation as R-I, R-II for (R-) ketoprofen, while S-I and S-II for (S-)ketoprofen, respectively. In binding orientation I, the carboxyl group is facing the secondary rim, while the carboxyl group stays at the primary rim in binding orientation II. Throughout the simulation, the stoichemistry of β-CD and ketoprofen is maintained to be 1:1 ratio, which has been suggested by many experimental works.
The obtained systems were firstly solvated in a pre-equilibrated TIP3P [24] water box. The typical size of the water box is calculated to be about 44 Å × 44 Å × 40 Å, consisting of 179 solute atoms and about 1800 solvent molecules. To prepare suitable force field for the ketoprofen molecule, we employed the standard Amber general amber force field (GAFF) generation procedure. First of all, the geometry optimizations of ketoprofen at HF/6-31G* level of theory were carried out using gaussian09 suite of program [25]. The partial atomic charges were then calculated using the restrained electrostatic potential (RESP) protocol after structure optimization and electrostatic potential calculations using B3LYP/6-31G* method. The force field parameters for the ligands generated using the Antechamber program were then described by GAFF module. The force field of β-cyclodextrin is expressed using Glycam06 carbohydrate parameters [26] in this work, which has been proved to accurately and efficiently represent carbohydrates. The periodic boundary conditions and an 8 Å cutoff for non-bond interactions were applied. The particle mesh ewald (PME) algorithm [27] was used to calculate the long-range electrostatic interactions. The positions of water molecules were relaxed by 500 steps of steepest descent (SD) and 1000 steps of conjugate gradient (CG) minimization approach with all of solute molecules fixed at their original positions. Further 10,000 steps of CG full minimization were carried out for total system. The obtained systems were gradually heated to 300 K in 20 ps in the NVT ensemble, followed by 20 ns equilibration simulation under 1 atm pressure. Subsequently, further 20 ns MD simulations in the isothermal–isobaric NPT ensemble were performed for data analysis. Newton’s equations of atomic motion were integrated by the Verlet algorithm with a 2-fs time step. SHAKE algorithm [28] was applied to constraint bond stretching of the covalent bonds involving hydrogen atoms. All of MD simulations were performed using PMEMD module of CUDA version implemented in AMBER 12 software package.
2.2 Binding free energy calculations
To quantitatively assess the binding affinity of β-CD in complex with ketoprofen molecule, it is necessary to calculate the binding free energy for the inclusion complex. Several methods have been proposed for calculating absolute binding free energy, e.g., linear response approximation (LRA) [29], linear interaction energy (LIE) [30–32], molecular mechanics Poisson Boltzmann (or generalized Born) surface area (MM-PB/GBSA) [33, 34], solvated interaction energy (SIE) [35], or free energy pathway method [36]. In this work, the binding free energy is calculated using MM-GBSA [33] method.
For the calculation of binding free energy in MM-GBSA framework, it has been discussed extensively [37–41]. Only a short description is summarized here:
\(\Delta G_{\text{complex}}\), \(\Delta G_{{\upbeta{\text{ - CD}}}}\) and \(\Delta G_{\text{ketoprofen}}\) are free energies of the complex, the β-CD and ketoprofen, respectively. Each term can be obtained according to Eq. (2). Practically, they are calculated as the statistical averages over frames extracted from MD trajectories. The solvation free energy (\(G_{\text{sol}}\)) can be divided into polar (\(G_{\text{GB}}\)) and nonpolar (\(G_{\text{np}}\)) contributions. The polar solvation contribution is calculated by solving the Generalized Born (GB) equation [42]. Dielectric constants (ε) for solute and solvent were selected to be 1 and 80, respectively. The nonpolar contribution due to cavity formation and van der Waals interactions between the solute and the solvent can be estimated by the equation of γ·SA + b, where γ = 0.0072 kcal/Å2, b = 0.0 kcal/mol. The SA is defined as the solvent accessible surface area, which was estimated using the program MSMS [43]. For each complex system, binding energies were averaged over 1,000 frames of the 20-ns MD trajectory. It has long been recognized that the inclusion of entropic effect in the calculation of total binding free energy can largely reduce the difference between theoretical and experimental values [44]. Entropy contributions are from changes in the degrees of freedom including translation, rotation and vibration. The translational, rotational and vibrational entropy terms are functions of the mass and moments of inertia of the molecule and thus can be calculated using the standard equations of statistical mechanics [45]. In this work, vibrational entropy contributions were estimated using the normal mode analysis approach [46].Due to the system size is just moderate, the –TΔS was averaged over 200 snapshots of the MD trajectory.
2.3 Formation of inclusion complex
As suggested in Scheme 2, two possible binding models with different position of the propionic acid end have been proposed for the molecules in profen family recognized by β-CD. Although the absolute binding free energy calculated using MM-GBSA method can provide some insights into the interaction affinity between β-CD and ligands, the detailed process to form the inclusion complex cannot be entirely revealed. Furthermore, the chiral recognition processes might be another interesting issue for the β-CD/ketoprofen inclusion complex. To address the formation of inclusion complexes, we further carried out extensive simulations of the inclusion processes using the adaptive biasing force (ABF) method [47]. MD simulations were performed using NAMD program [48], for which the initial system is constructed using the following strategy to avoid artificial impacts. As shown in Scheme 2, we randomly put the ketoprofen isomers outside the secondary (bigger) rim of the cavity of β-CD. We then simulate the translocation process through the cavity starting from the secondary rim, and finally moving out via the primary rim. The origin is defined as the center of mass of seven oxygen atoms (O4) of glycosidic linkage of the β-CD. In the present ABF calculations, we have two possible binding processes for each isomer: One is aromatic ring A enters the cavity first, and the second is the carboxylate group moves into the cavity first. Then, the order parameter or the inclusion coordinate should be defined correspondingly. For the first case, the orientation I, the order parameter (ξ), which ranges from −16 to 14 Å, was chosen as the projection of the distance between the origin and the center of mass of the aromatic ring A of ketoprofen on the z-axis. For the second case, the orientation II, the order parameter (ξ), which ranges from −16 to 14 Å, was chosen as the projection of the distance between the origin and the center of the mass of the carboxylate group of ketoprofen on the z-axis. The inclusion pathway was then divided into six consecutive windows to increase the computational efficiency. Each window was further divided into consecutive bins with bin width of 0.1 Å. For each window, up to 5 ns of MD trajectory was generated, resulted in a total of 30 ns simulation time. Instantaneous values of the forces, the first-order derivative of free energy with respect to the defined order parameter, were accrued in each bin to calculate the mean force value. Total of 1,500 force samples were accumulated in each bin prior to application of the adaptive bias. For the setup protocol of MD simulations, which is essentially the same as we did in previous section using Amber program, except the integration step is set as 1 fs time step in ABF calculations.
3 Results and discussion
3.1 Structures of the inclusion complexes
In this work, one of our major objectives is to understand the mechanism of chiral recognition of β-CD with ketoprofen enantiomers. The phenomenon called chiral discrimination in the host–guest interaction has been observed in experimental way [49]. Here, we performed extensive MD simulations to examine the dynamic effects of β-CD interacting with the enantiomers of the ketoprofen.
Throughout the molecular dynamics simulations, all four inclusion complexes formed between ketoprofen and β-CD were maintained very well judged by the distance between the C4 atom of ketoprofen and the center of mass of glycosidic oxygen atoms as shown in Fig. 1. It looks like that for the R-ketoprofen, the binding orientation I (R-I) is much more stable than the other binding orientation (R-II) according to different binding fluctuation, while the difference between S-I and S-II models is smaller. Therefore, it could be deduced that a little smaller difference of binding affinity in S-conformation than that in R-conformation is expected. To get a direct view of the structural change along the simulation time, we further plotted corresponding snapshots from 20 ns to 40 ns for each binding conformer in Figs. 2 and 3. In most of simulation time, the ligand molecule is inserted into the cavity in tilted direction, which would ensure that the sugar ring has more interactions with aromatic rings of guest molecule. Such phenomenon has been found in the simulation of β-CD/steroid drug systems investigated by Cai et al. [22]. At least one aromatic ring (A or B) of ketoprofen was found to be included in the cavity during the simulation, but some apparent fluctuations of the binding position could be observed. Such pattern could partially indicate the hydrophobic interaction plays important role in the guest molecule recognition by β-cyclodextrin. For clarity, the time course profiles of the distance between the center of mass of aromatic ring A (\(d_{\text{cm}}^{A}\)) or B (\(d_{\text{cm}}^{B}\)) and the center of mass of the glycosidic oxygen atoms of the host molecule are depicted in Fig. 4. It should be noted that this distance could only be used to elucidate the fluctuations for the binding status of the aromatic rings, but not for the estimation of binding stability, since this distance is different from the definition of order parameter (ξ) used in free energy pathway calculations as we gave above. Clearly, different binding performance for chiral isomers can be observed. For the orientation I, along the simulation trajectory, we can see that the carboxylate group always points outward and cannot be included into the cavity of β-CD. Such binding pattern requires that at least one of aromatic rings of ketoprofen is recognized by the hydrophobic interior environment of β-CD. According to the snapshots given in Fig. 2 and the relative position analysis between the benzene rings and β-CD in Fig. 4, the substantial fluctuation of the binding of the benzene rings is quite clear. That means both aromatic rings have chance to be included into the cavity. It thus suggests that β-CD cannot identify any specific aromatic ring of included ligand. If we recall the NMR spectroscopy of the inclusion complex of β-CD/ketoprofen [17], experimental work indicated that both rings (A and B) can be deeply embedded in the cavity with very close chemical shifts. Our results are then in reasonably agreement with this observation. For the binding orientation II, a little different binding pattern could be observed. Especially, we can see that ring A of R-II and ring B of S-II seem to be dominantly recognized by β-CD cavity. Such binding feature might be inconsistent with the experimental observations. This feature should be further examined by binding free energy calculations.

Distance between C4 atom and the center of mass of glycosidic oxygen atoms of β-cyclodextrin along the simulation time for four binding models

Snapshots of the inclusion complexes formed by the β-CD and (R-) and (S-)ketoprofen along the dynamics simulation time in orientation I, respectively. For clarity, the water molecules have been removed. The ketoprofen is plotted using stick style, while line style for β-CD

Snapshots of the inclusion complexes formed by the β-CD and (R-) and (S-)ketoprofen along the dynamics simulation time in orientation II, respectively. For clarity, the water molecules have been removed. The ketoprofen is plotted using stick style, while line style for β-CD

Distances between the aromatic rings of ketoprofen and the center of mass of seven glycosidic oxygen atoms of β-CD in binding orientation I (panel I) and in binding orientation II (panel II). For the binding of R-ketoprofen, \(d_{\text{cm}}^{A}\) is colored in black, and \(d_{\text{cm}}^{B}\) is colored in green. For the binding of S-ketoprofen, \(d_{\text{cm}}^{A}\) is colored in red, and \(d_{\text{cm}}^{B}\) is colored in blue
It is also interesting to examine the possibility of hydrogen bond formed between host and guest molecules, since there are a lot of hydroxyl groups in β-cyclodextrin and one carboxylate group in ketoprofen. We summarized the hydrogen bond occupancy analysis results in Table 1 over the MD trajectories for all four inclusion complexes. Quite low hydrogen bond occupancies were found between host and guest molecules. The biggest occupancy rate is just about 20 %. This supports from another angle that the hydrophobic interaction is possible the dominant factor in the stabilization of the inclusion complex. The electrostatic interactions should be considered to be a minor factor in the host–guest recognition. Indeed, the polar carboxylate group of ketoprofen is exposed to solvent molecules in almost most of simulation time no matter which rim it faces. However, the existence of the carboxyl group might be an issue to affect the formation of inclusion complex and the drug releasing, which we will discuss below using free energy calculations.
3.2 Binding free energy
Although binding models obtained from MD simulations can provide some insights into the guest molecule recognition, it is still difficult to identify which model is the favored one. Some quantitative measurements, such as absolute binding free energy calculation, can help us to get useful ideas. Due to chiral discrimination, different binding free energy for R- and S-enantiomers should be expected. Such results have been observed for the binding of ketoprofen, although researchers attributed the slight energetic difference into experimental uncertainty and thus cannot tell which the favored binding conformer is [17]. In this work, calculated absolute binding free energies are summarized in Table 2 for two binding orientations of both isomers, and the measured association free energies from different experimental sources [17, 18, 50–52] are included for comparison. The uncertainties for all of terms are included in the parentheses, which were calculated as the root mean square error for all of frames extracted in the MM/GBSA running. Overall, the calculated entropies for four binding models are negative, at the same time the enthalpies are also negative. This can suggest that the formation of inclusion complexes is an enthalpy driven process. For both R- and S-ketoprofen in binding orientation I, the calculated average binding free energy is around 3.8 kcal/mol, which is in excellent agreement with experimental values of ~4.0 kcal/mol listed in Table 2. On the other hand, for the orientation II, a little larger difference between experimental and theoretical values could be observed. This could suggest that for the inclusion complexes formed by β-CD and ketoprofen, the binding orientation I, the propionic acid group of ketoprofen facing the wider rim of β-CD, is more likely the favored conformer, no matter R- or S-isomer.
On the other hand, since marked ketoprofen is often in a molar ratio of 50:50 racemic nature, little efforts have been applied to examine the binding difference between two chiral models. Recently, to understand the chiral photoreaction properties, Marconi et al. [17] examined the stereoselective binding affinity of β-CD bound with ketoprofen enantiomers. It is not surprised that only very tiny difference for the binding free energies can be observed, −3.95 kcal/mol (S) versus 3.76 kcal/mol (R). The experimental work by Marconi et al. [17] suggests that β-CD can bind S-ketoprofen slightly stronger than its R-chiral conjugate, although they put this to be within experimental uncertainty. Our results are quite close this value, esp., the binding orientation I with the calculated absolute binding free energies are −3.87 kcal/mol for S-I, while −3.67 kcal/mol for R-I. For the binding orientation I, the difference of binding free energy between two enantiomers is calculated to be 0.2 kcal/mol, which agrees well with experimental difference of 0.19 kcal/mol [17] as shown in Table 2. Our results are also in consistent with a recent computational work, which suggests that the β-cyclodextrin binds (S-)ketoprofen a little tightly than (R-)ketoprofen [53].
Another interesting issue is that for both chiral isomers, the stronger binding affinity can be observed for orientation I than orientation II, esp. the R-isomer, which is consistent with our MD simulation result as described in above section. However, the negative binding free energies for the orientation II still suggest that such an inclusion conformation should not be totally neglected. Indeed, our simulation on the orientation II has confirmed that this conformer for both enantiomers is pretty stable as shown in Fig. 1. In addition, X-ray structure has proved the existence of the orientation II for the ibuprofen/β-CD inclusion complex [20]. It should be noted that the deviation of the calculated binding free energies of orientation II is a bit larger than those of orientation I. Based on the calculated absolute binding free energies, we then suggest that the preferred binding mode of ketoprofen by the natural β-cyclodextrin might take the orientation I for both isomers no matter it is in R- or S-isomer. Of course, such conclusion awaits more accurate experimental work to confirm. To this end, there is no contradiction between our results and a recent NMR study on the inclusion of R- and S-ketoprofen, in which Marconi et al. [17] concluded that the carboxyl group of ketoprofen exposed to the solvent from secondary edge of the host.
Usually, there are two major factors that could affect the substrate binding, i.e., the electrostatic (ΔE ele + ΔG sol, GB) and nonpolar (ΔE vdW + ΔG sol, np) terms. It has long been accepted that the recognition of lipophilic molecules by β-CD is mainly due to hydrophobic interactions. The structural feature of aromatic included in the cavity revealed by MD simulation can support this proposal. Further, from Table 2, it can be seen that all four conformers agree this very well. For example, the electrostatic terms are calculated to be about 6.1, 1.0, 8.2 and 0.5 kcal/mol for R-I, R-II, S-I and S-II inclusion complexes, respectively. The electrostatic interaction is canceled out by the polar part of solvation. Positive values for electrostatic contribution indicate that polar interactions between host and guest molecules clearly disfavor the molecular recognition in the β-CD based inclusion complexes. In contrast, van der Waals interactions have dominantly favorable contributions to the binding affinity, as does the nonpolar part of solvation. The calculated total nonpolar terms are −29.8, −22.1, −31.2 and −23.2 kcal/mol for R-I, R-II, S-I and S-II inclusion complexes, respectively. Quite interestingly, much larger nonpolar terms for the orientation I than the orientation II can be obtained in our simulations. This is also in consistent with our deduction of stronger binding affinity for orientation I than orientation II, since the hydrophobic interaction is the dominant factor in the formation of host–guest system we studied in this work.
3.3 ABF calculations
The binding free energies obtained using MM/GBSA method can explain the recognition driving force to some extent. However, it cannot tell us how the drug molecule is included into the cavity of host molecule. At the same time, it cannot identify the drug releasing direction and possible rate. To solve the problem, it is then necessary to examine the detailed energetic performance for the binding processes along the inclusion coordinates as depicted in Scheme 2. Calculated free energy profiles along the inclusion pathway for two binding orientations of R- and S-ketoprofen using ABF method are given in Figs. 5 and 6, respectively.

Free energy profiles via ABF calculations for the inclusion of ketoprofen into β-CD along the z-axis in binding orientation I, S-ketoprofen (solid line), R-ketoprofen (dash line)

Free energy profiles via ABF calculations for the inclusion of ketoprofen into β-CD cavity in binding orientation II, S-ketoprofen (solid line), R-ketoprofen (dash line)
The free energy profile derived from ABF calculation could be integrated along the order parameters (ξ) to obtain a quantitative estimation of the association constant, K a [54]. When ketoprofen penetrates through the β-CD cavity, the sampled volume was restricted to a cylinder defined by the area (A CD) available for in-plane movement of the guest molecule in the cavity (A CD = πr 2) and the linear trajectory perpendicular to this area [55].
where K a is the system association constant with an unit of M−1, N A = 6.02 × 1023 is the Avogadro constant and the temperature T is chosen at 300 K. For the choice of radius of the cylinder, it could be calculated to be the average radius along the ligand penetrating the cavity along the z-axis direction. In this work, we simply take the value of r = 2Å as suggested by Auletta et al. [55]. The integration ranged over the whole sampled pathway. The association free energy can be obtained again according to the relationship of ΔG = −RT ln K a . Calculated association constants and binding free energies of ΔG are listed in Table 3. The calculated association free energies are in excellent agreement with those values obtained using MM/GBSA method. Similarly, the association of S-isomer is systematically stronger than R-enantiomer, but with pretty small difference. For the binding orientation I, the results can be comparable to experimental values very well, with typical error less than 1.0 kcal/mol. These results could further indicate ABF method is one of suitable methods to address the energetic profiles for the formation of inclusion complexes. On the other hand, the results for the binding orientation II is not as good as those in orientation I. Considering the computational accuracy, our simulation results within both MM/GBSA and ABF frameworks seem to support that the inclusion complex formed in binding orientation I would be more likely the binding model observed in experimental way.
Since our simulations suggest that the model in orientation I would be the favored one, we will then focus on the free energy profiles depicted in Fig. 5. The overall topology of the free energy profiles for R-I and S-I is quite similar, which means that only a little difference can be observed in the chiral recognition. This is reasonable since there is only tiny difference for the chiral inclusion process revealed by NMR experiments [17]. On the other hand, there are two distinguished energy minima along the order parameter, ca. ξ = −3.6 and 1.9 Å for S-I, while ξ = −3.9 and 1.1 Å for R-I, respectively. We then plotted corresponding snapshots in Fig. 7 for the inclusion of S-ketoprofen, and Fig. 8 for the inclusion of R-ketoprofen, which are all extracted from ABF trajectories. For the second minimum of S-I (ξ = 1.9 Å) and R-I (ξ = 1.1 Å) models, two aromatic rings are fully inserted into the β-CD cavity to maximum the hydrophobic interactions, while the carboxyl group is facing the wider rim just as we have observed in MD simulations. Surprisingly, instead of the ring A included in the cavity, we can see that for the first minima at ξ = −3.6 Å (S-I) and −3.9 Å (R-I), the ring B is included by β-CD cavity. One barrier at ξ = −2.3 Å (S-I) and −2.6 Å (R-I) with a moderate barrier height can be observed to connect these two free energy minima, and the structures for two isomers are presented in Figs. 7 and 8, respectively. It looks like that this barrier stays at the stage of the ring A beginning entering the cavity, but not fully bounded. Such geometry transformation characteristics could explain why we can see significant fluctuations in the ligand binding dynamics presented above. However, if we carefully examine the structures, we may partially attribute this barrier to the carboxyl group of ketoprofen, which could form hydrogen bond network with hydroxyl groups around the secondary rim. To understand this issue, we then calculated the hydrogen bond occupancy between the carboxylate group and sugar hydroxyl groups within the ABF simulation window of [−6.0 and −1.0 Å]. The hydrogen bond occupancy for this oxygen atom is calculated to be about 35 %. Clearly, albeit the ring A is not included by the cavity, the existence of hydrogen bond between the carboxyl group and β-CD could still anchor the ketoprofen a little while. That will make the ring B has the chance to be bound in the cavity. Therefore, although the polar interaction has been proved to be a minor factor to stabilize the host–guest system, it still should be considered as one of the key factors to affect the formation of the inclusion complex.

Snapshots of the inclusion complexes of S-ketoprofen with β-CD at the inflection points along the free energy curve in orientation I. a Near the first minimum, ca. ξ = −3.6 Å, b near the maximum, ca. ξ = −2.6 Å, c near the second minimum, ca. ξ = 1.9 Å

Snapshots of the inclusion complexes of R-ketoprofen with β-CD at the inflection points along the free energy curve in orientation I. a Near the first minimum, ca. ξ = −3.9 Å, b near the maximum, ca. ξ = −2.3 Å, c near the second minimum, ca. ξ = 1.1 Å
Although we pull the ketoprofen from the wide edge to narrow rim in the ABF calculations, we can still take a look at the free energy in a recursive way. Quite interestingly, no barrier can be located for the ligand migration from the narrow rim into the cavity, for which the carboxyl group enters the cavity firstly. However, the barrier occurs along the z-axis positive direction, which means to enter the cavity from the wider edge, and it has to overcome a barrier height of 3.0 and 2.7 kcal/mol for S- and R-ketoprofen according to our calculations, respectively. In other words, we might safely conclude that to form the ketoprofen/β-CD inclusion complex, the polar carboxyl group will enter the cavity first, but finally two aromatic rings will be bound in the cavity as shown in Fig. 7. It should be emphasized here that the hydrophobic interactions constitute the major factor in the final formation of inclusion complex. Accounting for the ketoprofen releasing direction, it depends on the binding position of the carboxyl group, i.e., where the carboxyl group occurs, and the releasing the drug could move out more easily through this direction. About 10 kcal/mol energy has to be overcome for bounded ligand to migrate from the cavity through the narrow edge. We might dedicate this carboxyl group as one of manipulate factors for the drug releasing issue. This is quite smart of nature. According to our simulations, it is thus possible to design some new drugs with a desired releasing rate.
4 Conclusion
Correct understanding the inclusion mechanism of β-CD complexed with small drug molecules would be of particular importance in the drug delivery. We, in this work, systematically investigated the free energy profiles of the inclusion complexes formed between natural β-CD and ketoprofen enantiomers. Our results support that the R- and S-ketoprofen prefer binding β-CD with their carboxylate groups facing the wider edge of the cavity (binding orientation I). Additionally, both MM/GBSA and ABF approaches suggest that β-CD has stronger affinity with S- than R-ketoprofen, which is also consistent with NMR measurement. Calculated binding free energies for the model of S-I and R-I using MM/GBSA and ABF are very close to the experimental values, which further confirm that both methods are suitable to deal with the recognition problem related with host–guest supramolecular system. It has also been proved that natural β-CD might not be a good chiral separation matter due to similar binding performance of ketoprofen enantiomers into the cavity. Therefore, various substituted CDs might be good candidates to fulfill this object. Nonetheless, our simulations reveal that the ketoprofen might move out more easily from the cavity via the position of carboxyl group occurring. This carboxyl group can be denoted as one of manipulate factors in the drug releasing. It is our hope that this could be helpful for the further drug design with desired drug releasing rate.
References
Schneider H (2009) Angew Chem Int Ed 48:3924–3977
Fromming KH, Szejtli J (1994) Cyclodextrins in pharmacy. Kluwer, Dordrecht
Irie T, Uekama K (1999) Adv Drug Deliv Rev 36:101–123
Davis ME, Brewster ME (2004) Nature Rev Drug Discov 3:1023–1035
Del Valle EMM (2004) Process Biochem 39:1033–1046
Brewster ME, Loftsson T (2007) Adv Drug Deliv Rev 59:645–666
Loftsson T, Duchene D (2007) Int J Pharm 329:1–11
Nagaruju M, Sastry GN (2009) J Phys Chem A 113:9533–9542
Pandey S, Kumar B, Vijayendra Swamy SM, Gupta A (2010) Int J Pharm Technol 2:281–319
Stanier CA, O’Connell MJ, Anderson HL, Clegg W (2001) Chem Commun 5:493–494
Kuhnert N, Tang B (2006) Tetrahedron Lett 47:2985–2988
Avakyan VG, Nazarov VB, Alfimov MV, Bagatur’yants AA (1999) Russ Chem Bull 48:1833–1844
Kantor TG (1986) Pharmacotherapy 6:93–103
Neirinckx E, Croubels S, Remon JP, Devreese M, De Backer P, Vervaet C (2011) Vet J 190:290–292
Lu W, Zhang Q, Zheng L, Wang H, Li R, Zhang L, Shen W, Tu X (2004) Biol Pharm Bull 27:1515–1520
Maestrelli F, Gonzalez-Rodriguez ML, Rabasco AM, Mura P (2005) Int J Pharm 298:55–67
Marconi G, Mezzina E, Manet I, Manoli F, Zmabelli B, Monti S (2011) Photochem Photobiol Sci 10:48–59
Mura P, Bettinetti GP, Manderioli A, Faucci MT, Bramanti G, Sorrenti M (1998) Int J Pharm 166:189–203
Tayade PT, Vavia PR (2006) Ind J Pharm Sci 68:164–170
Braga SS, Goncalves IS, Herdtweck E, Teixeira-Dias JJC (2003) New J Chem 27:597–601
Brown G, Caira M, Nassimbeni L, Van Outtshoorn B (1996) J Incl Phenom Mol Recognit Chem 26:281–294
Cai W, Sun T, Liu P, Chipot C, Shao X (2009) J Phys Chem B 113:7836–7843
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) J Comput Chem 16:2785–2791
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) J Chem Phys 79:926–935
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski W, Martin RL, Morokuma K, Zakrzewski VG, Voth A, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision A.01. Gaussian, Inc., Wallingford CT
Kirschner KN, Yongye AB, Tschampel SM, Gonzάlez-outeiriño J, Daniels CR, Foley BLW, Wood RJ (2008) J Comput Chem 29:622–655
Darden TA, York D, Pedersen L (1993) J Chem Phys 98:10089
Ryckaert J-P, Ciccotti G, Berendsen HJC (1977) J Comput Phys 23:327–341
Lee FS, Chu ZT, Bolger MB, Warshel A (1992) Prot Eng 5:215–228
Åqvist J, Medina C, Samuelsson JE (1994) Prot Eng 7:385–391
Bren U, Martínek V, Florián J (2006) J Phys Chem B 110:10557–10566
Perdih A, Bren U, Solmajer T (2009) J Mol Model 15:983–996
Srinivasan J, Cheatham TE, Cieplak P, Kollman PA, Case DA (1998) J Am Chem Soc 120:9401–9409
Lee MS, Salsbury FR, Olson MA (2004) J Comput Chem 25:1967–1978
Naїm M, Bhat S, Rankin KN, Dennis S, Chowdhury SF, Siddiqi I, Drabik P, Sulea T, Bayly CI, Jakalian A, Purisima EO (2007) J Chem Inf Model 47:122–133
Gilson MK, Zhou H (2007) Annu Rev Biophys Biomol Struct 36:21–42
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Donini O, Wang W, Cieplak P, Srinivasan J, Case DA, Cheatham III TE (2000) Acc Chem Res 33:889-897
Hou T, Wang J, Li Y, Wang W (2011) J Comput Chem 32:866–877
Rizzo RC, Toba S, Kuntz ID (2004) J Med Chem 47:3065–3074
Strockbine B,Rizzo RC (2007) Proteins: Struct Funct Bioinform 67:630-642
Yang C, Sun H, Chen J, Coleska ZN, Wang S (2009) J Am Chem Soc 131:13709–13721
Onufriev A, Bashford D, Case DA (2000) J Phys Chem B 104:3712–3720
Sanner MF, Olson AJ, Spehner JC (1996) Biopolymers 38:305–320
Zoete V, Meuwly M, Karplus M (2005) Proteins 61:79–93
McQuarrie DA (2000) Statistical mechanics. University Science Books, Sausalito
Case DA (1994) Curr Opin Strut Biol 4:285–290
Darve E, Rodriigues-Gomez D, Pohorille A (2008) J Chem Phys 128:144120
Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel L, Schulten K (2005) J Comput Chem 26:1781–1802
Mason SF (1989) Chirality 1:183–191
Faucci MT, Melani F, Mura P (2002) Chem Phys Lett 358:383–390
Katritzky AR, Fara DC, Yang H, Karelson M, Suzuki T, Solovev VP, Varnek A (2004) J Chem Inf Comput Sci 44:529–541
Waters LJ, Bedford S, Parkes GMB, Mitchell JC (2010) Thermochim Acta 511:102–106
Felton LA, Popescu C, Wiley C, Esposito EX, Lefevre P, Hopfinger AJ (2014) AAPS Pharm Sci Technol 15:872–881
Shoup D, Szabo A (1982) Biophys J 40:33–39
Auletta T, de Jong MR, Mulder A, van Veggel FCJM, Huskens J, Reinhoudt DN, Zou S, Zapotoczny S, Schonherr H, Vancso GJ, Kuipers L (2004) J Am Chem Soc 126:1577–1584
Acknowledgments
This work was funded by National Science Foundation of China (Nos. 21073125 and 31170675) and the Program for New Century Excellent Talents in University (no. NCET-10-0606) to D. X., and by SRF for ROCS, SEM (No. 20111568-8-5) to C. Z. Parts of the results described in this paper are obtained on the Deepcomp7000 of Supercomputing Center, Computer Network Information Center of Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Dedicated to Professor Guosen Yan and published as part of the special collection of articles celebrating his 85th birthday.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shi, M., Zhang, C., Xie, Y. et al. Stereoselective inclusion mechanism of ketoprofen into β-cyclodextrin: insights from molecular dynamics simulations and free energy calculations. Theor Chem Acc 133, 1556 (2014). https://doi.org/10.1007/s00214-014-1556-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-014-1556-8