
Abstract
This paper reviews several pK a calculation strategies that are commonly used in aqueous acidity predictions. Among those investigated were the direct or absolute method, the proton exchange scheme, and the hybrid cluster–continuum (Pliego and Riveros) and implicit–explicit (Kelly, Cramer and Truhlar) models. Additionally, these protocols are applied in the pK a calculation of 55 neutral organic and inorganic acids in conjunction with various solvent models, including the CPCM-UAKS/UAHF, IPCM, SM6 and COSMO-RS, with a view to identifying a universal approach for accurate pK a predictions. The results indicate that the direct method is unsuitable for general pK a calculations, although moderately accurate results (MAD <3 units) are possible for certain classes of acids, depending on the choice of solvent model. The proton exchange scheme generally delivers good results (MAD <2 units), with CPCM-UAKS giving the best performance. Furthermore, the sensitivity of this approach to the choice of reference acid can be substantially lessened if the solvation energies for ionic species are calculated via the IPCM cluster–continuum approach. Reference-independent hybrid approaches that include explicit water molecules can potentially give reasonably accurate values (MAD generally ~2 units) depending on the solvent model and the number of explicit water molecules added.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Since the introduction of dielectric continuum models [1–4], a sizeable literature has appeared that utilizes solvation free energies obtained from these models for studying solvent effects on the thermodynamics of chemical reactions. Proton transfer reactions are by far the most studied because of their fundamental importance in biological systems and in synthetic applications [5, 6]. For example, the heterolytic cleavage of stable C–H bonds is the first step of many enzyme-catalyzed processes, including racemization, allylic isomerization, elimination, carboxylation and aldol condensation reactions [7–11]. The acidities of the active site residues in an enzyme are also of importance to the functionality of the enzyme since a catalytic reaction is usually initiated by transfer of a proton (or hydrogen bonding) from a protein residue to the substrate [12]. Accordingly, an understanding of how the chemical environment influences acidity could potentially lead to the development of novel synthetic strategies, and to important insights into the catalytic mechanism of enzymes.
The pK a is the most common measure of thermodynamic acidity, and is defined by the following equation:
where K a is the equilibrium constant for the deprotonation of the acid. Typically, this is determined through glass electrode measurements at half neutralization and/or spectrophotometry. However, accurate experimental measurements in aqueous solution are sometimes complicated by the very strong or weak acidities of these substrates (e.g. carbon acids have pK a values typically >20) due to the leveling effect of the solvent. The accuracy of pK a values obtained through extrapolations or the use of acidity functions in strongly protonating media may also be in considerable error [13, 14]. For example, alternative experimental values for H2SO4 range from −3 to −9, while those for CF3SO3H range from −6 to −14 [15, 16]. More recently, accurate aqueous pK a values for some very weak carbon acids have been determined from kinetic measurements using more sensitive techniques (such as NMR) [17–22], nonetheless these techniques cannot be applied universally and accurate data for problematic systems remain relatively scarce.
In this light, there has been significant effort targeted at making reliable predictions of aqueous pK a values using quantum chemical methods. Unfortunately, while the development of high-level composite methods, such as the Gaussian-n [23–27] and CBS composite methods [28–30], has facilitated the calculation of accurate gas-phase reaction energies, the accurate calculation of free energies in solution remains difficult. Dielectric continuum solvent models, such as the PCM family of continuum models: CPCM [31–33], IEF-PCM [34–37] and IPCM [38] and the SMx (x = 1–8) series [39–51], can normally deliver aqueous solvation free energies accurate to within 1 kcal/mol for typical neutral solutes [51, 52]. However, pK a calculations invariably involve ionic species, for which the errors incurred by continuum solvation calculations are considerably larger, typically around 4 kcal/mol [51, 52]. Moreover, continuum methods do not explicitly take account of direct solute–solvent interactions such as hydrogen bonding; in systems where such interactions are significant, much larger errors can be incurred in calculated solvation energies, and the pK a calculations that depend on them.
To address these problems, a wide variety of pK a calculation strategies have been developed, in which thermodynamic cycles are used to the improve the accuracy of solution-phase free energies, various types of proton exchange reactions are used to maximize systematic error cancellation, and/or water molecules are included in the various reaction schemes so as to improve the modeling of explicit solute–solvent interactions. When one considers that these protocols can be applied at various levels of theory, in conjunction with various solvation methods, the array of possible pK a calculation strategies can be bewildering. In this review, we examine these various protocols, with a view to evaluating their accuracy and identifying their potential limitations. The review is in two main parts. We first examine the existing literature, outlining the main protocols used to date, and summarizing the existing information on their performance. We then undertake our own benchmarking study of these methods on a consistent test set of 55 neutral species consisting of acids with various functionalities, including alcohols, phenols, carboxylic acids, inorganic acids and various carbon acids. In this way, we hope to identify whether any of the currently available methods offer a universal approach to accurate pK a calculations for neutral acids.
2 Overview of pK a calculation strategies
Before examining the individual pK a calculation strategies, it is first worth noting that, regardless of the reaction scheme adopted, the calculation of free energies in solution is usually performed via a thermodynamic cycle in which solution-phase reaction free energies are obtained as the sum of the corresponding gas-phase free energy and the free energy of solvation, as shown in Eq. 2.
where * denotes a standard state of 1 mol/L. Assuming ideal gas behavior, a correction corresponding to \( \Updelta nRT\ln (\tilde{R}T) \) must be added to the gas-phase reaction energy (denoted \( \Updelta G_{\text{gas}}^{ \circ } \)), as this is typically calculated for a standard state of 1 atm; ∆n refers to the change in number of species in the reaction and R and \( \tilde{R} \) are the gas constant in units of J/mol K and L atm/mol K, respectively. Typically, the gas-phase component is calculated using gas-phase optimized geometries and the solvation energies are calculated using solution-phase optimized geometries, though on occasion this can cause problems if, for example, there are stability issues associated with the solvent cavity model. In such cases, single point calculations are used instead. In the present work, we will examine the effect of geometry optimization on the accuracy of the calculated solvation energies.
The principal reason for using a thermodynamic cycle is that continuum solvation models are parameterized to produce accurate solvation energies, and the low levels of theory at which they are typically designed and implemented (such as small basis set HF or B3-LYP calculations) are not usually sufficiently accurate to reproduce accurate total free energies in solution. By using a thermodynamic cycle, one can make use of high-level ab initio calculations in the gas phase to improve the accuracy of the resulting free energies of reaction. As part of this work, we will examine the potential errors incurred by using instead the lower level methods for which continuum models are typically parameterized. Nonetheless, in general, we will assume that high levels of theory can be used for the gas-phase component, and the main challenge in any pK a calculation scheme is to maximize cancellation of errors incurred by the solvent model, i.e. make \( \Updelta \Updelta G_{\text{solv}}^{*} \) as accurate as possible.
2.1 The direct method
Invariably, all continuum solvent pK a calculations utilize a thermodynamic cycle such as those shown in Scheme 1 that combine accurate gas-phase acidity (experimental or computed via high level ab initio methods) with solvation free energies obtained from various solvent models. The directly calculated or absolute pK as may be obtained via cycles A and B through Eqs. 3 and 4, respectively.

pK a calculation via the direct or absolute method
where \( \Updelta \Updelta G_{\text{soln}}^{*} \) is obtained in the usual manner from Eq. 2. The correction term in Eq. 4 is necessary to correct for the standard state of liquid water, which is 55 mol/L. Bryantsev et al. [53] have recently highlighted that the incorrect assignment of standard state for water molecules has resulted in systematic errors in a number of reports, and the issue was also addressed in an earlier report by Pliego [54]. The confusion is a result of standard state for solutes in solution being 1 mol/L, and where water is acting as a co-reactant (e.g. cycle B), it is assumed to have a standard state for 1 mol/L. To verify that Eq. 4 is indeed the correct expression, we have evaluated pK a values for a number of acids by substituting their experimental gas-phase acidities and solvation free energies into Eqs. 3 and 4. The calculated pK a values from cycles A and B are shown in Table 1.
As shown, both cycles A and B in Scheme 1 give identical results, which are in very good agreement with the experimental pK a values. In fact, the excellent agreement with experiment is almost certainly guaranteed given that these “experimental” solvation free energies have been obtained through cycle A with experimental pK a and gas-phase reaction energies [55]. More importantly, it illustrates that if the correction factor in Eq. 4 were to be omitted, this would contribute a systematic error of 1.74 pK a units. The derivation of these corrections is straightforward and this has been presented elsewhere [53, 54]. In practice, one can simply subtract or add log[H2O] to the pK a value whenever a water molecule appears on the reactant or product side of the thermodynamic cycle, respectively. It is also worth adding that the choice of thermodynamic cycle is irrelevant if every quantity in cycles A and B is known with unlimited accuracy, i.e. they should give the exact same result. In other words, the energy associated with protonation of water to form the hydronium ion in cycle B is already included in the solvation free energy of the proton used in cycle A. Thus, the use of different thermodynamic cycles in theoretical pK a predictions is merely a computational strategy to remedy the uncertainties associated with solvation free energies from continuum models.
Cycle A [56–78] is by far the most commonly used because of its simplicity. For example, Schürmann et al. [79] have computed the pK a values of 16 aliphatic carboxylic acids in which gas-phase free energies were obtained at the SCF and MP2 levels of theory combined with solvation free energies obtained from the PCM-UAHF model. However, the approach was not sufficiently accurate for direct pK a calculations as a result of errors incurred in the gas-phase reaction energies. Liptak and Shields [59, 60] have subsequently utilized thermodynamic cycle A, but with gas-phase free energies obtained via high level ab initio methods (CBS-QB3 and G-n models) and found that very accurate results (within 1 pK a unit) may be obtained for phenols and aliphatic carboxylic acids, indicating the importance of accurate gas-phase reaction energies in a continuum solvent pK a calculation. This “prescribed protocol” has since been used in the pK a calculation of a range of acids, including carbenes [63, 64], carboxylic acid derivatives [66, 71, 80], inorganic acids [15, 67, 70, 81], carbon acids [82, 83] and amine compounds [65, 84]. For some of these classes of acids, including some carboxylic acids, phenols and some inorganic acids, the results are generally very good, but for some classes such as carbon acids, large errors in excess of 7 units occur. There are also a large number of studies that use gas-phase acidities computed using popular DFT methods such as the B3LYP method, most of which showed reasonable accuracies (1–2 kcal/mol) when compared with experiment [56, 58, 61, 62, 69, 72–74]. However, where possible, we advocate the use of high-level ab initio methods such as the CBS-X and G-n procedures over DFT methods because the latter’s performance is sometimes less consistent. These potential shortcomings are examined in greater detail in Sect. 3.5.
In spite of the partial successes reported for this protocol, there are some serious drawbacks that limit Scheme 1 as a general pK a calculation method. In particular, both cycles involve the generation of two ionic species and, since the associated uncertainties in their solvation energies are much larger, it can potentially lead to pK a predictions with very large errors. In our recent study of a set of biologically important carbon acids, we found that cycle A (using CPCM-UAKS and -UAHF solvent models) results in pK a values that overestimate experiment by 7 or more units [82]. The good performance observed in certain classes of acids such as carboxylic acids and phenols in earlier reports is presumably due to systematic cancellation of the errors incurred by the solvent model and/or because of the way the solvent model is parameterized. For example, the PCM-UAHF cavities have been parameterized to reproduce solvation free energies of set of 43 neutral solutes and 27 ions at the HF level with the 6-31G(d) and 6-31+G(d) basis sets, respectively [85]. The dataset is composed of typical organic species and their conjugate acids/bases, including alkanes, alcohols, amines, carboxylic acids and some hydrogen halides. However, the solvation patterns of carbanions are probably quite different from these organic acids and the parameterized cavities may not be accurate enough to account for these differences quantitatively.
Additionally, the experimental solvation free energy of the proton is required in the direct method (cycle A; Scheme 1). A brief literature search uncovered values that mainly lie in the range between −259 and −264 kcal/mol. The sources of some of these values as well as examples of studies that utilize them are shown in Table 2. Clearly, this scatter in the data contributes an additional source of uncertainty in the directly calculated pK a values. The range of these values is enough to contribute an error of ~3 pK a units. Furthermore, the standard state for which these values are quoted has also created some confusion in its usage in direct pK a calculations [86]. Experimental values are generally quoted under the standard state conventions of 1 bar in the gas phase and 1 molal in solution, which can be taken to differ negligibly from 1 atm in gas phase and 1 mol/L in solution (denoted ΔG solv without the asterisk). As noted in Eq. 1, the solvation free energies are defined for a standard state of 1 mol/L in the gas and aqueous phase, and accordingly, one must subtract 1.9 kcal/mol (\( RT\ln (\tilde{R}T) \) at 298.15 K) from these values to obtain the solvation free energy, \( \Updelta G_{\text{solv}}^{*} ({\text{H}}^{ + } ) \). As such, the misuse of these values has contributed a systematic error of 1.9 kcal/mol error in some of the values reported in the literature [67, 86]. Camaioni and Schwerdtfeger [86] have recently clarified the confusion over standard states and recommended using \( \Updelta G_{\text{solv}}^{*} ({\text{H}}^{ + } ) \) = −265.9 kcal/mol, a benchmark value derived by Tissandier et al. [87] by using the cluster pair approximation and recently reproduced to within 0.2 kcal/mol by Kelly et al. [88].
While cycle B [77, 79, 81, 89–94] has its fair share of the limitations as discussed above, it does offer several advantages over cycle A. Specifically, the use of water as a co-reactant is sometimes necessary for an accurate representation of the actual chemistry occurring in solution. As an example, Tossell has recently computed the pK a values of carbonic acid and boric acid using the protocol (cycle A) of Liptak and Shields [81]. However, this led to unreasonably large errors especially for boric acid where the deviation was in excess of 10 units. Closer examination and comparison with experimental spectral data suggests that the conjugate base of boric acid, B(OH)2O− does not exist in solution. Rather, it exists as B(OH) −4 , corresponding to the following reaction:
Using this reaction scheme, a vast improvement in the computed pK a value was obtained where the deviation was reduced to about 3 units. Likewise, carbonic acid (H2CO3) is better represented as CO2(aq)+H2O(aq), or as a hydrogen bonded complex. In this light, cycle B may be further generalized such that both the proton and the anionic conjugate base are solvated by water as shown in cycle C (Scheme 1). A related scheme, known as the implicit–explicit method is discussed in more detail in Sect. 2.3.
2.2 The proton exchange method
Given the above problems, the proton exchange method shown in Scheme 2 (also known as an isodesmic method or a relative pK a calculation) is considered more reliable because the number of charged species is conserved on both sides of equation thereby allowing for cancellation of some of the errors incurred in a continuum solvent calculation. Additionally, this approach also allows for further cancellation of errors in the gas-phase reaction free energy, especially when lower levels of theory (e.g. HF or DFT methods) are employed. The pK a is obtained through Eq. 5

pK a calculation via the proton exchange scheme. HRef is the reference acid
where the experimental value of the reference acid, HRef, is used. This approach also does not require an experimental value of \( \Updelta G_{\text{solv}}^{*} ({\text{H}}^{ + } ) \), which as discussed above, is a potential source of systematic error. Referring again to our earlier work, we found this approach significantly more accurate and delivered pK a values of various carbon acids, including acetamides, ketones, amines and small peptides that are within 1 unit of experiment (cf. 7 units in the direct method) [82]. In particular, this approach provides a useful comparison with the direct method and increasingly, the two approaches are being used together in the pK a calculation of various acids [52, 63, 72, 80, 84, 95–101] in aqueous and organic solvents, with generally good results.
Unfortunately, the success of this approach can depend heavily on the choice of reference acid, with best results expected if HRef is structurally similar to HA, since the errors incurred by the continuum solvent model are likely to be very similar and therefore should mostly cancel from \( \Updelta \Updelta G_{\text{solv}}^{*} \). To a certain extent, this depends on the solvent model used to evaluate the solvation energies. We found in the pK a calculation of neutral carbon acids of various functionalities that the CPCM-UAKS model worked particularly well using this scheme because the errors in this model are more systematic compared with the other solvent models examined [82]. Of course, the accuracy of the calculated value also depends on the accuracy of the experimental pK a of HRef. As a consequence, since accurate experimental pK a values of a structurally similar reference may not always be available, this may limit the proton exchange scheme as a universal pK a calculation method.
2.3 Hybrid cluster–continuum approaches
In this light, reference-independent methods that can deliver moderately accurate pK a values are highly desirable. Some success in this direction has been achieved through the inclusion of explicit solvent molecules in the acid dissociation process. There are several variants to this approach including the cluster–continuum model [53, 97, 99, 102–105] (Scheme 3) and the implicit–explicit solvent approach (Scheme 4) [106].

pK a calculation via the cluster–continuum model

pK a calculation via the implicit–explicit model
Pliego and Riveros have utilized Scheme 3 in combination with the IPCM solvent model to obtain pK a values that are accurate to within 2 units for a small test set of acids [103]. The pK a is obtained via Eq. 6
As noted in Table 1, using a standard state of 55 mol/L corresponds to a pK a of 14 for water as compared to the commonly quoted value of 15.74 which differs by a factor of log[H2O]. Thus, for consistency, this experimental value is adopted in Eq. 6. Scheme 3 is somewhat similar to a proton exchange scheme using water as a reference. However, it is strictly speaking not a proton exchange reaction because the number of moles of chemical species is not always conserved on both sides of the equation. Of course, in cases when n = 3, i.e. when the anion is solvated by three water molecules, then Eq. 6 collapses to Eq. 5 with an additional log[H2O] correction term, and HRef in this case is water. The number of water molecules (n) to include in an ion cluster, is determined using a “variational” cluster–continuum approach for solvation free energy calculations, as shown in Scheme 5. In this approach, the solvation of ionic species \( \Updelta \Updelta G_{\text{solv}}^{*} (A^{ \pm } ) \) corresponds to the free energy of the following process:

Thermodynamic cycle for calculation of cluster–continuum solvation free energy
Note that the standard states for each term in Eq. 7 are not the same, where \( \Updelta G_{\text{cluster}}^{ \circ } \) is in 1 atm, ΔG vap(H2O) corresponds to 1 atm in gas and 55 mol/L (pure water) in solution whereas \( \Updelta G_{\text{solv}}^{*} (A^{ \pm } ({\text{H}}_{ 2} {\text{O}})_{n} ) \) is 1 mol/L in both gas and solution. The vaporization free energy is related to the solvation free energy of water, \( \Updelta G_{\text{solv}}^{*} ({\text{H}}_{ 2} {\text{O}}) \)
where R and \( \tilde{R} \) are the gas constant in units of J/mol K and L atm/mol K, respectively. The cluster size is then determined on the basis of a “variational principle” that aims to maximize the stability of the ion in solution, i.e. find n that minimizes \( \Updelta G_{\text{solv}}^{*} \) [102]. Once the cluster size is determined, the solvation component, the solvation energy of each species in Scheme 3 is calculated using the IPCM continuum solvent model. In some sense, Scheme 3 works by decreasing the dielectric continuum contribution to the variation of \( \Updelta \Updelta G_{\text{solv}}^{*} \) in \( \Updelta G_{\text{soln}}^{*} \) through explicit introduction of short-range solute–solvent interactions in the first solvation shell around the ions. In this way, the accuracy in the calculated pK a value should be less sensitive to errors incurred by the dielectric continuum model. It is also possible that explicit solvation of ionic species also provides a more accurate representation of the actual chemistry occurring in solution. In their study, Pliego and Riveros have shown that \( \Updelta \Updelta G_{\text{solv}}^{*} \) contribution in a cluster–continuum model does not exceed 10 kcal/mol whereas pure dielectric continuum models contribute as much as 30–45 kcal/mol [102].
Kelly, Cramer and Truhlar have developed an implicit–explicit approach (Scheme 4), which, when used in conjunction with the SM6 model, offered significant improvement compared with the direct method [106]. The pK a is obtained from \( \Updelta G_{\text{soln}}^{*} \) in the same way as the direct method (cycle B) in Eq. 4, except in this case, the water molecule forms a complex with the anionic conjugate base of the acid. The standard state correction was also omitted in the original report [53, 106] although this does not affect the conclusions of that paper. In a similar spirit to the cluster–continuum approach, explicit solvation of the anionic conjugate base should compensate for some of the deficiencies inherent in continuum solvent models. The thermodynamic cycle also allows for short-range solvent–solute interactions to be calculated accurately in the gas phase.
We found in our recent assessment study that solvating the divalent conjugate bases of anionic carbon acids gives significantly improved results and in cases where it did not, the effect on the error was small [82]. However, as pointed out in the original study, there are issues relating to the number of water molecules to add, especially when the error in the directly calculated value is large, and in some cases (such as the bicarbonate anion) the addition of one water molecule is not always sufficient [106].
3 Benchmarking study
3.1 Experimental design
In view of the success reported for these various methods (Schemes 1, 2, 3, 4), we are interested in how their performance compares against a common dataset of acids. Specifically, which of these pK a calculation procedure(s) is most suitable for general pK a calculations? While all the above-mentioned procedures have been assessed to some extent, they are inevitably based on datasets composed of different numbers and types of acids. Furthermore, each approach is usually based on a specific solvent model applied at a specific level of theory and definition of solute cavities. To this end, we have compiled a dataset of 55 neutral species consisting of acids with various functionalities, including alcohols, phenols, carboxylic acids, inorganic acids and various carbon acids. Using this dataset, we have carried out a “grid search” using all possible combinations of thermodynamic cycles and solvent models to help identify the optimal combination of thermodynamic cycle and solvent model, capable of delivering chemically accurate pK a values. The definition of a chemically accurate pK a value is somewhat arbitrary and the general 1 kcal/mol definition for gas-phase calculations is unrealistic in view of the magnitude of the errors in an absolute continuum solvent calculation. The acceptable error margin for a directly calculated pK a value should be in the vicinity of 3.5 pK a units, and about 2 units for non-directly calculated values. The justification for these values is elaborated in Sect. 3.4.
3.2 Theoretical procedures
The pK a values have been computed using Eqs. 2–6 based on the various schemes. Experimental gas-phase acidities [107] were used in most of the directly calculated pK a values whereas gas-phase reaction energies corresponding to Schemes 2 to 4 were computed. The most recent experimental–theoretical values of −6.28 [59] and −265.9 [87] kcal/mol for the gas-phase Gibbs free energy, G(g, H+), and solvation free energy of the proton, \( \Updelta G_{\text{solv}}^{*} ({\text{H}}^{ + } ) \) as well as the experimental value of \( \Updelta G_{\text{solv}}^{*} ({\text{H}}^{ + } )({\text{H}}_{ 2} {\text{O}}) \) (6.32 kcal/mol) [86] were adopted in Schemes 1 and 4.
Ab initio gas-phase reaction energies were computed via the G3MP2(+) composite procedure [26] on geometries optimized at the B3LYP/6-31+G(d) level, denoted G3MP2(+)//B3. The G3MP2(+) is a modified version of G3MP2 in which calculations with 6-31G(d) have been replaced with 6-31+G(d) basis set, so as to allow for an improved description of anionic species. This approach has been demonstrated to deliver chemically accurate gas-phase reaction energies (i.e. 1 kcal/mol) in an earlier study [82]. To obtain the gas-phase free energies at 298.15 K, zero-point vibrational energy, thermal corrections and entropies were calculated from the B3LYP/6-31+G(d) geometries and frequencies, using the standard text book formulae for the statistical thermodynamics of an ideal gas under the rigid rotor/harmonic oscillator approximation [108]. Scale factors for the B3LYP/6-31G(d) frequencies were used for the free energy calculations [109]. For systems where multiple conformations exist, the lowest energy gas-phase conformer was used in the computations. The gas-phase optimized structures are provided in the Supporting Information. Additionally, gas-phase acidities for selected acids were also calculated using lower levels of theory for comparison, including HF, MP2 and the DFT methods, B3LYP [110], B97-1 [111], BMK [112] and BP86 [113, 114]. These gas-phase acidities were obtained as single point calculations with the G3MP2LARGE basis set on B3LYP/6-31+G(d) optimized geometries.
Solvation free energies obtained from the various solvent models are computed at levels of theories as recommended. The conductor-polarizable continuum model (CPCM) [31, 33] was applied at the B3LYP/6-31+G(d) and HF/6-31+G(d) levels of theory with the UAKS and UAHF [85] cavities to yield the CPCM-UAKS and CPCM-UAHF solvation energies respectively. In the directly calculated pK a values, we have also computed solvation free energies obtained from geometries optimized in vacuo as well as in the presence of solvent. In the cluster–continuum and implicit–explicit solvent models, the CPCM calculations were carried out on gas-phase optimized geometries.
The IPCM [38] calculation was carried out using an isodensity of 0.0004 and a dielectric constant of 78.39 at the MP2/6-31+G(d,p) level as recommended by earlier studies [102, 103]. In the IPCM continuum solvent calculation, only the electrostatic term (ΔGes) is considered and this is provided by the difference in electronic energies of the solute in vacuo and in the presence of solvent. These calculations were carried out on the B3LYP/6-31+G(d) gas-phase geometries. The CPCM and IPCM as well as all gas-phase calculations were all carried out using the Gaussian 03 software [115].
In addition, solvation free energies were also computed using the SM6 [49] and COSMO-RS [116–118] models. The SM6 model is based on a generalized Born approach which uses a dielectric continuum to treat bulk electrostatic effects combined with atomic surface tensions to account for first shell solvent effects, and it has been shown to deliver aqueous solvation free energies to within ~0.5 kcal/mol for neutral species [49]. Is also a density functional theory continuum model and can be used in conjunction with any good density functional, including the mPW0, B3LYP, and B3PW91 density functionals [49]. As such, the SM6 solvation free energies have been computed at the B3LYP/6-31+G(d) level of theory using the GAMESSPLUS program [119].
The COSMO-RS differs from a typical continuum solvent model in that the solvation free energies are derived from the statistical thermodynamics of interacting molecular surfaces, based on the polarization charge densities obtained from a COSMO calculation [116–118]. The parameterized model is capable of reproducing the solvation free energies of 163 neutral solutes to within 0.4 kcal/mol [117]. The ADF package [120] was used to compute the COSMO-RS solvation free energies on the gas-phase geometries at the BP/TZP level of theory (as it was parameterized for), and the rest of the parameters (e.g. atomic radii and cavity construction) were kept as default values [121].
Finally, we have also computed solvation free energies for the anions via the cluster–continuum approach of Pliego and Riveros using Eqs. 7 and 8 [102]. The solvation and vaporization free energies were computed using the various solvent models as mentioned above. As noted in Sect. 2.3, the number of solvent molecules (n) to include in the ion cluster is determined on the basis of a “variational principle” where the lowest value of \( \Updelta G_{\text{solv}}^{*} \) occurs. In the conformational sampling of these ion–water clusters, the solvent molecules are added to positions where they can directly hydrogen bond to the atom(s) bearing the charge in the anion; the maximum number of water molecules added generally corresponds to the number of electron lone pairs residing on that atom (usually three).
3.3 Comments on the calculation of solution reaction free energies, \( \Updelta G_{\text{soln}}^{*} \)
As shown in Eq. 2, the reaction free energy in solution is usually obtained as the sum of two components, the gas-phase reaction free energy, and a solvation contribution corresponding to the differences in the solvation free energies of the products and reactants. The equivalent expression for Eq. 2 in terms of solution free energies, \( \Updelta G_{\text{soln}}^{*} \), is shown in Eq. 9
To calculate the solution reaction free energy, \( \Updelta G_{\text{soln}}^{*} \), exactly, one would need to locate the equilibrium geometries of each reactant and product in both gas and solution phase. From these equilibrium geometries, one can compute the gas-phase reaction free energy as well as the solvation free energy of each species. In calculating \( \Updelta G_{\text{gas}}^{*} \), one would ideally also consider all conformers of each species and obtain its Gibbs free energy as a Boltzmann average of these conformers. However, it is also intuitively clear that in cases where the conformers are close in energies, the Boltzmann averaged Gibbs free energy should be very similar to that obtained on the global minimum structure. On the other hand, conformers which are significantly higher in energy would have very little contribution to the Boltzmann averaged Gibbs free energy. Thus, the use of equilibrium geometries should suffice for the calculation of \( \Updelta G_{\text{gas}}^{*} \).
In terms of the calculation of \( \Updelta G_{\text{solv}}^{*} \), this corresponds to the free energy change associated with the following process: A(g) ⇔ A(aq), where A is in its equilibrium geometry in the respective phases. In this view, computing solvation free energy as a single-point calculation on either the gas phase or solution equilibrium geometry implicitly assumes that the molecule undergoes little structural change between the two phases. This is probably true most of the time, especially for small rigid molecules with one dominating conformer. Indeed, several reports have found this choice makes little difference to the accuracy of the results. For example, Takano and Houk [52] have earlier compared the mean absolute deviations (MADs) in solvation free energies of 70 neutral and ionic species computed using geometries optimized in vacuo and in water, and found that the MADs were exceedingly similar; differing by no more than 0.5 kcal/mol.
However, a problem arises when the gas and solution equilibrium geometries are substantially different. The amino acids, which exist as zwitterions in solution but are neutral in the gas phase are one such example, and in such cases, the solvation free energies would need to be calculated via a cluster–continuum approach, i.e. by adding the number of explicit solvent molecules necessary to stabilize the zwitterion in the gas phase. Similarly, for larger molecules and in cases where one or more solvent molecule is treated explicitly, substantial changes in molecular geometries are usually associated with solvation. Specifically, in the ion–water clusters, one might expect the hydrogen-bonded structure to be less compact in solution. In such cases, there is an additional contribution to solvation free energy:
where the first three terms correspond to the standard electrostatic, dispersion–repulsion and cavitation contributions to the solvation free energy within the polarizable continuum model (PCM) framework and the last term refers to the contribution from changes in molecular structure during solvation. Equation 10 may alternatively be understood using a thermodynamic cycle:
where # denotes the solution equilibrium geometry. In this way, the single-point calculation is performed on the solution equilibrium geometry and ΔG conf may be approximately obtained as the difference in gas-phase electronic energies of the solution and gas-phase equilibrium structures, i.e. \( E_{\text{e}}^{\text{gas}} \;({\text{solution}}\;{\text{geometry}}) - E_{\text{e}}^{\text{gas}} \;({\text{gas}}\;{\text{phase}}\;{\text{geometry}}) \). Assuming that the contribution from changes in molecular geometry is small, the solvation free energy may be more conveniently obtained as just a single-point calculation on the solution equilibrium geometry. The effect of solution versus gas-phase geometries and the accuracy of the computed pK a values are examined in greater detail in Sects. 3.6 and 3.9.
3.4 Sources of error and definition of an acceptable margin
In comparing the performance of the various protocols in pK a calculations, it is useful to define an acceptable error margin. In Eq. 2, aqueous reaction free energies are composed of two components—a gas-phase term (\( \Updelta G_{\text{gas}}^{*} \)) and a solvation energy term (\( \Updelta \Updelta G_{\text{solv}}^{*} \)). As noted above, gas-phase reaction energies have an uncertainty of ~1 kcal/mol, and the errors in continuum solvent calculation for neutral and anionic species are typically about 1 and 4 kcal/mol [49, 51, 52]. The need to use the experimental solvation free energy of the proton, \( \Updelta G_{\text{solv}}^{*} ({\text{H}}^{ + } ) \) in the direct approach further contributes an additional source of error where the present benchmark value −265.9 kcal/mol has an estimated uncertainty of no less than 2 kcal/mol [88]. Assuming that these errors are additive, the uncertainty in a directly calculated pK a value can be as large as 6 units or more (1 pK a unit = 1.4 kcal/mol at room temperature). While the definition of an acceptable error margin is somewhat arbitrary, we propose that these should at least match the corresponding uncertainties associated with the experimental values. For neutral solutes, experimental \( \Updelta G_{\text{solv}} \) can be obtained directly by measuring partition coefficients of solutes between gas phase and dilute aqueous solutions in equilibrium, whereas “experimental” \( \Updelta G_{\text{solv}} \) for ionic species are usually obtained through the combination of experimental gas-phase acidities/basicities, pK a values and a thermodynamic cycle [55, 122, 123]. Pliego and Riveros have recently estimated that the associated errors for solvation free energies of ionic species are ~2 kcal/mol in aqueous solution [55]. Accordingly, the aim of any solvent model would be to achieve a directly calculated pK a value with an accuracy of 3.5 pK a units. On the other hand, the proton exchange scheme benefits from partial error cancellation. If we assume the residual error in the solvation component of a proton exchange reaction is about 2 kcal/mol, then a realistic error margin should be in the vicinity of 2 pK a units. In our subsequent discussion, this is the criterion we use for assessing the performance of any non-directly calculated pK a method (Schemes 2, 3, 4).
Finally, it should be mentioned that there are also errors associated with experimental pK a values, although these are usually not quoted. There are many factors that may affect the value of an experimental pK a measurement, and these include the ionic strength of the solution, the temperature, as well as the approximations used in kinetic measurements to derive these pK a values (e.g. protonation rate constants of carbanions that are combined with deprotonation rate constants via NMR spectroscopy [124]). Some of these values, especially for the very strong or weak acids (pK a < 0 or pK a > 15) are obtained via extrapolations. In this light, we have endeavored to compile a list of common organic and inorganic acids for which experimental pK a values are accurately known, most of which were used in developmental work for continuum solvent models [49, 55, 82, 125].
3.5 Assessment of gas-phase acidities
To evaluate the accuracy of the composite method G3MP2(+)//B3, we have computed gas-phase acidities for 30 of the acids in our test set and compared these with their experimental values as well as those obtained using various lower level methods. The methods B3LYP and MP2 are among the most commonly used for calculation of gas-phase acidities, whereas the BP86 and B97-1 have also been used in COSMO-RS [93, 126, 127] and SM6 [106] pK a calculations, respectively. The results are summarized graphically in Fig. 1 and full results are provided in Table S1 in the Supporting Information. As shown, the G3MP2(+)//B3 gas-phase acidities generally compare very well with the experimental values with a MAD of 1.43 kcal/mol. However, there is one notable exception, CF3SO3H, where the deviation is in excess of 5 kcal/mol across the seven levels of theory examined (data provided in Table S1 but omitted from Fig. 2). Gutowski and Dixon [15] have recently computed the gas-phase acidity of CF3SO3H and other strong acids (pK a < −10). Their gas-phase acidity of CF3SO3H (293 kcal/mol) computed at the CCSD(T)/CBS(+d) level of theory is in excellent agreement with our calculated value (cf. 293.2 kcal/mol). The deviation with the experimental result has been attributed to the large uncertainties associated with the gas-phase acidities of these strongly acidic species [15]. Omission of CF3SO3H results in an improved MAD for all the methods, although the HF method is clearly unsuitable for gas-phase acidity calculations, with MAD >6 kcal/mol and a maximum absolute deviation (ADmax) of 11 kcal/mol. The commonly used B3LYP and MP2 methods have reasonably small MADs of about 2 kcal/mol, but their ADmax values are considerably larger (6 or more kcal/mol). Interestingly, the ADmax (11 kcal/mol) in MP2 originates from HN3. The unusually large error in this system persisted even after the calculation was combined with the quadruple-zeta (aug-cc-pVQZ) basis set, indicating slow convergence towards the complete basis set limit for MP2.

The mean and maximum absolute deviations of gas-phase acidities (kcal/mol) computed at various levels of theory with the GTMP2LARGE basis set

The performance of directly calculated pK as (cycle A, Scheme 1) using various solvent models
Most of these errors can presumably be remedied by means of a proton exchange scheme or isodesmic reaction, although the HF residual errors are still likely to be significant. The two best-performing methods are G3MP2(+) and B97-1, with MAD values near chemical accuracy (~1 kcal/mol). The latter DFT method, however, has a slightly larger ADmax (4.1 vs. 3.2 kcal/mol). Nevertheless, where proton transfer reactions are concerned, we find B97-1 a reliable DFT method compared with other commonly used DFT methods, and this could provide a cost-effective alternative to the computationally more expensive G-n or CBS procedures.
3.6 Assessment of the direct method
The directly calculated and experimental pK a values for the 55 acids are provided in Table S2 in the Supporting Information and summarized in Fig. 2, where the acids in the test set have been broadly categorized according to their functionality. These pK a values have been computed by combining experimental gas-phase acidities (where available) with solvation free energies obtained from the five solvent models. Where more than one experimental pK a value is shown, the value with an asterisk was used to compute the errors in the calculations. Unsigned errors are shown in brackets.
As a useful aside, we have also examined if there were any significant difference in accuracy in the CPCM calculations if gas phase optimized geometries were employed. For both solvent models (CPCM-UAKS and UAHF), we found that re-optimization in aqueous solution generally performs better, although the overall gain in accuracy is only 0.6–0.8 pK a units in MAD, indicating the effect due to geometry changes in solution is reasonably small (full results in Table S16). In our directly calculated pK a values, all CPCM calculations use solution-optimized geometries unless stated otherwise.
A quick inspection of Fig. 2 (and Table S2) reveals that the performance of the direct method is somewhat inconsistent and can vary considerably depending on the solvent model and type of acid. As a whole, the CPCM-UAHF and SM6 methods are the best-performing continuum solvent models where the overall MADs are 3.8 units, close to the target accuracy of 3.5 units. Nonetheless, the unacceptably large maximum absolute deviations (ADmax) across all of the various solvent models, generally 10 units or more, questions the suitability of the direct method for general pK a calculations. In the CPCM-UAKS and UAHF models, there were stability issues associated with these cavity models and the errors incurred by the ammonia molecule (NH3) were unexpectedly large (>40 pK a units). The IPCM solvent model is also clearly unsuitable for direct pK a calculations with MAD and ADmax of 10 and 25 units respectively. Its poor performance is presumably due to the definition of the isodensity (0.0004) surface which has been applied universally for constructing the solute cavity of both neutral and ionic solutes.
Closer examination of how each solvent model performs with respect to the various classes of acids reveals some interesting trends. The CPCM-UAHF and SM6 models perform reasonably well with respect to alcohols, phenols and carboxylic acids (pK a values generally within 3 units of experiment) and this is consistent with results from earlier studies [59, 60, 71, 95]. However, the performance of the CPCM-UAHF model with respect to some inorganic and carbon acids is less ideal where some larger errors originate (e.g. HN3, HOOH and α-carbonyl carbon acids). On the other hand, the CPCM-UAKS model’s performance is slightly worse, but appears to be more consistent in that the calculated values are generally overestimated by 5–7 units across the various classes of acids. The SM6 and COSMO-RS models fair reasonably well for organic acids, but also appear to have problems with some inorganic acids (e.g. H2O, NH3 and HNO3). The poorer performance with respect to these species may be partly attributed to the uncertainty of associated with the experimental pK a values of these acids. As noted before, the pK as of very strong or weak acids (pK a < 0 and pK a > 14) may be subject to considerable error.
The selectively good performance of these solvent models within certain classes of acids is intriguing. As noted in Sect. 2.1, this is likely to be related to how these solvent models have been parameterized to account indirectly for short-range solvent–solute interactions. As a specific example, in the dataset used to parameterize PCM-UAHF, group 7 monovalent halide ions were used [85] and the model performed particularly well (errors <2 units) for these acids as shown in Table S2. On the other hand, the performance for related inorganic acids such as hydrogen peroxide and hydrogen azide were substantially worse, with deviations as large as 10 units. As such, an inherent deficiency in any parameterized model is that there is no guarantee that the accuracy of the calculated solvation energy will be carried over to species outside of the data set used to parameterize it. Unfortunately, all solvent models currently available have been parameterized to some extent. For example, the COSMO-RS method is composed of atomic radii, dispersion constants and other general parameters fitted against 642 data points corresponding to various properties such as solvation free energies, vapor pressure and partition coefficients [117], whereas the SM6 uses different parameters such as atomic surface tensions and a different set of atomic radii that are fitted against aqueous solvation free energies of 273 neutrals, 112 ions and 31 ion–water clusters [49]. This further reinforces our viewpoint that the direct method is currently unsuitable for general pK a predictions regardless of which solvent model is employed.
3.7 Assessment of pKa values via the proton exchange scheme
Using alcohols, phenols, carboxylic acids and carbon acids as examples, the pK a values for these molecules have been computed via a proton exchange scheme and results summarized in Fig. 3 (full details in Table S3). While these values can be computed via Eq. 5, they can be more simply obtained as the difference between the error in the directly calculated pK a of the reference acid and their directly calculated values. For example, the error in the directly calculated CPCM-UAKS pK a of methanol is 7.14. Using this as a reference, the CPCM-UAKS proton exchange pK a values of the remaining alcohols correspond to subtracting 7.14 from their directly calculated values in Table S2. This approach clearly results in significant improvement in accuracy; the overall MADs are mostly within the acceptable error margin of 2.5 pK a units. In particular, there is an approximately 4- to 5-fold reduction in MAD for CPCM-UAKS and IPCM compared with the direct method, bringing their overall MAD down to 1.8 and 3.3 units respectively. For the other models, where the MADs in the directly calculated values are already reasonably small (<3 units), the proton exchange scheme provided further improvement of 1–2 units.

The MAD in pK a values obtained using the proton exchange method for the various classes of acids. CH3OH, Ph-OH, HCOOH and CH3CONH2 are used as reference acids for alcohols, phenols, carboxylic acids and carbon acids, respectively
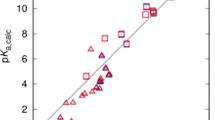
It should be emphasized that the improvement observed in the proton exchange scheme is strongly dependent on the nature of the errors incurred in a direct pK a calculation. As noted above (see also Table S2), CPCM-UAKS consistently overestimates directly calculated pK a values of (-carbonyl carbon acids and this is illustrated in Fig. 4 where the graph of directly calculated pK a values is essentially a vertical translation from the line y = x by 7 pK a units. In this example, acetamide was chosen as the reference because its pK a is accurately known, and the proton exchange scheme works exceptionally well (MAD = 0.9 and ADmax = 2.45), where the data points are clustered along the line y = x. In the COSMO-RS and SM6 methods, we note that the errors are less systematic to begin with, and, as a consequence, the use of the proton exchange scheme did not necessarily give an improvement over the directly calculated values.

The correlation between experimental and calculated (direct and proton exchange methods using CPCM-UAKS model) aqueous acidities of carbon acids at 298 K. Least squares correlation for direct method (filled triangle): pK a(Calc) = 1.03pK a(Expt) + 7.11; r 2 = 0.98 and proton exchange method (filled square): pK a(Calc) = 1.03pK a(Expt) − 0.12; r 2 = 0.98
3.8 Assessment of the cluster–continuum approach
We have examined the performance of the cluster–continuum approach (Scheme 3) for the 5 solvent models using a selection of 32 acids from Table S2. The cluster–continuum solvation free energies, calculated as a function of cluster size, are provided in Table S4. Interestingly, our cluster number (n) deviates slightly from those reported in an earlier study by Pliego and Riveros. For O-centered anions, the recommended the number of solvent molecules to add is usually 3, while we find that this number can vary between 2 and 3. This is presumably due to the different levels of theory employed in the gas-phase energetics and geometry optimization calculations. In the original paper, MP2/6-311+G(2df,2p) single point calculations were performed on HF/6-31+G(d,p) optimized geometries whereas in this work, the benchmarked G3MP2(+) composite procedure was applied to B3LYP/6-31+G(d) optimized geometries. Using HCl as an example, we note that the optimized geometry of the Cl(H2O) −2 cluster is substantially different at the two levels of theory. We have also neglected to apply anharmonic corrections, which were carried out in the original study on selected ion clusters, as it is not clear when anharmonicity is likely to be significant for the present test set. The pK a for each acid has been determined for n = 1–3 and the full results are shown in Tables S5–S9, and summarized in Fig. 5.

The performance of cluster–continuum pK as (Scheme 3) as a function of cluster size (n) using various solvent models
In the CPCM calculations, there were convergence problems associated with the optimization of certain ion clusters, such as the OH−(H2O)3 cluster which led to the dissociation of the hydroxide O–H bond, presumably due to stability issues associated with the definition of atomic radii used to construct the molecular cavity (UAHF and UAKS). Furthermore, the OH−(H2O)3 cluster is a recurring species in the cluster–continuum pK a calculation scheme. Thus, for consistency, all CPCM calculations were carried out using gas phase optimized geometries. As noted in Sect. 3.3, solution and gas-phase equilibrium geometries for ion–water clusters are likely to be quite different, and the computation of solvation free energies on gas-phase geometries is likely to introduce additional errors to the pK a calculations. The effect of molecular geometry on the accuracy of the implicit–explicit model (Scheme 4) is examined in greater detail in Sect. 3.9.
An immediate observation from Fig. 5 is that addition of a water molecule generally improves the result for all solvent models with the optimum number of water molecules (n) being 1 or 2. The best-performing solvent models were CPCM and COSMO-RS, where the lowest MAD is about 2 units whereas the SM6 and IPCM models also performed reasonably well with the lowest MADs around 3.5 units. The good performance of the COSMO-RS model is quite remarkable in view of the fact that it does not involve any experimental pK a values or other ion data in its parameterization [117].
While it seems physically more reasonable to consider each acid individually, i.e. assign the number of water molecules required to maximize its stability in solution on the basis of the “variational principle” and Eq. 7, we find that this does not necessarily give a better result. In Table S8, the values in bold refer to pK a values that would have been predicted if the number of water molecules added were determined based on Eq. 7. As shown, this approach can still lead to some rather large errors (e.g. HNO3 and CF3COOH). On the other hand, it is possible to achieve our target accuracy using a “one-size-fits-all” n = 2 in the CPCM and COSMO-RS models, where the MAD is about 2 units and the performance is reasonably consistent across the various types of acids (see Fig. 5, and also Tables S5, S6 and S9). Presumably, at this value of n, the contribution of \( \Updelta \Updelta G_{\text{solv}}^{*} \) to \( \Updelta G_{\text{soln}}^{*} \) is sufficiently small and therefore the effect on \( \Updelta G_{\text{soln}}^{*} \) of variations in \( \Updelta G_{\text{solv}}^{*} \) between the different acids is minimal. The large increase in MAD of these models when n = 3 is clearly undesirable, and as noted before, it is possible that at this coordination number, their solution equilibrium geometries may differ appreciably from the gas phase which could be a potential source of error. On the other hand, the SM6 model is significantly more stable with respect to n, and Fig. 5 shows a monotonic decrease in MAD as n increases from 0 to 3. Presumably, the empirical corrections in this model are sufficient to partially account for differences in gas and solution phase geometries.
3.9 Assessment of pKa values via the implicit–explicit model
The pK a values computed using Scheme 4 are shown in Tables S10–S14 and the results are summarized in Fig. 6. As shown, the success of this method appears to be limited to the SM6 and COSMO-RS solvent models. Addition of 1 water molecule generally reduced the error in the directly calculated pK a values (n = 0) for these models. Conversely, the errors in the CPCM pK a values increases with n, and there is a dramatic increase in MAD as three water molecules are added. The IPCM values improved by 3 units as two water molecules were added but the absolute errors were still substantial, with an MAD of 8 units. Best results were observed in the SM6 and COSMO-RS models when the ion is solvated by one water molecule (n = 1), bringing the MADs to about 3 units. On closer examination (Tables S12, S14), it appears that the errors associated with the organic acids are generally much smaller, about 2 units or less, indicating that these approaches might be more suitable for the pK a predictions of these species.

The performance of implicit–explicit model pK a s (Scheme 4) as a function of cluster size (n) using various solvent models
This raises two questions: (1) Why does addition of water molecules not improve the accuracy for the CPCM models? (2) For the SM6 and COSMO-RS models, why do the errors not improve with the addition of more water molecules? With respect to the first question, the use of gas-phase equilibrium geometries for computing solvation free energies of ion–water clusters is a potential source of error, since their solution and gas-phase equilibrium structures are expected to be quite different. To investigate this, a selection of ten acids from Table S10 were selected and the gas-phase geometries of these species and their associated ion–water clusters were re-optimized in the presence of solvent for the CPCM-UAKS and CPCM-UAHF models. Additionally, the COSMO-RS solvation energies were also computed on the CPCM-UAKS solution-optimized geometries. Inspection of the molecular geometries reveal that re-optimization in solution has the greatest effect on the structures of ion–water clusters, where they tend to adopt more “open” hydrogen bonded clusters. For example, the gas phase and solution optimized equilibrium structures for the HCOO−(H2O)2 and OCl−(H2O)3 clusters are shown in Fig. 7. As such, the pK as were recalculated using CPCM-UAHF, CPCM-UAKS and COSMO-RS solvation free energies obtained from the solution equilibrium geometries, and the results are summarized in Table 3. As shown, using solution-optimized structures improves the stability of the CPCM model, where the large errors associated with the addition of three solvent molecules have dropped by 4–5 pK a units. The inclusion of contribution for geometry changes, \( \Updelta G_{\text{Conf}} \), in Eq. 9, for the CPCM-UAKS solvation free energies, made little difference to the results. However, the improved performance is still not accurate enough for quantitative pK a calculations and addition of explicit solvent molecules does not lead to further improvement.

The solution (CPCM-UAKS) and gas-phase equilibrium structures of selected ion–water clusters
Another possibility relates to the parameterization of the CPCM models. The UAHF and UAKS atomic radii are optimized as functions of connectivity, hybridization state and formal charge [85], to reproduce the solvation free energies obtained from the continuum solvent calculations on a bare solute and, to a certain extent, this indirectly accounts for the short-range solvent–solute interactions in the continuum model. This unsystematic approach could counteract the systematic treatment of these errors through the introduction of explicit solvent–solute interactions. On the other hand, other solvent models are parameterized differently and in COSMO-RS and SM6, the atomic radii are functions of only atomic number. As shown in an earlier study, introduction of an explicit solvent molecule results in further improvement in the predicted solvation free energies for the SM6 model [49]. Since most solvent models have been parameterized to some extent, the addition of more water molecules is unlikely to systematically improve the errors in a continuum solvent pK a calculation. Cramer and Truhlar have also highlighted that addition of explicit solvent molecules changes the non-electrostatic contributions to solvation free energy (e.g. cavitation and dispersion free energies) as well as the solute’s translational, vibrational and rotational free energies, and the parameterized surface tensions may not be accurate enough to account for these changes quantitatively [106]. As noted before, the poorer performance of the unparameterized IPCM model is presumably due to the definition of molecular cavity based on the 0.0004 isodensity surface which have been applied universally to neutral and ionic solutes.
3.10 Towards a universal proton exchange scheme based on cluster continuum solvation energies
The results so far indicate that direct methods are only suitable for the pK a predictions of certain classes of acids for which the solvent models have been parameterized, whereas cluster–continuum hybrid models show more promise in terms of providing a universal pK a prediction approach. Still, these models are not without limitations; for example, their performance are somewhat sensitive to the ion-cluster size (n) and the rules for automating this choice, empirical or otherwise, still require further refinement. The proton exchange scheme is significantly more straightforward and its performance is comparable, if not slightly better, to the cluster–continuum models. However, an obvious limitation of a proton exchange scheme is clearly the need for a structurally similar reference. In both CPCM-UAKS/UAHF, the use of formic acid (HCOOH) as a reference was clearly unsuitable for the pK a calculation of trifluoroacetic and trichloroacetic acids where the deviations increased by more than 3 units (Table S3). Furthermore, there is some ambiguity pertaining to the experimental data of some strong inorganic acids where experimental pK a values that differ by 5 or more units have been reported for the same species (Table S2). As a consequence, since accurate experimental pK a data for a structurally similar reference may not always be available, this limits the applicability of the proton exchange scheme as a general pK a calculation method.
In a separate study, Pliego and Riveros have used the cluster–continuum approach (Scheme 5) for the computation of IPCM solvation energies of ionic species at the MP2/6-31+G(d,p) level of theory using Eq. 7 [102]. While the results typically underestimate experiment by ~9 kcal/mol [102], this systematic error could potentially lessen the sensitivity of the proton exchange method to the choice of reference acid. In particular, we note that there should be substantial cancellation of these errors in the solvation contribution (\( \Updelta \Updelta G_{\text{solv}}^{*} \)) to the aqueous reaction energy. To investigate this possibility, the solvation free energies for the anionic conjugate bases of a selection of 32 acids from Table S2 were computed via Eq. 7. These cluster–continuum solvation free energies for ionic species were combined with pure continuum IPCM solvation free energies for neutral species for the calculation of direct pK a values and are labeled “Direct(Cluster IPCM)” in Figs. 8 and 9. For comparison, the results for directly calculated pK as using solely the pure IPCM solvation energies are also shown and labeled “Direct(Pure IPCM)”. Complete pK a values are provided in Table S15. As shown in Fig. 8, using cluster–continuum solvation free energies for ionic species, results in a substantial reduction in the MAD of the directly calculated pK a values by about 4 units (6.5 cf. 11), and the ADmax was more than halved (10 cf. 22). While the errors in the former are still relatively large, it is important to note that there is also a significant reduction in standard deviation in the errors (2.3 cf. 5). This indicates that the cluster–continuum solvation free energies have a leveling effect on the errors in a direct pK a calculation and this is illustrated graphically in Fig. 9. As shown, cluster–continuum solvation free energies bring the directly calculated pK a values closer to the line y = x. More importantly, a least squares fit of these data points gives an equation: pK a(Calc) = 1.07 pK a(Expt)+5.39; r 2 = 0.98, where the gradient is close to unity and is almost a vertical translation of the line of unit gradient upwards by 5.4 units. Thus, using methanol as the reference acid, the aqueous acidity constants of the remaining acids were computed via a combined cluster continuum-proton exchange approach and the resulting MAD was 1.8 units, which is a further improvement from the direct-cluster method (MAD 6.5 units). This is particularly promising because Table S15 includes a diverse range of acids, such as alcohols, carboxylic acids, various carbon acids and inorganic acids. To achieve an average accuracy of 2 units by merely using methanol as a reference is a very good result. Specifically, Fig. 8 shows that the large errors typically originate from very strong inorganic acids where the pK as are <0. As noted in our earlier discussion, this is presumably due to the considerable uncertainty associated with some of the pK as of these acids. On the other hand, this method is much more stable with respect to organic acids were the errors are generally <2 units. In practice, one would select the closest possible reference (as opposed to methanol in this case), which should give an even better result. In this light, the combined IPCM cluster–continuum proton exchange method is effectively a reference-independent approach and should be useful for general pK a predictions of neutral organic acids.

The performance of the directly calculated pK a values using pure IPCM solvation free energies versus IPCM cluster–continuum solvation free energies, and the corresponding proton exchange pK a values of the latter approach

The performance of the directly calculated pK a values using pure IPCM solvation free energies versus IPCM cluster–continuum solvation free energies, and the corresponding proton exchange pK a values of the latter approach. Least squares correlation for Direct(Pure IPCM) (filled diamond): pK a(Calc) = 1.14 pK a(Expt) + 9.15; r 2 = 0.92. Direct(Cluster IPCM) method (filled square): pK a(Calc) = 1.07 pK a(Expt) + 5.39; r 2 = 0.98. PEX(Cluster IPCM) (filled triangle): pK a(Calc) = 1.07pK a(Expt) − 2.2; r 2 = 0.98
4 Summary and concluding remarks
In this paper, we have reviewed several commonly used pK a calculations methods (Schemes 1, 2, 3, 4) and examined their performance in conjunction with several popular solvent models, namely CPCM-UAKS/UAHF, SM6, IPCM and COSMO-RS, in the pK a predictions of a common dataset of neutral organic and inorganic acids with a view to identifying a universal approach that can deliver pK a values with chemical accuracy (defined here as 2.5 pK a units). Several promising pK a calculation protocols have been short-listed, including the proton exchange scheme and its IPCM combined cluster–continuum analog, the COSMO-RS and CPCM cluster–continuum approach and the COSMO-RS and SM6 implicit–explicit model, where accuracies of 2 units can be achieved. In particular, a proton exchange scheme based on the cluster continuum model appears to be much less sensitive to the chosen reference than traditional continuum model based approach, and shows promise as a universal approach to accurate pK a values. We advocate the use of these short-listed protocols over the direct method, as this work has further confirmed that the success of the direct approach is mainly limited to species with identical or similar structures to those used in the original parameterization of the chosen solvation models. Furthermore, because these protocols are complementary to one another, they should provide useful comparisons when used in general pK a predictions.
On the other hand, there is certainly no guarantee that they will always deliver pK a values with 2 units accuracy; the safest gauge is probably given by their ADmax values, which are still unacceptably large (>5 units), indicating that further refinements to present solvent models are still needed. There is no need to be discouraged by these less than ideal results. In fact, considering accuracies of typical continuum solvent model calculations, the present pKa calculation protocols are already in a relatively good place. However, it is important to acknowledge that there will always be inherent difficulties in trying to model solvation, a dynamic and complex phenomenon, based on a dielectric continuum.
References
Tomasi J, Persico M (1994) Chem Rev 94:2027
Cramer CJ, Truhlar DG (1999) Chem Rev 99:2161
Orozo M, Luque FJ (2000) Chem Rev 100:4187
Tomasi J, Mennucci B, Cammi R (2005) Chem Rev 105:2999
Bell RP (1973) The proton in chemistry. Chapman and Hall, London
Stewart R (1985) The proton: applications to organic chemistry. Academic Press, Orlando
Richard JP, Amyes TL (2001) Curr Opin Chem Biol 5:626
Houk RJT, Monzingo A, Anslyn EV (2008) Acc Chem Res 41:401
Gerlt JA (1993) Biochemistry 32:11943
Toney MD (2005) Arch Biochem Biophys 433:279
Fitzpatrick PF (2001) Acc Chem Res 34:299
Nielsen JE, McCammon JA (2003) Protein Sci 12:1894
Arnett EM, Mach GW (1966) J Am Chem Soc 88:1177
Arnett EM, Scorrano G (1976) Adv Phys Org Chem 13:83
Gutowski KE, Dixon DA (2006) J Phys Chem A 110:12044
Guthrie JP (1978) Can J Chem 56:2342
Wong FM, Capule C, Chen DX, Gronert S, Wu W (2008) Org Lett 10:2757
Amyes TL, Richard JP (1996) J Am Chem Soc 118:3129
Richard JP, Williams G, Gao J (1999) J Am Chem Soc 121:715
Rios A, Richard JP, Amyes TL (2002) J Am Chem Soc 124:8251
Sievers A, Wolfenden R (2002) J Am Chem Soc 124:13986
Wong FM, Capule C, Wu W (2006) Org Lett 8:6019
Curtiss LA, Redfern PC, Raghavachari K (2007) J Chem Phys 127:124105
Baboul AG, Curtiss LA, Redfern PC, Raghavachari K (1999) J Chem Phys 110:7650
Curtiss LA, Raghavachari K, Redfern PC, Rassolov V, Pople JA (1998) J Chem Phys 109:7764
Curtiss LA, Redfern PC, Raghavachari K, Rassolov V, Pople JA (1999) J Chem Phys 110:4703
Curtiss LA, Redfern PC, Raghavachari K (2007) J Chem Phys 126:84108
Ochterski JW, Petersson GA, Montgomery JA (1996) J Chem Phys 104:2598
Montgomery JA Jr, Frisch MJ, Ochterski JW, Petersson GA (1999) J Chem Phys 110:2822
Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA (2000) J Chem Phys 112:6532
Klamt A, Schurmann G (1993) J Chem Soc Perkin Trans 2:799
Barone V, Cossi M (1998) J Phys Chem A 102:1995
Cossi M, Rega N, Scalmani G, Barone V (2003) J Comp Chem 24:669
Cances MT, Mennucci B, Tomasi J (1997) J Chem Phys 107:3032
Mennucci B, Tomasi J (1997) J Chem Phys 106:5151
Mennucci B, Cances MT, Tomasi J (1997) J Phys Chem B 101:10506
Tomasi J, Mennucci B, Cances MT (1999) J Mol Struct (Theochem) 464:211
Foresman JB, Keith TA, Wiberg KB, Snoonian J, Frisch MJ (1996) J Phys Chem 100:16098
Hawkins GD, Cramer CJ, Truhlar DG (1998) J Phys Chem B 102:3257
Cramer CJ, Truhlar DG (1991) J Am Chem Soc 1991:8552
Cramer CJ, Truhlar DG (1992) Science 256:213
Cramer CJ, Truhlar DG (1992) J Comput Chem 13:1089
Storer JW, Giesen DJ, Hawkins GD, Lynch GC, Cramer CJ, Truhlar DG, Liotard DA (1994) A solvation modeling in aqeous and nonaqueous solvents: new techniques and a re-examination of the Claisen rearrangement. In: Cramer CJ, Truhlar DG (eds) Structure and reactivity in aqueous solution. American Chemical Society, Washington, pp 24–49
Hawkins GD, Cramer CJ, Truhlar DG (1996) J Phys Chem 100:19824
Hawkins GD, Cramer CJ, Truhlar DG (1997) J Phys Chem B 101:7147
Hawkins GD, Liotard DA, Cramer CJ, Truhlar DG (1998) J Org Chem 63:4305
Thompson JD, Cramer CJ, Truhlar DG (2005) Theor Chem Acc 113:107
Thompson JD, Cramer CJ, Truhlar DG (2004) J Phys Chem A 108:6532
Kelly CP, Cramer CJ, Truhlar DG (2005) J Chem Theory Comput 1:1133
Marenich AV, Olson RM, Kelly CP, Cramer CJ, Truhlar DG (2007) J Chem Theor Comput 3:2011
Cramer CJ, Truhlar DG (2008) Acc Chem Res 41:760
Takano Y, Houk KN (2005) J Chem Theory Comput 1:70
Bryantsev VS, Diallo MS, Goddard WA III (2008) J Phys Chem B 112:9709
Pliego JR (2003) Chem Phys Lett 367:145
Pliego JR Jr, Riveros JM (2002) Phys Chem Chem Phys 4:1622
Kallies B, Mitzner R (1997) J Phys Chem B 101:2959
Shapley WA, Backsay GB, Warr GG (1998) J Phys Chem B 102:1938
Topol IA, Tawa GJ, Caldwell RA, Eissenstat MA, Burt SK (2000) J Phys Chem A 104:9619
Liptak MD, Shields GC (2001) J Am Chem Soc 123:7314
Liptak MD, Gross KC, Seybold PG, Feldgus S, Shields GC (2002) J Am Chem Soc 124:6421
Chipman DM (2002) J Phys Chem A 106:7413
Klicic JJ, Friesner RA, Liu S-Y, Guida WC (2002) J Phys Chem A 106:1327
Magill AM, Cavell KJ, Yates BF (2004) J Am Chem Soc 126:8717
Magill AM, Yates BF (2004) Aust J Chem 57:1205
Murlowska K, Sadlej-Sosnowska N (2005) J Phys Chem A 109:5590
Krol M, Wrona M, Page CS, Bates PA (2006) J Chem Theory Comput 2:1520
da Silva G, Kennedy EM, Dlugogorski BZ (2006) J Phys Chem A 110:11371
Lu H, Chen X, Zhan CG (2007) J Phys Chem B 111:10599
Bryantsev VS, Diallo MS, Goddard WA (2007) J Phys Chem A 111:4422
Zimmermann MD, Tossell JA (2009) J Phys Chem A 113:5105
Namazian M, Zakery M, Noorbala MR, Coote ML (2008) Chem Phys Lett 451:163
Lopez X, Schaefer M, Dejaegere A, Karplus M (2002) J Am Chem Soc 124:5010
Saracino GAA, Improta R, Barone V (2003) Chem Phys Lett 373:411
Schmidt n, Knapp EW (2004) Chem Phys Chem 5:1513
Tossell JA, Sahai N (2000) Geochim Cosmochim Acta 64:4097
Lee C, Yang W, Parr RG (1988) Phys Rev B Condens Matter 37:785
Sadlej-Sosnowska N (2007) Theor Chem Acc 118:281
Lim C, Bashford D, Karplus M (1991) J Phys Chem 95:5610
Schuurmann G, Cossi M, Barone V, Tomasi J (1998) J Phys Chem A 102:6706
Dong H, Du H, Qian X (2008) J Phys Chem A 112:12687
Tossell JA (2995) Geochim Cosmochim Acta 69:5647
Ho J, Coote ML (2009) J Chem Theory Comput 5:295
Gao D, Wong PK, Maddalena D, Hwang J, Walker H (2005) J Phys Chem A 109:10776
Caballero NA, Melendez FJ, Munoz-Caro C, Nino A (2006) Biophys Chem 124:155
Barone V, Cossi M, Tomasi J (1997) J Chem Phys 107:3210
Camaioni DM, Schwerdtfeger CA (2005) J Phys Chem A 109:10795
Tissandier MD, Cowen KA, Feng WY, Gundlach E, Cohen MH, Earhart AD, Coe JV, Tuttle TR Jr (1998) J Phys Chem A 102:7787
Kelly CP, Cramer CJ, Truhlar DG (2006) J Phys Chem B 110:16066
da Silva EC, Silva CO, Nascimento MAC (1999) J Phys Chem A 103:11194
Silva CO, da Silva EC, Nascimento MAC (2000) J Phys Chem A 104:2402
Namazian M, Halvani S (2006) J Chem Thermodyn 38:1495
Namazian M, Heidary H (2003) THEOCHEM 620:257
Klamt A, Eckert F, Diedenhofen M, Beck ME (2003) J Phys Chem A 107:9380
Yang W, Qian Z, Miao Q, Wang Y, Bi S (2009) Phys Chem Chem Phys 11:2396
Toth AM, Liptak MD, Phillips DL, Shields GC (2001) J Chem Phys 114:4595
Namazian M, Kalantary-Fotooh F, Noorbala MR, Searles DJ, Coote ML (2006) THEOCHEM 758:275
Fu Y, Liu L, Li R-Q, Liu R, Guo Q-X (2004) J Am Chem Soc 126:814
Gomez-Bombarelli R, Gonzalez-Perez M, Perez-Prior MT, Calle E, Casado J (2009) J Org Chem 74:4943
Ding F, Smith JM, Wang H (2009) J Org Chem 74:2679
Mujika JI, Mercero JM, Lopez X (2003) J Phys Chem A 107:6099
Almerindo GI, Tondo DW, Pliego JR Jr (2004) J Phys Chem A 108:166
Pliego JR Jr, Riveros JM (2001) J Phys Chem A 105:7241
Pliego JR Jr, Riveros JM (2002) J Phys Chem A 106:7434
Wang X, Fu H, Du D, Zhou Z (2008) Chem Phys Lett 460:339
Smiechowski M (2009) J Mol Struct 924–926:170
Kelly CP, Cramer CJ, Truhlar DG (2006) J Phys Chem A 110:2493
Linstrom PJ, Mallard WG (eds) NIST Chemistry WebBook, NIST Standard Reference Database Number 69. National Institute of Standards and Technology, Gaithersburg MD, 20899. http://webbook.nist.gov. Retrieved 12 August 2009
Atkins PW (1998) Physical chemistry, 6th edn. Oxford University Press, Oxford
Scott AP, Radom L (1996) J Phys Chem 100:16502
Becke AD (1993) J Chem Phys 98:5648
Hamprecht FA, Cohen AJ, Tozer DJ, Handy NC (1998) J Chem Phys 109:6264
Boese AD, Martin JML (2004) J Chem Phys 121:3405
Becke AD (1988) Phys Rev A Gen Phys 38:3098
Perdew JP (1986) Phys Rev B 33:8822
Frisch MJT GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, JohnsonB, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, Revision C.02. Gaussian, Inc., Wallingford CT
Klamt A (1995) J Phys Chem 99:2224
Klamt A, Jonas V, Burger T, Lohrenz JCW (1998) J Phys Chem A 102:5074
Klamt A (2005) COSMO-RS: from quantum chemistry to fluid phase thermodynamics and drug design. Elsevier Science Ltd., Amsterdam, The Netherlands
Higashi MMAV, Olson RM, Chamberlin AC, Pu J, Kelly CP, Thompson JD, Xidos JDL Jr, Zhu T, Hawkins GD, Chuang Y-Y, Fast PL, Lynch BJ, Liotard DA, Rinaldi DG Jr, Cramer CJ, Truhlar DG, GAMESSPLUS—version 2008–2, University of Minnesota M, 2008, based on the General Atomic and Molecular, Electronic Structure System (GAMESS) as described in Schmidt MWB KK, Boatz JA, Elbert STG MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TLD M, Montgomery JA (1993) J Comp Chem 14:1347
Louwen JN, Pye C, Lenthe Ev (2008) ADF2008.01 COSMO-RS, SCM. Theoretical chemistry. Vrije Universiteit, Amsterdam. http://www.scm.com
Pye CC, Ziegler T, van Lenthe E, Louwen JN (2009) Can J Chem 87:790
Florian J, Warshel A (1997) J Phys Chem B 101:5583
Pearson RG (1986) J Am Chem Soc 108:6109
Amyes TL, Richard JP (2007) Proton transfer to and from carbon in model reactions. In: Hynes JT, Kilnman JP, Limbach H, Schowen RL (eds) Hydrogen-transfer reactions. Wiley, Weinheim
Williams R. http://research.chem.psu.edu/brpgroup/pKa_compilation.pdf
Eckert F, Klamt A (2006) J Comput Chem 27:11
Eckert F, Leito I, Kaljurand I, Kütt A, Klamt A, Diedenhofen M (2009) J Comp Chem 30:799
Topol IA, Burt SK, Rashin AA, Erikson JW (2000) J Phys Chem A 104:866
Tawa GJ, Topol IA, Burt SK, Caldwell RA, Rashin AA (1998) J Chem Phys 109:4852
Zhan CG, Dixon DA (2001) J Phys Chem A 105:11534
Acknowledgments
We gratefully acknowledge support from the Australian Research Council under their Centres of Excellence program, and generous allocations of computing time on the National Facility of the Australian Partnership for Advanced Computing.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Ho, J., Coote, M.L. A universal approach for continuum solvent pK a calculations: are we there yet?. Theor Chem Acc 125, 3–21 (2010). https://doi.org/10.1007/s00214-009-0667-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00214-009-0667-0