
Abstract
Rationale
Evaluation of pharmacotherapies for acute stress disorder (ASD) or post-traumatic stress disorder (PTSD) is challenging due to robust heterogeneity of trauma histories and limited efficacy of any single candidate to reduce all stress-induced effects. Pursuing novel mechanisms, such as the nociceptin/orphanin FQ (NOP) system, may be a viable path for therapeutic development and of interest as it is involved in regulation of relevant behaviors and recently implicated in PTSD and ASD.
Objectives
First, we evaluated NOP receptor antagonism on general behavioral performance and again following a three-species predator exposure model (Experiment 1). Then, we evaluated effects of NOP antagonism on fear memory expression (Experiment 2).
Methods
Adult, male rats underwent daily administration of NOP antagonists (J-113397 or SB-612,111; 0–20 mg/kg, i.p.) and testing in acoustic startle, elevated plus maze, tail-flick, and open field tests. Effects of acute NOP antagonism on behavioral performance following predator exposure were then assessed. Separately, rats underwent fear conditioning and were later administered SB-612,111 (0–3 mg/kg, i.p.) prior to fear memory expression tests.
Results
J-113397 and SB-612,111 did not significantly alter most general behavioral performance measures alone, suggesting minimal off-target behavioral effects of NOP antagonism. J-113397 and SB-612,111 restored performance in measures of exploratory behavior (basic movements on the elevated plus maze and total distance in the open field) following predator exposure. Additionally, SB-612,111 significantly reduced freezing behavior relative to control groups across repeated fear memory expression tests, suggesting NOP antagonism may be useful in dampening fear responses. Other measures of general behavioral performance were not significantly altered following predator exposure.
Conclusions
NOP antagonists may be useful as pharmacotherapeutics for dampening fear responses to trauma reminders, and the present results provide supporting evidence for the implication of the NOP system in the neuropathophysiology of dysregulations in fear learning and memory processes observed in trauma- and stress-related disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Trauma- and stress-related disorders, such as acute stress disorder (ASD) and post-traumatic stress disorder (PTSD), are multifaceted disorders that affect a small percentage of those who experience a traumatic stressor, and are of concern for individuals that may be more likely to be exposed to trauma, such as combat-exposed military personnel. ASD and PTSD are typically accompanied by behavioral symptoms (e.g., hypervigilance, avoidance of trauma reminders, deficits in fear learning and memory, and anxiety-like behaviors) which can range in severity and can ultimately disrupt day-to-day functioning (Holowka and Marx 2012). Currently, the risk factors, developmental course, and treatment of ASD and/or PTSD are not well understood. Further it is difficult to ethically study traumatic experiences in a controlled environment at acute and chronic time points. Therefore, animal models have been designed to model and study symptoms of trauma- and stress-related disorders observed in humans. These models vary in the type and severity of behavioral impairments used to infer face validity of the model and no single model recapitulates the entirety of the symptomatology of the disorders (for review, see Lowery-Gionta et al. 2019). One preclinical model involves the procedure of predator odor exposure (such as soiled litter from a cat) to the rodent test subject which capitalizes on the use of cue- or context-associated fear memory retrieval (Blanchard et al. 2001; for review, Takahashi et al. 2008; Takahashi et al. 2005). Additionally, fear conditioning paradigms that incorporate inescapable footshock are sometimes regarded in the literature as a model, and particularly so if they are designed to produce robust fear-associated learning effects via freezing behavior (Olson et al. 2011; Pynoos et al. 1996). Although both of these models can explore fear-associated learning and memory, they do not incorporate a perceived direct threat-to-life to the test subject, which limits their translational value. To address this, our lab has developed a model of traumatic stress which involves a perceived direct-threat-to-life via a protected exposure to three species of predators: cats, ferrets, and snake (Moore et al. 2016). Together, these models can be used to evaluate the efficacy of novel potential pharmacotherapies for the treatment of trauma- and stress-related disorders, as there are at present only two FDA-approved drugs for PTSD and no FDA-approved drugs for ASD. Over the past two decades, psychiatrists have relied on a single class of psychotropic medications to treat the heterogeneous presentation of acute and post-traumatic stress symptoms. These approved compounds are both selective serotonin reuptake inhibitors, leaving a gap for evaluating compounds that possess other putative downstream targets, including the nociceptin/orphanin FQ (NOP) receptor system (Genovese and Dobre 2017; Zhang et al. 2012; Zhang et al. 2018; Zhang et al. 2015).
Since the discovery of the NOP receptor system (Mollereau et al. 1994; Reinscheid et al. 1995), evidence has emerged implicating it in stress-related disorders. The NOP receptor is similar to the classical opioid receptors, but does not bind any of the classical mu, delta, or kappa ligands with high affinity (Bunzow et al. 1994; Mollereau et al. 1994; Reinscheid et al. 1995). Early evidence demonstrated the endogenous peptide-mediated anxiolytic-like effects while homozygous knockout mouse lines demonstrated increased anxiety-like behavior as inferred on behavioral assays such as the elevated plus maze and open field tests (Jenck et al. 1997; Koster et al. 1999). In preclinical rodent models of stress-related disorders, increased central levels of the endogenous peptide have been found as early as 9 days and as late as 28 days following traumatic stress exposure (Zhang et al. 2012) that were reduced after administration of an NOP antagonist (Zhang et al. 2015). Moreover, findings of a single-nucleotide polymorphism within the encoding gene (OPR1) have been observed in preclinical models and are also correlated with increased symptoms of PTSD in people with a history of childhood abuse (Andero et al. 2013). Recent reports of correlational PET-imaging data suggest an increased number of NOP receptors corresponds to an increase in PTSD symptom severity in human patients, and although this first study found significant correlations only for select brain regions (midbrain and cerebellum), the authors suggested these findings indicate NOP receptors may play a role in the adaptive response to traumatic stress exposure (Narendran et al. 2019).
Further, there is a general consensus in the preclinical literature that fear memory consolidation is impaired following administration of NOP agonists which can be blocked by NOP antagonists (see review, Andero 2015). Enhanced retention of avoidance behavior in rats following post-avoidance training infusions of the NOP antagonist [Nphe1]nociceptin(1-13)NH2 into the basolateral amygdala has been observed (Roozendaal et al. 2007). Similar improved learning effects have been reported in murine NOP peptide (N/OFQ) knockout lines (Higgins et al. 2002), suggesting that blockade of NOP receptors enhances memory consolidation. However, the effects of NOP antagonism on fear extinction are unknown. Preclinical assessment of fear extinction is particularly relevant as it has been suggested to be a model of prolonged exposure therapy, a commonly used treatment for PTSD, in the clinic (Paredes and Morilak 2019). Together, the available research indicates that the NOP receptor system is dysregulated in stress-related disorders. Therefore, we hypothesized that systemic pharmacological antagonism of NOP receptors would reduce behavioral impairments induced by traumatic stress in rodent models.
In the present study, we first evaluated a battery of behavioral tasks in rats that model various symptom domains which are commonly affected in trauma-associated disorders, such as PTSD, but may vary in severity across cases. The behavioral tasks were selected for their use in the preclinical literature and translational value, and included evaluation of: reflexive behavior (acoustic startle), anxiety-related and exploratory behavior (elevated plus maze), pain sensitivity (warm-water tail immersion test), and locomotor activity (total distance traveled in the open field test). Behavioral performance effects of pharmacological blockade of the NOP receptor have been previously reported; however, their characterization has been mostly limited to blocking or mitigating agonist-induced effects rather characterization of their inherent effects on behavior. In Experiment 1, behavioral performance trajectories were tracked for all performance measures following predator exposure and acute NOP antagonism in rats. In Experiment 2, we evaluated fear-associated learning and memory via fear memory expression tests in rats. We hypothesized that fear responses (i.e., freezing behavior) would be dampened following systemic NOP antagonism.
Methods
Subjects
Adult (10–14 weeks at baseline), male Sprague-Dawley rats (Charles River Laboratories; n = 7–12/group) were kept on a 12-h/12-h light/dark cycle (lights on from 0600 to 1800 h) in an AAALACi-accredited animal facility and allowed to acclimate to the facility for at least 7 days prior to any testing. Rats were then singly-housed and handled for at least 5 days prior to experimental manipulations. For the duration of the study, rats were maintained on a mild food restriction procedure in which daily allotments of food were provided to maintain bodyweights at approximately 330 g (± 15 g). Water was provided ad libitum. All procedures were conducted during the light phase and were in accordance with the “Guide for the Care and Use of Laboratory Animals” (Institute of Laboratory Animal Resources, National Academy Press, 2011) and were approved by the Institutional Animal Care and Use Committee of Walter Reed Army Institute of Research.
Experimental design
The present study was separated into two experiments. In Experiment 1, the dose-response behavioral effects after acute and repeated NOP antagonism were characterized (1a), in which select doses were then used to characterize behavioral performance following traumatic stress exposure (1b). In Experiment 2, the effects of NOP antagonism on fear memory expression after initial fear conditioning were measured. The experimental timelines are shown in Fig. 1. Experiments 1a, 1b, and 2 were conducted in separate cohorts of animals that were naïve to experimentation before the start of procedures described below.

Experimental timeline. For experiments 1a and 1b (a, b), upward arrows indicate when behavioral tests (ASR, EPM, tail-flick, and open field test) were performed. For experiment 2 (c), upward arrows indicate when behavioral tests (ASR, EPM) were performed, while downward arrows indicate when freezing tests were performed
Experiment 1a: Acute and repeated dose-response relationship of NOP antagonism on general behavioral performance
On day 1, vehicle (VEH), J-113397 (2, 7.5, or 20 mg/kg, i.p.), or SB-612, 111 (0.1, 0.4, 1, or 4 mg/kg, i.p.) was administered 30-min prior to collecting a blood sample from the lateral tail vein (approx. 0.1 mL) followed by behavioral assessments in acoustic startle response (ASR), elevated plus maze (EPM), open field, and warm-water tail immersion (i.e., “tail-flick”) tests. Daily injections were then administered from days 2 through 7, while behavioral testing only occurred on days 2 and 7 (see Fig. 1 for timeline).
Experiment 1b: Effects of NOP antagonism on general behavioral performance after a three-species predator exposure procedure
As shown in Fig. 1, on day 1, a blood sample from the lateral tail vein (approx. 0.1 mL) was collected prior to behavioral assessments in ASR, EPM, open field, and tail-flick tests. On day 2, all rats underwent a three-species predator exposure procedure as described below. VEH, J-113397 (7.5 or 20 mg/kg, i.p.), or SB-612, 111 (0.4 or 1 mg/kg, i.p.) was then administered 24-h post-predator exposure, with a repeat of day 1 behavioral tests beginning approximately 30-min later. Behavioral tests were again repeated on days 4 and 10 (i.e., 2-, and 8-day post-predator exposure) but without drug treatment.
Experiment 2: Effects of NOP antagonism on fear memory expression
As shown in Fig. 1, on day 1, a blood sample from the lateral tail vein (approx. 0.2 mL) was collected prior to behavioral performance assessment (EPM and ASR). On day 3, all rats were exposed to either a fear conditioning session (i.e., inescapable foot shock, IES; as described in the following section) or sham. On day 5, a repeat of day 1 behavioral tests was performed to track possible changes in performance measures following IES (or sham) exposure. On days 8–11, rats were administered either VEH or SB-612,111 (1 or 3 mg/kg, i.p.) and tested 1 h later in locomotor freezing chambers. For this experiment, SB-612,111 was chosen because of findings indicating a lack of nonspecific effects in Experiment 1, and a reported greater potency at the NOP receptor compared to J-113397 (Spagnolo et al. 2007). Moreover, J-113397 has been reported to bind to other receptors, such as sigma receptors, despite having a high selectivity for the NOP receptor (Chiou et al. 2007). Finally, on day 12, a tail vein blood sample was collected followed by behavioral tests.
Traumatic stress procedures
Predator exposure procedure
A three predator exposure procedure (PRED) was performed by exposing rats to three live predators in sequence within a single day. This procedure has been previously characterized in our lab (Moore et al. 2016), and was modified slightly for the present study. Predator species used (in order of exposure) were a snake (ball python), black-footed ferrets, and two pairs of domestic cats. PRED exposures took place in two neighboring rooms that housed all three species, with the snake being housed in one room and the ferrets and cats in the other. Rats were placed in a protective non-restraining enclosure specifically designed to allow perception of visual, olfactory, and auditory cues associated with the predators without the possibility of direct physical contact. Enclosures were placed within the feeding chamber of the snake and the housing area of the ferrets or cats. To maximize odor presence for the snake exposure, pieces of dried shed snake skin were placed inside the enclosure with the rats. To maximize odor cue in cat exposures, the enclosure holding the rat was placed on a bed of soiled (wet) cat litter with cat toys hanging above the enclosure to increase engagement of the cats. Exposure lasted 10 min for the snake, followed by 5 min for ferrets, and finally 10 min for cats. After the cat exposure, rats were placed into fresh, clean home cages.
Fear conditioning using inescapable shock
Two computer-controlled fear conditioning chambers (Med Associates Inc., St. Albans, VT) were used to establish fear conditioning in rats. Each chamber (12 in. × 9.5 in. × 8.25 in.) contained lights, a sound-generating device, and stainless steel rod flooring in which brief programmed footshocks were delivered. Chambers were placed in sound- and light-attenuated, ventilated cabinets (22 in. × 23.5 in. × 16 in.). For animals in the IES groups, a 35-min session was programmed to deliver 20 randomly programmed presentations (excluding the first and last 5 min) of a light and tone cue (i.e., conditioned stimulus, CS) that co-terminated with a 2-s, 1.0 mA footshock (i.e., unconditioned stimulus, US). For animals in the SHAM groups, an identical conditioning session was programmed, however, there was no US. Fear expression was measured by freezing tests which later occurred (see Fig. 1) in identical chambers in a separate room than where conditioning sessions took place, in a different-sized sound- and light-attenuated, ventilated cabinet (23.5 in. × 12.5 in. × 28 in.) fixed with a near-infrared camera on the interior door to record freezing behavior (Med Associates, Inc., St. Albans, VT). Freezing sessions were 10 min in length, in which a single CS was delivered 6 min into the session. Freezing behavior was measured via a near-infrared camera and microcomputer software and was defined as the absence of locomotor activity with the exception of breathing. Following each test, the chamber was cleaned immediately (PURE Hard Surface Spray; PURE Bioscience, Inc., El Cajon, CA) following each test to mitigate the possibility of introducing pheromonal cues across subjects.
Behavioral outcome measures
Acoustic startle response
Eight computer-controlled acoustic startle response (ASR) chambers (Kinder Scientific, Poway, CA) were used to measure acoustic startle response and related measures (e.g., prepulse inhibition of the startle reflex). Each rat was placed in an enclosure within a sound- and light-attenuated, ventilated chamber, where the enclosure was rested on a platform that measured changes in force as a result of a startle response. For Experiment 1, the session began with a 5-min acclimation period to a background level of noise (70 dB), followed by 30 pseudorandomized trials of pulse-only trials (115 dB; 40 ms). For Experiment 2, the session began with a 5-min acclimation period to a background level of noise (70 dB), followed by five repeats of pulse-only trials (115 dB; 20 ms). The session then presented eight pseudorandomized repeats of five various trial types: no pulse, pulse-only, and prepulse-containing trials (73, 76, and 82 dB as prepulses; 20-ms duration, with an inter-stimulus interval of 100-ms). The session then ended with five repeats of pulse-only trials. The inter-trial interval for all experiments was on average 20 s (range, 10–30 s) and the recording window was 250 ms which began at the beginning of each trial. Startle response was recorded for each trial in Newtons (N).
For data analysis, the max startle magnitudes for “pulse-only” trial types were averaged across replicates and were used to infer the startle response, with the first five pulse-only trials being used as a measure of startle reactivity. For Experiment 2, prepulse inhibition of the startle reflex (PPI) was calculated as a percentage using averaged max startle magnitudes of the eight repeats of pulse-only and 73 dB prepulse-containing trial types, with the following formula: 100 × [(pulse-only trials − 73 dB prepulse-containing trials) / (pulse-only trials)]. To avoid ceiling effects that may inherently occur due to the direct relationship between PPI and prepulse intensity level, PPI was only analyzed for the 73 dB prepulse intensity level. For both experiments, within-session habituation was assessed by comparing the last five trials of the session to the first five trials, using the formula: 100 × [(AVG last five trials – AVG first five trials)/(AVG first five trials)].
Elevated plus maze
The elevated plus maze (EPM) consisted of four tinted plastic arms (50 cm × 10 cm) segmented into two separate parts in a “plus” configuration (Kinder Scientific, Poway, CA). Two arms were closed with walls 40 cm in height and the other two arms were open with no walls. Testing consisted of 5-min sessions with dim lighting (lux: 7 for open arms, 1 for closed arms, 2 for intersection) and a white noise generator (70 dB). Each test started with the rat being placed at the intersection of the maze pointed towards an open arm, and the apparatus was cleaned immediately (PURE Hard Surface Spray; PURE Bioscience, Inc., El Cajon, CA) following each test to mitigate the possibility of introducing pheromonal cues across subjects. Animal movement was measured using Kinder Scientific automated software and photobeam tracking.
For data analysis, basic movements (i.e., the count of consecutive photobeam breaks) were used as a measure of exploratory behavior. We have previously reported that basic movements are well-correlated to total distance traveled, and are a useful measure to track within-subject behavioral performance on the EPM (Schrader et al. 2018). In addition, a measure widely considered to represent anxiety-like behavior (i.e., open arm distance) was analyzed.
Open field test
The open field apparatus (100 cm × 100 cm × 35 cm) placed on a table (height, 29 in.) was used to capture locomotor activity via a ceiling-mounted video camera equipped with a range night light illuminator and microcomputer connected to commercial software (ANY-MAZE, Stoelting Co., Wood Dale, IL). Sessions were run under no visible light but with infrared light source in the presence of a white-noise generator (70 dB). The apparatus was segmented into three concentric rectangle sections: center, middle, and edge zones which measures 816 cm2, 2449 cm2, and 6735 cm2 respectively. A 10-min session began when the rat was placed in the center of the enclosure. The apparatus was cleaned immediately following each test (PURE Hard Surface Spray; PURE Bioscience, Inc., El Cajon, CA). Total distance traveled in the open field was measured and analyzed.
Warm-water tail immersion test (tail-flick)
Tail-flick tests were conducted by heating water in a large beaker to approximately 50–55 °C. Rats were gently wrapped in a cloth and held to expose the tail only. The distal third end of the tail was then lowered into the water and the latency to withdraw the tail was measured. A cut-off time of 20 s was used to ensure no physical damage was inflicted to the tail.
Freezing behavior
The first 2 min of the 10-min freezing test session was used to infer baseline performance. Freezing behavior was then analyzed for the 4-min prior to and after the single CS delivery (i.e., Pre-CS and Post-CS), excluding a 3-s window immediately before and after the CS presentation due to software difficulties inferring accurate freezing behavior during the CS in which the cue-light is being flashed on/off. Pre-CS freezing was used in addition to findings from Experiment 1a to assess nonspecific effects of SB-612,111.
Drugs
J-113397 ((±)-1-[(3R*,4R*)-1-(Cyclooctylmethyl)-3-(hydroxymethyl)-4-piperidinyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one; purity > 98%) and SB-612, 111 (7-[[4-(2,6-Dichlorophenyl)-1-piperidinyl]methyl]-6,7,8,9-tetrahydro-1-methyl-5H-benzocyclohepten-5-ol hydrochloride; purity > 97%) were supplied from R&D Systems (Minneapolis, MD). Working solutions of vehicle and drug were prepared fresh the day of injection. All drugs were prepared in a solution of 5% ethanol/5% emulsifier (Alkamuls EL-620, Solvay Chemicals, Princeton, NJ)/90% sterile, non-heparinized 0.9% saline. All injections were administered intraperitoneally (needle size 28G, 0.5 in.) at an injection volume of 2 mL/kg for Experiment 1 and 1 mL/kg for Experiment 2.
Data analysis
All data are represented as the mean ± SEM. GraphPad Prism 7.0 microcomputer software was used to graph and analyze all data. Data were analyzed using two-way ANOVA tests, with a between-subject factor of group (see Table 1 for test groups for each experiment) and a within-subject factor of time. For Experiment 2, a within-subject factor of test or Post-CS minute was used for fear learning results. Dunnett’s post hoc tests were used for comparisons to baseline or the VEH-treated group in Experiment 1, while Tukey post hoc tests were used for between-group comparisons in Experiment 2. An alpha level of 0.05 was used to infer statistical significance. For Experiment 1, data for the VEH group are presented and analyzed with each NOP antagonist (i.e., the same VEH data is shown twice in graphs) to minimize the number of subjects needed to complete experiments and to better present the effects of each drug on the behavior outcome measure relative to VEH treatment.
Results
General behavioral performance in rats is not altered by acute or repeated systemic NOP antagonism
For all behavioral outcome measures evaluated (ASR, EPM, tail-flick, open field test), there was a lack of a main effect of either NOP antagonist treatment as shown in Fig. 2. In general, startle reactivity remained stable following administration of J-113397 or SB-612,111 (as shown in Fig. 2a and b), although a significant main effect of time was observed (Time, F(2, 176) = 3.453, p = 0.0338) but not of group or an interaction of time and group (Group, F(7,88) = 0.8459, p = 0.5525; Time × Group, F(14, 176) = 0.7278, p = 0.7445). For open arm distance on the EPM (Fig. 2c, d), a significant main effect of time was observed (Time, F(2, 176) = 17.06, p < 0.0001) but not of group or an interaction of time and group (Group, F(7,88) = 0.8193, p = 0.5738; Time × Group, F(14, 176) = 1.545, p = 0.0995). Basic movements on the EPM, an index of general exploratory behavior, showed a significant main effect of time (F(2, 176) = 40.49, p < 0.0001) and group (F(7,88) = 2.346, p = 0.0303) but not a significant interaction of time and group (F(14, 176) = 1.212, p = 0.2704), although Dunnett’s post hoc analyses did not reveal any significant between-group differences at any timepoint (Supplemental Table 1). The latency to tail-flick in the warm-water tail immersion test decreased over time for all groups as shown in Fig. 2e and f, where a significant main effect of time was observed (Time, F(2, 160) = 9.351, p = 0.0001; Group, F(7, 80) = 0.2905, p = 0.9559; Time × Group: F(14, 160) = 1.001, p = 0.4549). For total distance traveled in the open field test (shown in Fig. 2g and h), a significant main effect of time was observed while a main effect of group was near significance (Time, F(2, 176) = 56.04, p < 0.0001; Group, F(7, 88) = 2.053, p = 0.0571). A significant interaction of time and group was also observed (Time × Group, F(14, 176) = 1.887, p = 0.0304). Dunnett’s post hoc analyses revealed select significant between-group comparisons at the day 7 timepoint, where distance traveled for the VEH-treated group was significantly greater than the 7.5 mg/kg J-113397 (p = 0.0127) and the 1 mg/kg SB-612,111 (p = 0.0349) treatment groups; however, total distance trended in a positive direction on day 7 relative to day 2 for all groups and therefore limited interpretation of these select differences.

Behavioral dose-response relationship of two NOP antagonists, J-113397 and SB-612,111 (n = 10–12/group). Overall, there were no significant off-target, adverse effects of either NOP antagonist on behavior (Startle reactivity in ASR (a, b); open arm distance in EPM (c, d); latency to tail-flick (e, f); total distance traveled in the open field test, (g, h). *p < 0.05 vs VEH for the 7.5 mg/kg J-113397-treated group and the 1.0 mg/kg SB-612,111-treated group
A significant main effect of time for most behavioral performance measures suggest these measures may be sensitive to behavioral habituation or sensitization (depending on measure; see Supplemental Table 2). Importantly, with the exception of total distance traveled in the open field test, there was a lack of between-group differences for all behavioral outcome measures, suggesting a lack of significant behavioral effects in the assays used following acute or repeated administration of the NOP antagonists J-113397 and SB-612,111.
NOP antagonism does not alter most general behavioral performance measures following predator exposure in rats
The trajectory of behavioral performance on various tests (ASR, EPM, tail-flick, and open field test) following traumatic stress via a three-species predator exposure procedure and either VEH or NOP antagonist treatment is shown in Fig. 3. In general, startle reactivity was largely unaltered following PRED exposure (Fig. 3a, b), where a two-way ANOVA found no significant main effect of time or group, nor an interaction of time and group (Time, F(3, 162) = 1.383, p = 0.25; Group, F(4, 54) = 0.392, p = 0.8135; Time × Group, F(12, 162) = 1.199, p = 0.2881). In general, open arm distance traveled in the EPM decreased for all groups following PRED exposure as shown in Fig. 3c and d, where a significant main effect of time was observed (Time, F(3, 162) = 12.7, p < 0.0001). Dunnett’s post hoc analyses revealed a significant decrease from baseline at the 1-day timepoint for the 20 mg/kg J-113397-treated group (p = 0.0252) and the 0.4 mg/kg SB-612,111-treated group (p = 0.0192). Significant decreases from baseline at the 2-day timepoint was also observed for SB-612,111-treated groups (0.4 mg/kg, p = 0.0019; 1 mg/kg, p = 0.0047). Although open arm distance decreased at the 1- and 2-day timepoints for the VEH-treated group, there were no significant within-group changes as compared to baseline. Further, there was no significant main effect of group or a significant interaction of time and group (Fig. 3c, d; Group, F(4, 54) = 2.106, p = 0.0927; Time × Group, F(12, 162) = 0.62, p = 0.8231). For the warm-water tail immersion test (shown in Fig. 3e and f), the latency to tail-flick was unaltered following PRED exposure, where there were no significant main effects of time or group, or a significant interaction of time and group (Time, F(3, 162) = 0.1277, p = 0.9436; Group, F(4, 54) = 1.102, p = 0.3651; Time × Group, F(12, 162) = 1.513, p = 0.124). Total distance traveled in the open field is shown in Fig. 3g and h, where a significant main effect of time was observed (Time, F(3, 162) = 15.26, p < 0.0001; Group, F(4, 54) = 0.6213, p = 0.6493; Time × Group, F(12, 162) = 0.5441, p = 0.8831). Dunnett’s post hoc analyses revealed a significant difference from baseline at 1-day post-PRED for the VEH-treated group (p = 0.0266; Fig. 3g and h, symbolized by asterisk) that did not extend past 2-day post-PRED (2 days, p = 0.0633). This significant difference from baseline was not observed for groups treated with either NOP antagonist at the 1-day timepoint, although the group treated with 0.4 mg/kg SB-612,111 showed near significance (1 day, p = 0.0533) and later showed a significant difference from baseline at the 2 day timepoint (p = 0.0028; Fig. 3h, symbolized as “$$”), suggesting behavioral restoration via NOP antagonism is mostly observed at acute timepoints and with a certain level of circulating drug.

Behavioral performance following PRED exposure and NOP antagonism (n = 11–12/group). In general, NOP antagonism did not alter behavior relative to the VEH-treated group at the tested doses and timepoints following PRED exposure (Startle reactivity in ASR (a, b); open arm distance in EPM (c, d); latency to tail-flick (e, f); total distance traveled in the open field test (g, h). The vertical hatched line designates when subjects underwent PRED exposure and the upward arrow designates when the NOP antagonist or VEH was administered. *p < 0.05 vs baseline for VEH-treated group; $$ p < 0.01 vs baseline for 0.4 mg/kg SB-612,111-treated group. See Results for significant within-group changes in open arm distance
Interestingly, basic movements in the EPM appeared to decrease at acute timepoints following PRED exposure for all groups (Fig. 4), where a significant main effect of time was observed (Time, F(3, 162) = 28.03, p < 0.0001; Group, F(4, 54) = 1.362, p = 0.2594; Time × Group, F(12, 162) = 0.7936, p = 0.6565). Dunnett’s post hoc analyses revealed significant differences from baseline for most groups at the 1-day timepoint, with the exception for the group treated with 7.5 mg/kg J-113397 (see Table 2 for p values from Dunnett’s multiple comparisons test), suggesting J-113397 may have moderate efficacy on ameliorating PRED exposure-induced effects on exploratory behavior. However, by the 8-day timepoint, there were no significant differences from baseline for any group. For the ASR-related measure of habituation (Fig. 4c, d), a two-way ANOVA found a significant main effect of time (F(3, 162) = 6.953, p = 0.0002), but not of group (F(4, 54) = 1.206, p = 0.3189) or a significant interaction of time and group (F(12, 162) = 1.678, p = 0.0760). Dunnett’s post hoc analyses revealed significant increases in habituation from baseline for only the group treated with 20 mg/kg J-113397 (1 day, p = 0.0038; 2 days, p = 0.0021; 8 days, p = 0.0115). Further, Dunnett’s post hoc analyses revealed a significant between-group difference at the 8-day timepoint between the VEH- and 7.5 mg/kg J-113397-treated groups (p = 0.0384), suggesting possible restoration of habituation by NOP antagonism at later timepoints.

Trajectory of basic movements on the EPM (a, b) and habituation of ASR startle reactivity (c, d) following NOP antagonism and PRED exposure (n = 11–12/group). The vertical hatched line designates when subjects underwent PRED exposure and the upward arrow designates when the NOP antagonist or VEH was administered. See Table 2 for significant within-group changes in basic movements for (a) J-113397 and (b) SB-612,111. *p < 0.05 vs VEH for the 7.5 mg/kg J-113397-treated group
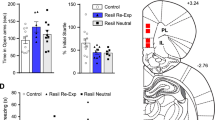
NOP antagonism reduces fear memory expression in rats
The effects of NOP antagonism (via treatment with SB-612,111; SB) on fear expression were characterized. Fear memory expression was inferred by freezing behavior to a footshock-paired conditioned stimuli (i.e., CS; a light + tone cue) across four daily test sessions beginning 5 days after the fear conditioning session, as shown in Figs. 1 and 5. Baseline freezing behavior prior to CS presentation (during the first 2 min of the freezing test session) was minimal, as shown in Fig. 5a, and did not show a significant main effect of group (F(4, 39) = 0.7808, p = 0.5445), although a significant main effect of test was observed (F(3, 117) = 4.177, p = 0.0075). Despite this, there was no significant interaction of test and group (F(12, 117) = 0.8468, p = 0.6025) and no significant between-group comparisons were found at any timepoint. Further inference of the nonspecific effects of SB-612,111 was evaluated by analyzing Pre-CS freezing behavior, as shown in Fig. 5b. In general, Pre-CS freezing was minimal for all groups and was maintained at minimal levels across tests; however, a significant main effect of group was observed (Group, F(4, 39) = 4.653, p = 0.0036; Test, F(3, 117) = 1.024, p = 0.3846; Group × Test, F(12, 117) = 0.8067, p = 0.6430). Tukey’s post hoc analyses revealed the IES-VEH group had significantly greater Pre-CS freezing behavior than the Sham-VEH group on the first and second freezing test (Test 1, p = 0.0154; Test 2, p = 0.0402), suggesting there may have been mild contextual freezing behavior as the chambers were similar to those in which fear conditioning took place. Tukey’s post hoc analyses further revealed the IES-VEH group had significantly greater Pre-CS freezing than the IES-exposed, SB-treated groups at the second test (IES-VEH vs IES-1 mg/kg SB, p = 0.0251; IES-3 mg/kg SB, p = 0.0087) and greater Pre-CS freezing than the IES-3 mg/kg SB group at the fourth test (p = 0.0244), suggesting SB-612,111 may help to dampen contextual freezing behavior as well, although this needs to be formally evaluated. Post-CS freezing behavior, an averaged measure of the freezing behavior during the 4 min following CS presentation and shown in Fig. 5c, was used to infer whether fear-associated learning took place in IES-exposed groups. A significant main effect of freezing test (F(3, 117) = 7.587, p = 0.0001) and group (F(4, 39) = 16.37, p < 0.0001), as well as a significant test and group interaction (F(12, 117) = 2.236, p = 0.0141) was observed for Post-CS percent of time spent freezing. Tukey’s post hoc analyses revealed a significant difference in Post-CS freezing between the IES-VEH and Sham-VEH groups occurred over the first three freezing tests before resolving by the last freezing test (Test 4, p = 0.0541). For IES-exposed groups, freezing behavior was highest on the first freezing test and subsequently decreased across the remaining three freezing tests, indicating that extinction learning had occurred. Tukey post hoc comparisons revealed a significant reduction in Post-CS freezing in the IES-exposed, SB-treated groups as compared to IES-exposed, VEH-treated groups as early as the first freezing test (VEH vs 1 mg/kg SB, p = 0.0033; 3 mg/kg SB, p = 0.0002). This significant difference from the IES-exposed, VEH-treated group was observed across the remaining freezing tests for the IES exposed, 3 mg/kg SB-treated group (Test 2, p = 0.0005; Test 3, p = 0.0147; Test 4, p = 0.0226). For the IES-exposed group treated with the low dose of SB (1 mg/kg), this significant difference from the IES-exposed, VEH-treated group was observed up until the second freezing test (p = 0.0006). Importantly, Sham- and IES-exposed 3 mg/kg SB-treated groups were not significantly different from one another after the initial freezing test (Test 1, p = 0.0367), suggesting a lack of off-target effects of SB-612,111 on freezing behavior. To better understand the immediate effects of NOP antagonism on Post-CS freezing behavior, we analyzed the individual 4 min following the CS presentation at the first test (Fig. 5d). A two-way ANOVA revealed a significant main effect of group but not of Post-CS minute, nor a significant interaction of group and Post-CS minute (Group, F(4, 39) = 8.523, p < 0.0001; Post-CS minute, F(3, 117) = 1.788, p = 0.1533; Group × Post-CS minute, F(12, 117) = 0.5592, p = 0.8705). Tukey’s post hoc analyses revealed that freezing was significantly greater in the IES-VEH group compared to the Sham-VEH group (p < 0.001 for all four Post-CS minutes). Tukey’s post hoc analyses further revealed significant differences between IES-VEH and IES-3 mg/kg SB groups after the first minute (p < 0.05 for Post-CS minutes 2–4). For all IES-exposed groups, freezing behavior was maintained at a steady level for the full 4 min following the CS presentation at Test 1, suggesting that SB-612,111 did not expedite the rate of return to normal activity. Due to this steady behavior, linear regression analyses on the rate of return to normal behavior were not performed and inferences regarding facilitated extinction are limited.

NOP antagonism significantly dampened fear responses (n = 7–10/group). Freezing behavior was minimal during the first 2 min of fear memory expression tests (a) and prior to the CS presentation (b), indicating a lack of nonspecific effects on activity. Post-CS freezing was significantly reduced in IES, SB-treated groups relative to the IES, VEH-treated group across all four freezing tests (c), *p < 0.05, ***p < 0.001 vs IES-VEH for the IES-3 mg/kg SB group. Freezing behavior on Test 1 did not significantly decrease across the four Post-CS minutes for any IES-exposed group, but was significantly different between the IES-VEH and IES-3 mg/kg SB groups at the later timepoints (d), ***p < 0.001, ****p < 0.0001 vs Sham-VEH; $p < 0.05 vs IES-VEH for the IES-3 mg/kg SB group. See Results for other significant group comparisons
General behavioral performance on ASR and the EPM was also tracked in this experiment without the utilization of the PRED exposure procedure to infer performance effects following fear conditioning or the combination of fear conditioning and previous NOP antagonism and is shown in Supplemental Table 3. In general, startle reactivity did not change over time (Time, F(2, 78) = 0.2097, p = 0.8113), although a significant main effect of group was observed (Group, F(4, 39) = 2.944, p = 0.0322; Group × Time, F(8, 78) = 0.9277, p = 0.4986), where Tukey’s post hoc analyses indicated a significant difference between groups treated with 3 mg/kg SB (i.e., Sham- vs IES-exposed) on day 5 (p = 0.0073) and day 12 (p = 0.0380). Despite these differences, the Sham-3 mg/kg SB group had significantly greater startle reactivity at baseline compared to the IES-3 mg/kg SB group (Tukey’s post hoc, p = 0.0354) as found in a separate one-way ANOVA (F(4,39) = 2.71, p = 0.0439), suggesting the difference observed at the later timepoints were not indicative of an effect of the IES procedure or drug administration history. For the ASR-related measure of prepulse inhibition of the startle reflex, or %PPI, at the 73 dB prepulse intensity, a significant main effect of time, but not group or a significant interaction of time and group (Time, F(2, 78) = 4.524, p = 0.0138; Group, F(4, 39) = 1.268, p = 0.2992; Time × Group, F(8, 78) = 0.3635, p = 0.9366). Tukey’s post hoc analyses, however, did not reveal any significant between-group comparison at any timepoint. For open arm distance traveled on the EPM, a significant interaction of time and group was observed (Time × Group, F(8, 78) = 2.748, p = 0.0100; Time, F(2, 78) = 2.089, p = 0.1307; Group, F(4, 39) = 1.559, p = 0.2045) which appeared to be driven by near-significant differences between groups treated with 3 mg/kg SB (i.e., Sham- vs IES-exposed) at day 12 (Tukey’s post hoc, p = 0.0522), suggesting a possible residual effect of the NOP antagonist on open arm distance. For the measure of basic movements on the EPM, a significant main effect of time was observed (Time, F(2, 78) = 13.33, p < 0.0001; Group, F(4, 39) = 1.219, p = 0.3186; Time × Group, F(8, 78) = 0.6043, p = 0.7716); however, no significant between-group comparisons were observed.
Discussion
In the present study, we demonstrated that systemic NOP receptor antagonism via administration of SB-612,111 effectively reduced fear expression in rats after prior fear conditioning using cue-paired inescapable footshocks. The effects of NOP antagonism on general behavioral performance trajectories following predator exposure or fear conditioning, however, were modest. Moreover, there was a lack of nonspecific effects of NOP antagonism on general behavioral performance alone, suggesting the effects on fear expression were specific to NOP receptor antagonism. Therefore, the present data suggest these receptors play a role in the normal processes underlying fear learning and memory. The results from the present study suggest that NOP antagonists may be a potential pharmacotherapy for trauma- and stress-related disorders such as ASD or PTSD in which fear memory processes are dysregulated, whereas additional research is needed to understand if they would be effective for mitigating other behavioral impairments subsequent to traumatic stress exposure.
Under the present experimental conditions, a lack of effects on general behavioral performance by the tested NOP antagonists was not surprising. While there is growing interest in the effects of NOP antagonists alone as a candidate pharmacotherapy (Awwad et al. 2018; Gavioli and Calo 2013; Rorick-Kehn et al. 2016; Zhang et al. 2015), the majority of published literature surrounds NOP agonist-induced effects and their blockade, rather than reversal, via antagonists. Under acute conditions (i.e., day 1 of Fig. 2) and repeated administration conditions, we observed a lack of significant differences between the VEH-treated group and NOP antagonist-treated groups for most behaviors tested. The exception was for total distance traveled measure in the open field at the day 7 timepoint in which the VEH-treated group traveled significantly more than middle doses of J-113397 and SB-612,111, suggesting a possible U-shaped dose effect of the NOP antagonists on motor activity. However, for the group treated with the middle dose of J-113397 (7.5 mg/kg), distance traveled was also the lowest of all groups at baseline suggesting the difference from the VEH group at day 7 was not strong evidence of a drug-mediated effect. Moreover, higher doses did not greatly dampen distance traveled at any timepoint, indicating NOP antagonists do not cause significant alterations in motor activity. Because of the similarities of the NOP receptor to the classical opioid receptors, we were interested in characterizing any modulation of pain sensitivity imposed by NOP antagonism. In the warm-water tail immersion test, the latency to withdraw the tail did not differ after NOP antagonism alone for either J-113397 or SB-612,111, suggesting a lack of effect of NOP antagonism on altering pain sensitivity to a thermal stimulus (i.e., warm water).
Others have reported null or minimal effects of pharmacological blockade of the NOP receptor on locomotor activity (Calo et al. 2002; Gavioli et al. 2003; Genovese and Dobre 2017; Kuzmin et al. 2004; Redrobe et al. 2002). Better performance on the rotarod assay after administration of the NOP antagonist UFP-101 or J-113397 was suggested to indicate hyperlocomotive effects of NOP antagonism (Marti et al. 2004); however, rotarod performance over time incorporates learning and therefore may not reflect inherent motor activity-specific effects of a drug. However, this was challenged by the authors as they observed better rotarod performance in unhabituated NOP knockout mice at the first test compared to wildtype mice (Marti et al. 2004). Null or minimal effects of NOP antagonism on acoustic startle response (Ces et al. 2012) have been reported, while effects on alteration of pain sensitivity have been mixed (Calo et al. 2002; Chiou et al. 2007; Shinkai et al. 2000; Zhang et al. 2015). Open arm measures on the elevated plus maze have been shown to be unaltered by NOP antagonism as well (Genovese and Dobre 2017; Uchiyama et al. 2008; Vitale et al. 2006). Interestingly, increased anxiety-like behavior was observed after genetic deletion of the N/OFQ peptide precursor but not the encoding gene of the parent receptor (Reinscheid and Civelli 2002). This was opposite of previous findings of anxiogenic behavior in NOP receptor knockout mice (Gavioli et al. 2007). In sum, the results from Experiment 1a of the present study are in agreement with previous evidence suggesting a lack of nonspecific or off-target effects of NOP antagonists which support them for further testing as a candidate pharmacotherapy for disorders in which behavioral performance is impacted.
NOP antagonism following exposure to a traumatic stress procedure (three-species predator exposure) generally did not alter general behavioral performance trajectories under the conditions tested in the current study. Our lab has previously reported significant behavioral deficits after a three-species predator exposure in male adolescent and adult rats (Moore et al. 2016) in particular for basic movements, a measure of exploration on the EPM that correlates well with total distance traveled (Schrader et al. 2018). In the present study, we again observed significant decreases in basic movements following predator exposure (see Fig. 4 and Table 2). Interestingly, administration of 7.5 mg/kg J-113397 1 day following predator exposure did prevent the decrease in basic movements on the EPM but this did not extend to the 2-day or 8-day behavioral timepoints. Total distance traveled in the open field test was also significantly reduced 1 day following predator exposure and was prevented by J-113397 and SB-612,111, although this did not extend to the 2-day or 8-day timepoints. Moreover, results from Experiment 1a suggest this effect at the 1-day timepoint was not due to a nonspecific increase in locomotor activity. In fact, daily administration of NOP antagonists for 7 days in Experiment 1a showed select significant decreases in total distance traveled relative to a VEH-treated control group, suggesting differential behavioral effects of acute and repeated administration of NOP antagonists. Together, these data suggest that a certain level of circulating drug is needed to observe behavioral protection; however, this interpretation is limited as the half-life of J-113397 or SB-612,111 has not yet been reported. In sum, results from Experiment 1b provide moderate evidence to suggest that behavioral performance following predator exposure was restored at acute timepoints with the NOP antagonists J-113397 and SB-612,111.
Our lab has previously reported on the effects of the NOP antagonist J-113397 following predator exposure, but with a single species (cat) acting as the main stressor (Genovese and Dobre 2017). In that study, significant stress-induced decreases in basic movements in the EPM was blocked by J-113397 in a dose-dependent manner. However, it is important to distinguish that J-113397 was administered prior to the predator exposure procedure and therefore represented prophylactic efficacy of the NOP antagonist. In the present study, significant decreases in basic movements in the EPM was observed at the 1 and 2-day timepoints for VEH-treated group, whereas these decreases were observed at later timepoints (3-day and 7-day) in the Genovese et al. study. Further, outcome differences from our previous findings in regard to efficacy of the NOP antagonist may be due to differences in the stress procedure (single predator species versus multiple species, exposure duration, etc.) and/or study testing conditions (timepoints assessed, behavioral test parameters). Despite these differences, both of these studies observed efficacy of NOP antagonism in blocking these stress-induced decreases in exploratory behavior.
In the present study, a high degree of variability between groups observed at baseline in the measure of EPM open arm distance limited interpretations. While future studies can be designed to balance groups on baseline performance, it is important to consider challenges involved with animal models of trauma- and stress-related disorders in regard to variability and individual differences in responsivity to stress. Several recent literature reviews highlight this challenge and remind readers that the incidence of PTSD, for example, is only observed in a small percentage of people that have been exposed to one or more traumatic stress event(s) indicating that it may bode well for preclinical traumatic stress research studies to begin subtyping subjects based on behavioral performance (Deslauriers et al. 2018; Lowery-Gionta et al. 2019; Richter-Levin et al. 2019; Zhang et al. 2019). Differences in responding can be observed in the same lab across time, as one group was unable to replicate earlier findings of increased anxiety-like behavior after single-prolonged stress in male rats which was suggested to be due to differences of strain and/or housing conditions (Zhang et al. 2012; Zhang et al. 2018; Zhang et al. 2015). Interestingly, they observed increased levels of the NOP receptor peptide ligand, N/OFQ, and anxiety-like behavior following single-prolonged stress (Zhang et al. 2012) that was blocked after administration of the NOP antagonist JTC-801 (Zhang et al. 2015). The current data should be interpreted with behavioral variability in mind and it may be beneficial to include subject subtyping in future research evaluating pharmacologic targets.
There is a general consensus in the literature that the NOP agonists possess basal anxiolytic-like properties (for review, see Gavioli et al. 2019). The nociceptin receptor system interacts with the stress-related corticotropin-releasing factor (CRF) receptor system, as high expression of both receptors are found in regions implicated in the stress response such as the central amygdala and bed nucleus of the stria terminalis (BNST) where N/OFQ acts as a putative CRF antagonist (Ciccocioppo et al. 2014). Differences in mRNA expression levels of the precursor peptide (ppN/OFQ) in the central amygdala and BNST following acute or repeated restraint stress in rats have been observed, suggesting stress-mediated alterations in underlying mechanisms regulating the NOP receptor system (Delaney et al. 2012). In the same study, researchers found increased activity of the hypothalamic-pituitary-adrenal axis following administration of the NOP antagonist JTC-801 to home-cage control rats. In contrast, reduced anxiety-like behavior associated with alcohol withdrawal has been observed after intraperitoneal administration of the NOP agonist SR-8993 or intracerebroventricular administration of the endogenous peptide (Aujla and Nedjadrasul 2015; Aziz et al. 2016). Together, these findings highlight the complex relationship of the NOP receptor system and responsivity and/or adaptation to stress. The present findings add to this growing body of literature and are in support of stress-induced effects being alleviated by NOP receptor antagonism.
Our major finding is the significant efficacy of the NOP antagonist SB-612,111 in dampening fear responses in rats. It has been shown that NOP agonists impair fear memory acquisition, consolidation, and reconsolidation (Andero et al. 2013; Fornari et al. 2008; Goeldner et al. 2009; Rekik et al. 2017) although their effect on acquisition has been variable (Andero et al. 2013). Interestingly, freezing behavior was maintained over time in knockout mice for the encoding gene (ORL1) which was interpreted as improved fear memory retention rather than impaired fear extinction (Mamiya et al. 2003). In contrast, we repeatedly observed a decrease in freezing behavior following pharmacological blockade of the NOP receptor, with no evidence of motor impairment. This agrees with previous literature that indicates pharmacological blockade or genetic deletion of the NOP receptor helps to enhance or block agonist-mediated impairments in learning (Adem et al. 2017; see review, Andero 2015; Han et al. 2019; Rekik et al. 2017), although biphasic effects of N/OFQ or the agonist Ro 64-6198 have been seen in the passive avoidance task (Adem et al. 2017). An alternative interpretation might be that NOP antagonism impaired fear memory retention, as freezing was dampened as early as the first test. However, if the second and subsequent tests are indicative of retention of the newly-formed extinction memory, we would hypothesize that an impairment in extinction memory retention would be demonstrated by increased freezing behavior at the second and subsequent tests. Therefore, because freezing decreased over the repeated tests and was significantly lower in groups treated with the NOP antagonist, we suggest memory retention was not impaired by NOP antagonism. The current study did not test whether distinct mechanisms of action exist for fear memory retention and extinction memory retention. Future studies are needed to better discern if effects of NOP antagonism are distinguishable between expression of the initial fear memory and the extinction memory or if it is generalizable. Furthermore, facilitation of fear extinction was not observed under the tested conditions as the slope of freezing responses over the four fear memory expression tests were similar between VEH- and NOP antagonist-treated groups. Further studies are therefore needed where extension of the duration following the CS presentation could allow formal testing of differences in the rate of return to normal activity. Alternatively, extinction within a single test session could likely be tracked via multiple presentations of the CS. This would limit the pharmacological history to a single pre-session administration of drug, which would be informative as pharmacokinetic data for SB-612,111 is lacking. Future studies are also warranted to determine if dampened fear responses following NOP antagonism are sustained under drug-free conditions.
The present study provides an initial characterization of NOP antagonism in a rodent model of traumatic stress via predator exposure and fear memory expression. While sex differences are beyond the scope of the current study, a potential limitation in the interpretation of these results is the use of only male subjects. Sex differences in PTSD-related behaviors have been well established in several animal models (Kornfield et al. 2018). However, much remains to be discovered regarding the role of sex differences in NOP antagonism. Zhang et al. (2018) demonstrated sex differences in NOP receptor knockout rats following single prolonged stress (2-h restraint followed by group forced swimming and exposure to diethyl ether). In that study, male NOP receptor knockout rats demonstrated decreased traumatic stress-induced allodynia and thermal hyperalgesia compared to female NOP receptor knockout rats. Notably, all female rats (wild type and NOP receptor knockout) developed allodynia and hyperalgesia at the same rates as wild type males. Further research is therefore needed to determine if the present results from pharmacologic NOP receptor antagonism demonstrate similar effects in female subjects.
The present results demonstrated that NOP antagonism via acute and repeated administration of SB-612,111 significantly dampened fear responses in rats while administration of J-113397 or SB-612,111 restored performance on some, but not all, behavioral measures following traumatic stress exposure and particularly at acute timepoints. Further research is needed to determine the efficacy of NOP antagonists as a potential pharmacotherapy to reduce behaviors associated with ASD or PTSD, as Experiment 1 found moderate evidence that J-113397 restored behavioral performance on the EPM (basic movements) and both J-113397 and SB-612,111 restored performance in the open field test (total distance) acutely following predator exposure. Evidence of reduction in behaviors associated with traumatic stress would give credence to the use of this compound in future clinical pharmaceutical interventions following exposure to traumatic stress. The present results on fear memory expression are pertinent to animal models of traumatic stress, as a significant reduction of freezing behavior following repeated “reminders” to the fear-associated context or cue is a favorable outcome of a potential candidate pharmacotherapy. While inescapable footshock may not be a model of trauma- and stress-related disorders entailing a true and direct threat-to-life, the confluence of results with predator exposure may help determine the broader effects of NOP antagonism on a traumatic stress-induced impairment in fear extinction or other outcome measures. Indeed, rodent models of exposure therapy incorporate this thought (Paredes and Morilak 2019). In conclusion, the present study suggests efficacy of NOP antagonists as a potential pharmacotherapy for use in targeting dysregulated fear memory processes in trauma- and stress-related disorders, such as acute stress disorder or post-traumatic stress disorder.
References
Adem A, Madjid N, Kahl U, Holst S, Sadek B, Sandin J, Terenius L, Ögren SO (2017) Nociceptin and the NOP receptor in aversive learning in mice. Eur Neuropsychopharmacol 27:1298–1307. https://doi.org/10.1016/j.euroneuro.2017.09.005
Andero R (2015) Nociceptin and the nociceptin receptor in learning and memory. Prog Neuro-Psychopharmacol Biol Psychiatry 62:45–50. https://doi.org/10.1016/j.pnpbp.2015.02.007
Andero R et al (2013) Amygdala-dependent fear is regulated by Oprl1 in mice and humans with PTSD. Sci Transl Med 5:188ra173. https://doi.org/10.1126/scitranslmed.3005656
Aujla H, Nedjadrasul D (2015) Low-dose nociceptin/orphanin FQ reduces anxiety-like performance in alcohol-withdrawn, but not alcohol-naive, male Wistar rats. Neuropharmacology 93:1–6. https://doi.org/10.1016/j.neuropharm.2015.01.006
Awwad HO, Durand CD, Gonzalez LP, Tompkins P, Zhang Y, Lerner MR, Brackett DJ, Sherry DM, Awasthi V, Standifer KM (2018) Post-blast treatment with nociceptin/orphanin FQ peptide (NOP) receptor antagonist reduces brain injury-induced hypoxia and signaling proteins in vestibulomotor-related brain regions. Behav Brain Res 340:183–194. https://doi.org/10.1016/j.bbr.2016.10.041
Aziz AM, Brothers S, Sartor G, Holm L, Heilig M, Wahlestedt C, Thorsell A (2016) The nociceptin/orphanin FQ receptor agonist SR-8993 as a candidate therapeutic for alcohol use disorders: validation in rat models. Psychopharmacology 233:3553–3563. https://doi.org/10.1007/s00213-016-4385-8
Blanchard RJ, Yang M, Li CI, Gervacio A, Blanchard DC (2001) Cue and context conditioning of defensive behaviors to cat odor stimuli. Neurosci Biobehav Rev 25:587–595. https://doi.org/10.1016/s0149-7634(01)00043-4
Bunzow JR, Saez C, Mortrud M, Bouvier C, Williams JT, Low M, Grandy DK (1994) Molecular cloning and tissue distribution of a putative member of the rat opioid receptor gene family that is not a mu, delta or kappa opioid receptor type. FEBS Lett 347:284–288. https://doi.org/10.1016/0014-5793(94)00561-3
Calo G, Rizzi A, Rizzi D, Bigoni R, Guerrini R, Marzola G, Marti M, McDonald J, Morari M, Lambert DG, Salvadori S, Regoli D (2002) [Nphe1,Arg14,Lys15]nociceptin-NH2, a novel potent and selective antagonist of the nociceptin/orphanin FQ receptor. Br J Pharmacol 136:303–311. https://doi.org/10.1038/sj.bjp.0704706
Ces A, Reiss D, Walter O, Wichmann J, Prinssen EP, Kieffer BL, Ouagazzal AM (2012) Activation of nociceptin/orphanin FQ peptide receptors disrupts visual but not auditory sensorimotor gating in BALB/cByJ mice: comparison to dopamine receptor agonists. Neuropsychopharmacology 37:378–389. https://doi.org/10.1038/npp.2011.175
Chiou LC, Liao YY, Fan PC, Kuo PH, Wang CH, Riemer C, Prinssen EP (2007) Nociceptin/orphanin FQ peptide receptors: pharmacology and clinical implications. Curr Drug Targets 8:117–135. https://doi.org/10.2174/138945007779315605
Ciccocioppo R, de Guglielmo G, Hansson AC, Ubaldi M, Kallupi M, Cruz MT, Oleata CS, Heilig M, Roberto M (2014) Restraint stress alters nociceptin/orphanin FQ and CRF systems in the rat central amygdala: significance for anxiety-like behaviors. J Neurosci 34:363–372. https://doi.org/10.1523/JNEUROSCI.2400-13.2014
Delaney G, Dawe KL, Hogan R, Hunjan T, Roper J, Hazell G, Lolait SJ, Fulford AJ (2012) Role of nociceptin/orphanin FQ and NOP receptors in the response to acute and repeated restraint stress in rats. J Neuroendocrinol 24:1527–1541. https://doi.org/10.1111/j.1365-2826.2012.02361.x
Deslauriers J, Toth M, Der-Avakian A, Risbrough VB (2018) Current status of animal models of posttraumatic stress disorder: behavioral and biological phenotypes, and future challenges in improving translation. Biol Psychiatry 83:895–907. https://doi.org/10.1016/j.biopsych.2017.11.019
Fornari RV, Soares JC, Ferreira TL, Moreira KM, Oliveira MG (2008) Effects of nociceptin/orphanin FQ in the acquisition of contextual and tone fear conditioning in rats. Behav Neurosci 122:98–106. https://doi.org/10.1037/0735-7044.122.1.98
Gavioli EC, Calo G (2013) Nociceptin/orphanin FQ receptor antagonists as innovative antidepressant drugs. Pharmacol Ther 140:10–25. https://doi.org/10.1016/j.pharmthera.2013.05.008
Gavioli EC, Marzola G, Guerrini R, Bertorelli R, Zucchini S, de Lima TCM, Rae GA, Salvadori S, Regoli D, Calo G (2003) Blockade of nociceptin/orphanin FQ-NOP receptor signalling produces antidepressant-like effects: pharmacological and genetic evidences from the mouse forced swimming test. Eur J Neurosci 17:1987–1990. https://doi.org/10.1046/j.1460-9568.2003.02603.x
Gavioli EC, Rizzi A, Marzola G, Zucchini S, Regoli D, Calo G (2007) Altered anxiety-related behavior in nociceptin/orphanin FQ receptor gene knockout mice. Peptides 28:1229–1239. https://doi.org/10.1016/j.peptides.2007.04.012
Gavioli EC, Holanda VAD, Ruzza C (2019) NOP ligands for the treatment of anxiety and mood disorders. Handb Exp Pharmacol 254:233–257. https://doi.org/10.1007/164_2018_188
Genovese RF, Dobre S (2017) Mitigation of adverse behavioral impact from predator exposure by the nociceptin/orphanin FQ peptide antagonist J-113397 in rats. Behav Pharmacol 28:521–530. https://doi.org/10.1097/FBP.0000000000000329
Goeldner C, Reiss D, Wichmann J, Kieffer BL, Ouagazzal AM (2009) Activation of nociceptin opioid peptide (NOP) receptor impairs contextual fear learning in mice through glutamatergic mechanisms. Neurobiol Learn Mem 91:393–401. https://doi.org/10.1016/j.nlm.2008.12.001
Han RW, Tian AW, Lin HR, Chang M, Wei J, Deng KY, Xin HB (2019) Nociceptin impairs acquisition of novel object recognition memory in perirhinal cortex. Neurobiol Learn Mem 162:9–14. https://doi.org/10.1016/j.nlm.2019.04.015
Higgins GA, Kew JNC, Richards JG, Takeshima H, Jenck F, Adam G, Wichmann J, Kemp JA, Grottick AJ (2002) A combined pharmacological and genetic approach to investigate the role of orphanin FQ in learning and memory. Eur J Neurosci 15:911–922. https://doi.org/10.1046/j.1460-9568.2002.01926.x
Holowka DW, Marx BP (2012) Assessing PTSD-related functional impairment and quality of life. In: The Oxford handbook of traumatic stress disorders. Oxford University Press, New York, pp 315–330
Jenck F, Moreau JL, Martin JR, Kilpatrick GJ, Reinscheid RK, Monsma FJ, Nothacker HP, Civelli O (1997) Orphanin FQ acts as an anxiolytic to attenuate behavioral responses to stress. Proc Natl Acad Sci U S A 94:14854–14858. https://doi.org/10.1073/pnas.94.26.14854
Kornfield SL, Hantsoo L, Epperson CN (2018) What does sex have to do with it? The role of sex as a biological variable in the development of posttraumatic stress disorder. Curr Psychiatry Rep 20(6):39. https://doi.org/10.1007/s11920-018-0907-x
Koster A, Montkowski A, Schulz S, Stube EM, Knaudt K, Jenck F, Moreau JL, Nothacker HP, Civelli O, Reinscheid RK (1999) Targeted disruption of the orphanin FQ/nociceptin gene increases stress susceptibility and impairs stress adaptation in mice. Proc Natl Acad Sci U S A 96:10444–10449. https://doi.org/10.1073/pnas.96.18.10444
Kuzmin A, Sandin J, Terenius L, Ogren SO (2004) Evidence in locomotion test for the functional heterogeneity of ORL-1 receptors. Br J Pharmacol 141:132–140. https://doi.org/10.1038/sj.bjp.0705583
Lowery-Gionta EG, May MD, Taylor RM, Bergman EM, Etuma MT, Jeong IH, Simmons LP, Ventura MC, Capaldi VF, Matson LM, Moore NLT (2019) Modeling trauma to develop treatments for posttraumatic stress. Transl Issues Psychol Sci 5:243–275. https://doi.org/10.1037/tps0000199
Mamiya T, Yamada K, Miyamoto Y, Konig N, Watanabe Y, Noda Y, Nabeshima T (2003) Neuronal mechanism of nociceptin-induced modulation of learning and memory: involvement of N-methyl-D-aspartate receptors. Mol Psychiatry 8:752–765. https://doi.org/10.1038/sj.mp.4001313
Marti M et al (2004) Blockade of nociceptin/orphanin FQ receptor signaling in rat substantia nigra pars reticulata stimulates nigrostriatal dopaminergic transmission and motor behavior. J Neurosci 24:6659–6666. https://doi.org/10.1523/JNEUROSCI.0987-04.2004
Mollereau C, Parmentier M, Mailleux P, Butour JL, Moisand C, Chalon P, Caput D, Vassart G, Meunier JC (1994) ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett 341:33–38. https://doi.org/10.1016/0014-5793(94)80235-1
Moore NL, Altman DE, Gauchan S, Genovese RF (2016) Adulthood stress responses in rats are variably altered as a factor of adolescent stress exposure. Stress 19:295–302. https://doi.org/10.1080/10253890.2016.1191465
Narendran R, Tollefson S, Fasenmyer K, Paris J, Himes ML, Lopresti B, Ciccocioppo R, Mason NS (2019) Decreased nociceptin receptors are related to resilience and recovery in college women who have experienced sexual violence: therapeutic implications for posttraumatic stress disorder. Biol Psychiatry 85:1056–1064. https://doi.org/10.1016/j.biopsych.2019.02.017
Olson VG, Rockett HR, Reh RK, Redila VA, Tran PM, Venkov HA, DeFino MC, Hague C, Peskind ER, Szot P, Raskind MA (2011) The role of norepinephrine in differential response to stress in an animal model of posttraumatic stress disorder. Biol Psychiatry 70:441–448. https://doi.org/10.1016/j.biopsych.2010.11.029
Paredes D, Morilak DA (2019) A rodent model of exposure therapy: the use of fear extinction as a therapeutic intervention for PTSD. Front Behav Neurosci 13:46. https://doi.org/10.3389/fnbeh.2019.00046
Pynoos RS, Ritzmann RF, Steinberg AM, Goenjian A, Prisecaru I (1996) A behavioral animal model of posttraumatic stress disorder featuring repeated exposure to situational reminders. Biol Psychiatry 39:129–134. https://doi.org/10.1016/0006-3223(95)00088-7
Redrobe JP, Calo G, Regoli D, Quirion R (2002) Nociceptin receptor antagonists display antidepressant-like properties in the mouse forced swimming test. Naunyn Schmiedeberg's Arch Pharmacol 365:164–167. https://doi.org/10.1007/s00210-001-0511-0
Reinscheid RK, Civelli O (2002) The orphanin FQ/nociceptin knockout mouse: a behavioral model for stress responses. Neuropeptides 36:72–76. https://doi.org/10.1054/npep.2002.0901
Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma FJ, Civelli O (1995) Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science 270:792–794. https://doi.org/10.1126/science.270.5237.792
Rekik K, Faria da Silva R, Colom M, Pacifico S, Zaveri NT, Calo' G, Rampon C, Frances B, Mouledous L (2017) Activation of nociceptin/orphanin FQ receptors inhibits contextual fear memory reconsolidation. Neuropharmacology 125:39–49. https://doi.org/10.1016/j.neuropharm.2017.07.006
Richter-Levin G, Stork O, Schmidt MV (2019) Animal models of PTSD: a challenge to be met. Mol Psychiatry 24:1135–1156. https://doi.org/10.1038/s41380-018-0272-5
Roozendaal B, Lengvilas R, McGaugh JL, Civelli O, Reinscheid RK (2007) Orphanin FQ/nociceptin interacts with the basolateral amygdala noradrenergic system in memory consolidation. Learn Mem 14:29–35. https://doi.org/10.1101/lm.403607
Rorick-Kehn LM, Ciccocioppo R, Wong CJ, Witkin JM, Martinez-Grau MA, Stopponi S, Adams BL, Katner JS, Perry KW, Toledo MA, Diaz N, Lafuente C, Jiménez A, Benito A, Pedregal C, Weiss F, Statnick MA (2016) A novel, orally bioavailable nociceptin receptor antagonist, LY2940094, reduces ethanol self-administration and ethanol seeking in animal models. Alcohol Clin Exp Res 40:945–954. https://doi.org/10.1111/acer.13052
Schrader AJ, Taylor RM, Lowery-Gionta EG, Moore NLT (2018) Repeated elevated plus maze trials as a measure for tracking within-subjects behavioral performance in rats (Rattus norvegicus). PLoS One 13:e0207804. https://doi.org/10.1371/journal.pone.0207804
Shinkai H, Ito T, Iida T, Kitao Y, Yamada H, Uchida I (2000) 4-Aminoquinolines: novel nociceptin antagonists with analgesic activity. J Med Chem 43:4667–4677. https://doi.org/10.1021/jm0002073
Spagnolo B, Carrà G, Fantin M, Fischetti C, Hebbes C, McDonald J, Barnes TA, Rizzi A, Trapella C, Fanton G, Morari M, Lambert DG, Regoli D, Calò G (2007) Pharmacological characterization of the nociceptin/orphanin FQ receptor antagonist SB-612111 [(−)-cis-1-methyl-7-[[4-(2,6-dichlorophenyl)piperidin-1-yl]methyl]-6,7,8,9-tetrah ydro-5H-benzocyclohepten-5-ol]: in vitro studies. J Pharmacol Exp Ther 321:961–967. https://doi.org/10.1124/jpet.106.116764
Takahashi LK, Nakashima BR, Hong H, Watanabe K (2005) The smell of danger: a behavioral and neural analysis of predator odor-induced fear. Neurosci Biobehav Rev 29:1157–1167. https://doi.org/10.1016/j.neubiorev.2005.04.008
Takahashi LK, Chan MM, Pilar ML (2008) Predator odor fear conditioning: current perspectives and new directions. Neurosci Biobehav Rev 32:1218–1227. https://doi.org/10.1016/j.neubiorev.2008.06.001
Uchiyama H, Toda A, Hiranita T, Watanabe S, Eyanagi R (2008) Role of amygdaloid nuclei in the anxiolytic-like effect of nociceptin/orphanin FQ in rats. Neurosci Lett 431:66–70. https://doi.org/10.1016/j.neulet.2007.11.023
Vitale G, Arletti R, Ruggieri V, Cifani C, Massi M (2006) Anxiolytic-like effects of nociceptin/orphanin FQ in the elevated plus maze and in the conditioned defensive burying test in rats. Peptides 27:2193–2200. https://doi.org/10.1016/j.peptides.2006.04.003
Zhang Y, Gandhi PR, Standifer KM (2012) Increased nociceptive sensitivity and nociceptin/orphanin FQ levels in a rat model of PTSD. Mol Pain 8:76. https://doi.org/10.1186/1744-8069-8-76
Zhang Y, Simpson-Durand CD, Standifer KM (2015) Nociceptin/orphanin FQ peptide receptor antagonist JTC-801 reverses pain and anxiety symptoms in a rat model of post-traumatic stress disorder. Br J Pharmacol 172:571–582. https://doi.org/10.1111/bph.12701
Zhang Y, Schalo I, Durand C, Standifer KM (2018) Sex differences in nociceptin/orphanin FQ peptide receptor-mediated pain and anxiety symptoms in a preclinical model of post-traumatic stress disorder. Front Psychiatry 9:731. https://doi.org/10.3389/fpsyt.2018.00731
Zhang L, Hu XZ, Li H, Li X, Yu T, Dohl J, Ursano RJ (2019) Updates in PTSD animal models characterization. Methods Mol Biol 2011:331–344. https://doi.org/10.1007/978-1-4939-9554-7_19
Acknowledgments
The authors thank Sangeeta Gauchan, Mahder Etuma, and Matthew Ventura for their excellent technical assistance. The authors also thank the Veterinary Services Program of Walter Reed Army Institute of Research for their veterinary support, guidance, and animal care for the present study.
Funding
This work was supported by the Military Operational Medicine Research Program, US Army Medical Research and Development Command. RMT was supported by an NRC Research Associateship award at Walter Reed Army Institute of Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Disclaimer
The views expressed in this article are those of the author and do not necessarily reflect the official policy or position of the Department of the Army, Department of Defense, nor the U.S. Government. This is a U.S. Government work. There are no restrictions on its use. There were no financial conflicts of interests among any of the authors. Material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the author, and are not to be construed as official, or as reflecting true views of the Department of the Army or the Department of Defense. Research was conducted under an approved animal use protocol in an AAALACi accredited facility in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, NRC Publication, 2011 edition
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Liana M. Matson and Emily G. Lowery-Gionta share joint last authorship
Electronic supplementary material
ESM 1
(DOCX 17 kb)
Rights and permissions
About this article

Cite this article
Taylor, R.M., Jeong, I.H., May, M.D. et al. Fear expression is reduced after acute and repeated nociceptin/orphanin FQ (NOP) receptor antagonism in rats: therapeutic implications for traumatic stress exposure. Psychopharmacology 237, 2943–2958 (2020). https://doi.org/10.1007/s00213-020-05582-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-020-05582-0