
Abstract
Aim
Indoleamine 2,3-dioxygenase 1 (IDO) is responsible for the progression of the kynurenine pathway, which has been implicated in the pathophysiology of inflammation-induced depression. It has been reported that asperosaponin VI (ASA VI) could play a neuroprotective role through anti-inflammatory and antioxidant. In this study, we examined the antidepressant effect of ASA VI in lipopolysaccharide (LPS)-treated mice and further explored its molecular mechanism by looking into the microglial kynurenine pathway.
Methods
To generate the model, LPS (0.83 mg/kg) was administered intraperitoneally to mice. The mice received ASA VI (10 mg/kg, 20 mg/kg, 40 mg/kg, and 80 mg/kg, i.p.) 30 min before LPS injection. Depressive-like behaviors were evaluated based on the duration of immobility in the forced swim test. Microglial activation and inflammatory cytokines were detected by immunohistochemistry, real-time PCR, and ELISA. The TLR4/NF-κB signaling pathway and the expression of IDO, GluA2, and CamKIIβ were also measured by western blotting.
Results
ASA VI exhibited significant antidepressant activity in the presence of LPS on immobility and latency times in the forced swim test. The LPS-induced activation of microglia and inflammatory response were inhibited by ASA VI, which showed a dose-dependent pattern. TLR4/NF-κB signaling pathway also was suppressed by ASA VI in the hippocampus and prefrontal cortex of LPS-treated mice. Furthermore, ASA VI inhibited the increase in IDO protein expression and normalized the aberrant glutamate transmission in the hippocampus and prefrontal cortex caused by LPS administration.
Conclusion
Our results propose a promising antidepressant effect for ASA VI possibly through the downregulation of IDO expression and normalization of the aberrant glutamate transmission. This remedying effect of ASA VI could be attributed to suppress microglia-mediated neuroinflammatory response via inhibiting the TLR4/NF-κB signaling pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Depression is highly prevalent worldwide and a leading cause of disability. However, the medications currently available to treat depression fail to adequately relieve depressive symptoms for a large number of patients. Inflammation-induced depression is a debilitating psychiatric disorder that is caused by abnormal tryptophan metabolism in the central nervous system (CNS) (Dantzer et al. 2008; Kohler et al. 2016). This type of depression is refractory to conventional medications such as selective serotonin reuptake inhibitors (SSRIs) (Al-Harbi 2012; Hashmi et al. 2013). Research on the aberrant over-activation of the kynurenine pathway and the production of various active metabolites has brought new insight into the progression of depression (Ogyu et al. 2018; Oxenkrug 2010; Savitz 2017). In the non-challenged brain, kynurenine (KYN) is predominantly metabolized to kynurenic acid (KYNA) by astrocytes (Schwarcz et al. 2012). However, disruption of neuroendocrine or neuroinflammatory balance, which involves activation of microglia, can make KYN metabolized to 3-hydroxykynurenine (3HK) in microglia (Schwarcz et al. 2012; Schwarcz and Stone 2017). The 3HK is further metabolized into quinolinic acid (QUIN), the agonist of the N-methyl-D-aspartate (NMDA) receptor, which leads to neuronal dysfunction and aberrant glutamate transmission (Duman and Aghajanian 2012; Kubicova et al. 2013).
Microglia, the principal immune cells in the brain, plays a central role in immune surveillance and inflammatory-related neuropathology (Ginhoux et al. 2010; Hanisch and Kettenmann 2007; Yirmiya et al. 2015). The dysfunction of microglia has been associated with depression (Brites and Fernandes 2015; Frick et al. 2013; Yirmiya et al. 2015). The cascade of microglial activation can promote the synthesis and secretion of a large number of inflammatory mediators, resulting in neuroinflammatory response (Zhang et al. 2017; Zhang et al. 2016). Inflammatory cytokines such as IL-1β and TNF-α have been shown to enhance this pathway in microglia by increasing the expression of the kynurenine pathway’s main controller enzyme, indoleamine 2,3-dioxygenase (IDO), through STAT1 activation (Hemmati et al. 2019). Similarly, lipopolysaccharide (LPS) upregulates IDO levels through activating the TLR4/NF-κB signaling pathway and inducing depressive-like behaviors (O’Connor et al. 2009b).
A growing body of research suggests that natural products have a good antidepressant effect via inhibiting microglia-mediated neuroinflammation (Lee et al. 2013; Zhang et al. 2017). The active ingredients of traditional Chinese medicine have great potential in anti-depression research. Radix Dipsaci, a well-known traditional Chinese herb, not only is found to play an important role in promoting osteoblast formation (Zhang et al. 2019) but also is used in the clinical treatment of depression in traditional Chinese medicine. Asperosaponin VI (ASA VI) is a natural compound isolated from Radix Dipsaci (Ke et al. 2016). Studies showed that ASA VI has a neuroprotective effect through anti-inflammatory and antioxidant effects (Wang et al. 2018; Yang et al. 2019; Yu et al. 2012). In this study, we explored the antidepressant effects of ASA VI on LPS-treated mice, and corresponding molecular mechanisms by detecting the activation of microglia, TLR4/NF-κB pathway, IDO protein expression, and GluA2 and CamKIIβ.
Materials and methods
Animals
Male C57BL/6 mice (8 weeks old) were purchased from the Biotechnology Centre (Changsha, China). All the animals were raised in a single cage in a suitable environment of 25 ± 1 °C and 65% humidity, with free drinking water and activities. All animal experiments were carried out by the China code for the care and use of animals for scientific purposes at Guizhou University of Traditional Chinese Medicine, Guiyang, China, with local animal ethics committee approval.
LPS administration and pharmacological intervention
The ASA VI standard (purity = 99.92%) was purchased from Chengdu Alfa Biotechnology Co., Ltd. The ASA VI was dissolved in saline. To generate the model, LPS (0.83 mg/kg) was administered intraperitoneally to mice. The control mice were administered with equivoluminal saline. Both control mice and model mice were received ASA VI (10 mg/kg, 20 mg/kg, 40 mg/kg, and 80 mg/kg, i.p.) 30 min before LPS injection. The dose selected in this study is determined by both the dosage of Radix Dipsaci in traditional Chinese medicine (0.2 g–1.6 g/kg/d) and the content of ASA VI in Radix Dipsaci (5%).
Body weight test, measurement of food consumption, and open field test were performed for mice 24 h after LPS administration. And depressive-like behaviors were measured using the forced swim test for mice 2 h after open field test. After the behavioral analysis, animals are sacrificed for immunocytochemistry, RT-PCR analysis, enzyme-linked immunosorbent assay (ELISA), and western blot analysis.
Open field test
The open field test was conducted to examine whether the alterations in forced swim test (FST) immobility duration were related to changes in locomotor function. For this test, each mouse was placed in an acrylic plastic box (50 × 50 × 30 cm). The locomotor activity, the time in center, and the travel distance during a 5-min period were measured by the open field test (OFT) 100 software (Taimen, Chengdu, China).
Forced swim test
Each mouse was placed in a cylinder containing water with 15-cm depth for 15 min. Twenty-four hours later, the animals were placed in the cylinder again and the duration of immobility (including movements to keep afloat, but not active swimming) was recorded for a 6-min period.
Primary microglia culture and treatment
Primary microglia were derived from brains of neonatal (P0–P3) C57BL/6 mice. After removing the meninges and blood vessels, brains were minced and centrifuged at 800g for 5 min. The sediment was dissociated with 0.0125% trypsin for 10 min, and then, one volume of DMED-F12 culture medium (Gibco, USA) was added to inactivate the trypsin. The suspension was passed through a 70-μm cell strainer (Koch Membrane Systems, USA). Cell pellets were harvested, washed, and cultured in DMED-F12 culture medium with 10% fetal calf serum (FBS) at 37 °C. The medium was changed after 24 h. On day 14, mixed glial cultures were briefly shaken by hand for 2 s, and floating cells were collected and transferred to a new plate with fresh cultured in DMED-F12 culture medium with 10% FBS. At 7 days later, microglia were pretreated with either ASA VI (10 μM, 50 μM, 100 μM) or PBS (control). After 30 min, these cells were treated with LPS (100 ng/mL) and incubated for 24 h. These cells are then used for immunocytochemistry and RT-PCR analysis.
Immunocytochemistry and image analysis
Mice were anesthetized with 1% pentobarbital (50 mg kg−1 i.p.), perfused with 4% paraformaldehyde (0.1 M phosphate buffer configuration, PBS) for 10 min, and then the whole brain of mice was isolated and soaked into 4% paraformaldehyde for 48 h. After dehydrated in 30% sucrose for 48 h, the frozen section was 30 μm. Primary microglia were fixed by 4% paraformaldehyde for 30 min. Brain slices or cultured cells were placed in 24-well plates, washed with PBS for 3 times, 5 min/time, permeated with 0.5% Triton X-100 for 15 min, and then washed with PBS for 3 times again, 5 min/time. The brain slices were incubated for 2 h at room temperature in a blocking solution (10% donkey serum prepared with PBS) and incubated for 24 h at 4 °C in the solution containing the primary antibodies (mouse anti-Iba1 antibody, Abcam, 1:400; rabbit anti-inducible nitric oxide synthase (iNOS) antibody, Abcam, 1:50; mouse anti-GFAP antibody, Cell Signaling Technology, 1:400; rabbit anti-NeuN antibody, Cell Signaling Technology, 1:400). Finally, the brain slices were incubated with the secondary antibodies (anti-rabbit IgG-conjugated Alexa fluorochrome or anti-mouse IgG-conjugated Alexa fluorochrome, Invitrogen; 1:500) for 2 h at room temperature.
Microglia and astrocyte were photographed using fluorescence microscopy (Olympus BX51). Image analysis was used to quantify the area, perimeter, and fluorescence intensity of microglia. The images of microglia were imported to the ImageJ software (version 1.45 J) and used to determine a threshold for positive staining while excluding background staining. The amount, average percent area, and fluorescence intensity of the positive threshold for all representative pictures were reported.
Quantitative PCR
Mice were sacrificed under aseptic conditions, and the hippocampus and cortex were isolated and placed in 1.5-mL aseptic centrifuge tubes; total RNA was extracted by Trizol (TaKaRa, Japan), using the reverse transcription kit (Invitrogen Life Technologies, USA) to get the cDNA in strict accordance with the manufacturer’s instructions. The RT-PCR reaction mixture contains 1 μL of template cDNA, 5 μL MasterMix, and 1 μL primer (Sangon Biotech, Sichuan, China); add DEPC water to a total reaction volume of 10 μL. After mixing, put it into the 7500 Real-Time PCR System (Applied Biosystems, USA). The internal reference gene is β-actin and the expression of related genes is calculated according to the method of −ΔΔ Ct. Primer sequences have been listed in Table S1.
Enzyme-linked immunosorbent assay
Mice were sacrificed under aseptic conditions, and the hippocampus and cortex of the mice were placed in a 1.5-mL centrifuge tube. After homogenization, the supernatant was centrifuged for testing the secretion of inflammatory cytokines. BCA (Sangon Biotech, Sichuan, China) was used to determine the concentration of total protein. The total protein levels were diluted to the same concentration and then operated in strict accordance with the instructions of the ELISA Kit (Boster, Wuhan, China) to determine the levels of IL-1β and TNF-α.
Western blot analysis
Mice were sacrificed under aseptic conditions, and the hippocampus and cortex of the mice were placed in a 1.5-mL centrifuge tube; RIPA lysis buffer (Solarbio, China) was used for lysis. The lysis solution was centrifuged at 1000×g for 30 min. The concentration was measured by the BCA method. Equal amount of protein was resolved using 12% SDS polyacrylamide gel. The proteins were transferred onto PVDF membranes. Membranes were blocked for 120 min with 5% non-fat skimmed milk and incubated with the following primary antibodies overnight: phosphorylated NF-κB (pNF-κB) (p65) (rabbit anti-pNF-κB antibody, Abcam, 1:800), NF-κB (rabbit anti-NF-κB antibody, Abcam, 1:1000), IDO1 (mouse anti-IDO1 antibody, Santa Cruz, 1:600), AMPA Receptor 2 (GluA2) (rabbit-GluA2 antibody, Cell Signaling Technology, 1:1000), CaMKIIβ (rabbit-CaMKII beta antibody, Abcam, 1:1000), GAPDH (mouse-GADPH antibody, Abcam, 1:10000), and β-actin (mouse-beta Actin antibody, Abcam, 1:10000). Membranes were then washed 3 times with TBST (TBS + Tween 80) and incubated for 1 h at room temperature with secondary antibodies. Bands were visualized using the BM Chemiluminescence Western Blotting Kit acquired from Roche Diagnostics GmbH (Mannheim, Germany). Proteins were tested using the chemiluminescence detection system (Amersham, Berkshire, UK). Finally, the bands were analyzed using the ImageJ software (version 1.45 J).
Statistical analysis
The statistical analyses were performed using the SPSS 17.0 software (SPSS Inc., Chicago, USA). Differences between the mean values were evaluated by a paired Student’s t test for independent samples, one-way or two-way analysis of variance (ANOVA) followed by a protected Fisher’s least significance difference (LSD) test for post hoc comparisons of multiple groups. A P value of 0.05 or less was considered indicative of statistical significance. Each sample was repeated 3 times for qPCR, ELISA, and western blot; 5 immunofluorescence images of each sample were used to image analysis. The mean value of the parallel repeated data was used for significance statistical analysis.
Results
ASA VI alleviates LPS-induced depressive-like behaviors
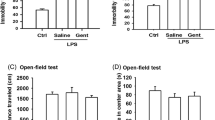
Motor function, immobility, and latency times were compared between study groups of 8–15 mice. The two-way ANOVA showed that changes in mobility and stationary time, and moved distance in the OFT were not significant following ASA VI pretreatment regardless of LPS or saline administration (Fig. 1b and c). The changes in the time in center were not significant following ASA VI pretreatment in control mice when compared with saline-treated mice. LPS treatment markedly decreased the time in the center of mice in the OFT, while ASA VI pretreatment reversed the LPS-induced increase in the time in center, which depended on the applying dose of ASA VI. All ASA VI treatments except the dose of 10 mg/kg significantly increased the time in center when compared with LPS-treated mice without pharmacological intervention (Fig. 1d).

Effects of ASA VI on LPS-induced depressive-like behaviors. a The timeline of our experimental process. b–d Changes in mobility and stationary time, moved distance and time in the center of mice in open field test (OPT) following ASA VI pretreatment before LPS or saline administration. e, f Changes in immobility and latency times of mice in forced swim test (FST) following ASA VI pretreatment before LPS or saline administration. g Changes in food consumption of mice during 24 h following ASA VI pretreatment before LPS or saline administration. h, i Changes in body weight of mice during 24 h following ASA VI pretreatment before LPS or saline administration. Data of Fig. 1b are mean ± SEM, data of Fig. 1c–i is displayed individually (n = 8–15 per group), *P < 0.05, **P < 0.01, ***P < 0.005 (two-way ANOVA with LSD test)
In the FST, changes in immobility and latency times were not significant following ASA VI pretreatment in control mice when compared with saline-treated mice (Fig. 1e and f). LPS treatment markedly increased the immobility time and decreased the latency times in the TST. The extent that ASA VI pretreatment reversed the LPS-induced increase in the immobility time or decrease in latency times in a dose-dependent manner. In the LPS-treated mice, 40 mg/kg and 80 mg/kg but not 10 mg/kg and 20 mg/kg of ASA VI significantly decreased the immobility time and increased the latency times in FST when compared with the mice without pharmacological intervention (Fig. 1e and f).
The food consumption and body weight of LPS- or saline-treated mice were determined when pretreated with ASA VI. The results showed that LPS treatment markedly decreased the food consumption and body weight of mice, while ASA VI generally had no effect on the LPS-induced decreases in food consumption and body weight (Fig. 1g–i).
Antidepressant effect of ASA VI is potentially mediated by inhibiting LPS-induced activation of microglia and neuroinflammation
In the brain, glia could respond positively to LPS stimulation. We examined the effects of ASA VI on the astrocyte and microglia in the hippocampus and PFC of LPS-treated mice. The two-way ANOVA results showed that both the changes in GFAP mRNA expression and staining area of GFAP+ were not significantly affected by ASA VI pretreatment regardless of LPS or saline administration (Fig. 2a–c). LPS administration resulted in an increase in microglial marker (Iba1 and CD11b) expression and changes in the microglial morphology, including the increased area of microglia and decreased number and length of branches in the hippocampus and PFC (Fig. 2). The ASA VI inhibited LPS-induced activation of microglia in a dose-dependent manner. When the mice were pretreated with higher dosage of ASA VI (40 mg/kg, 80 mg/kg) before LPS administration, the mRNA expression of microglial markers (Iba1 and CD11b) were significantly inhibited in hippocampus and PFC (Fig. 2d–g). While when 10 mg/kg ASA VI was pretreated, no significant effect on the mRNA expression of microglial markers was found. It was noteworthy that the effects of 20 mg/kg ASA VI on the mRNA expression of microglial markers were wobbly in the hippocampus and PFC of LPS-treated mice (Fig. 2d–g).

Effects of ASA VI on LPS-induced activation of microglia in the hippocampus and prefrontal cortex. a Immunohistochemistry detects the changes of astrocyte (GFAP, green) and microglia (IBA1, red) in the hippocampus and prefrontal cortex of mice following ASA VI pretreatment before LPS or saline administration. Scale bar, 50 μm. b Quantitative PCR detects the mRNA expression of GFAP in the hippocampus. Data showed the fold change relative to control group. c Quantification of the area of GFAP+ cells in the hippocampus of mice. d–g Quantitative PCR detects the mRNA expression of CD11b and IBA1 in the hippocampus and prefrontal cortex. Data showed the fold change relative to control group. h–m Quantification of the area, number, and length of branches of IBA1+ cell in the hippocampus and prefrontal cortex. Data are mean ± SEM (n = 4–6 per group); each sample was repeated 3 times for qPCR; 5 immunofluorescence images of each simple were used to analysis. ***P < 0.005 when compared with control group, #P < 0.05, ##P < 0.01, ###P < 0.005 when compared with LPS group (two-way ANOVA with LSD test)
The LPS-induced increase in the area of IBA1+ cells in the hippocampus and PFC was suppressed by 40 mg/kg and 80 mg/kg of ASA VI. Nevertheless, 10 mg/kg and 20 mg/kg of ASA VI pretreatment did not significantly affect the area of IBA1+ cells in the hippocampus and PFC of LPS-treated mice (Fig. 2h and i). The LPS-induced decrease in number and length of branches of microglia in the hippocampus and PFC was reserved by 40 mg/kg of ASA VI. The 80 mg/kg ASA VI only increased the number and length of branches of microglia in PFC (Fig. 2j–m).
The expression of inflammatory cytokines was usually synchronized with changes in microglial morphology; therefore, we also examined the effects of ASA VI on the expression of pro-inflammatory cytokines in the hippocampus and PFC of LPS-treated mice. The results showed that there were no significant changes in mRNA expression (IL-1β, TNF-α, IL-6, and iNOS) and protein expression (IL-1β and TNF-α) of inflammatory cytokines following ASA VI pretreatment in the hippocampus and PFC of control mice as determined by two-way ANOVA (Fig. 3a and b). LPS administration led to an increase of IL-1β, TNF-α, IL-6, and iNOS expression in the hippocampus and PFC (Fig. 3). Conversely, ASA VI decreased the expression of IL-1β, TNF-α, IL-6, and iNOS in both the hippocampus and PFC of LPS-treated mice, which was determined by the pretreatment dose (Fig. 3a). The expression of IL-1β and TNF-α was further validated at the protein level by ELISA. The results were consistent with those of mRNA expression (Fig. 3b). The result from immunohistochemistry showed that LPS-induced iNOS was mainly expressed in microglia (Fig. 3c).

Effects of ASA VI on neuroinflammatory response in the hippocampus and prefrontal cortex of LPS-treated mice. a Quantitative PCR detects the mRNA expression of IL-1β, TNF-α, IL-6, and iNOS in the hippocampus and prefrontal cortex. Data showed the fold change relative to control group. Data are mean ± SEM (n = 4–6 per group); each sample was repeated 3 times. *P < 0.05, ***P < 0.005 when compared with control group, #P < 0.05, ##P < 0.01, ###P < 0.005 when compared with LPS group (two-way ANOVA with LSD test). b ELISA detects the protein expression of IL-1β and TNF-α in the hippocampus and prefrontal cortex. Data were displayed individually (n = 5 per group); each sample was repeated 3 times.*P < 0.05, **P < 0.01, ***P < 0.005 (two-way ANOVA with LSD test). c Immunohistochemistry detects the sources of iNOS. iNOS (green) was located in the IBA1+ cell (red) in prefrontal cortex of mice following LPS administration. Scale bar, 50 μm
ASA VI suppressed microglia-mediated neuroinflammation by inhibiting the TLR4/NF-κB signaling pathway
To further explore the molecular mechanism of the anti-inflammatory activity of ASA VI, we examined the total NF-κB and pNF-κB by western blotting in the hippocampus and PFC of mice received LPS administration and ASA VI pretreatment. The results showed that LPS administration promoted the phosphorylation of NF-κB in both the hippocampus and PFC when compared with control mice (Fig. 4a–d). ASA VI inhibited the phosphorylation of NF-κB in a dose-dependent manner. The 40 mg/kg and 80 mg/kg ASA VI significantly inhibited LPS-induced phosphorylation of NF-κB in both the hippocampus and PFC (Fig. 4a–d).

Effects of ASA VI on phosphorylation of NF-κB in the hippocampus and prefrontal cortex of LPS-treated mice. a, b Western blotting detects NF-κB and pNF-κB in the hippocampus and prefrontal cortex of mice following ASA VI pretreatment before LPS administration. c, d Quantification of the phosphorylation level of NF-κB in the hippocampus and prefrontal cortex. NF-κB was normalized β-actin, and pNF-κB was normalized total NF-κB. e Quantitative PCR detects the mRNA expression of TLR4 in LPS-treated primary microglia. f Immunohistochemistry detects the effect of ASA VI on p65 transposition to nucleus in LPS-treated primary microglia. p65 (green), nucleus (DAPI, blue), microglia (Iba1, red); scale bar, 10 μm. Data were displayed individually (n = 3–4 per group); each sample was repeated 3 times. ***P < 0.005 when compared with control group, #P < 0.05, ###P < 0.005 when compared with LPS group (one-way ANOVA with LSD test)
To determine the inhibitory effect of ASA VI on the microglial TLR4/NF-κB signaling pathway, we examined the effect of ASA VI on TLR4 gene expression and p65 transposition to nucleus in LPS-treated primary microglia. The results showed that 50 μM and 100 μM of ASA VI significantly inhibited LPS-induced increases in TLR4 gene expression (Fig. 4e). At the same time, 50 μM and 100 μM of ASA VI inhibited the p65 transposition to nucleus in LPS-treated microglia (Fig. 4f).
IDO expression is downregulated in ASA VI–treated mice in a dose-dependent manner
Based on the effect of microglia-mediated inflammatory response on tryptophan metabolism, we examined the expression of key enzymes (IDO) in the kynurenine pathway. The protein expression of IDO1 was detected by western blotting in the hippocampus and PFC of mice received LPS administration and ASA VI pretreatment. The results showed that LPS administration increased the expression of IDO1 in both the hippocampus and PFC when compared with control mice (Fig. 5a–d). When the mice were pretreated with higher dosage of ASA VI (40 mg/kg, 80 mg/kg) before LPS administration, the expression of IDO1 in the hippocampus and PFC was inhibited when compared with those treated with LPS alone (Fig. 5a–d).

Effects of ASA VI on IDO expression in the hippocampus and prefrontal cortex of LPS-treated mice. a, b Western blotting detects IDO1 in the hippocampus and prefrontal cortex of mice following ASA VI pretreatment before LPS administration. c, d Quantification of the protein level of IDO1 in the hippocampus and prefrontal cortex. IDO1 was normalized GADPH. Data were displayed individually (n = 3–4 per group); each sample was repeated 3 times. ***P < 0.005 when compared with control group, #P < 0.05, ##P < 0.01, ###P < 0.005 when compared with LPS group (one-way ANOVA with LSD test)
ASA VI normalized the aberrant glutamate transmission in the hippocampus and prefrontal cortex of LPS-treated mice
To test the effect of neuroinflammatory cytokines and the IDO-mediated kynurenine pathway on neurons, we first examined the morphological and quantitative changes of mature neurons by immunohistochemistry (Fig. 6a). The result showed that the area of NeuN+ cell in both the hippocampus and PFC was not significant when pretreated with ASA VI regardless of LPS administration as determined by one-way ANOVA (Fig. 6b and c). Considering that the effect of the IDO-mediated kynurenine pathway on glutamate transmission may be the main cause of depression-like behavior, we further examined the expression of synaptic proteins (GluA2 and CamKIIβ). Our analysis of synaptic proteins in mice after LPS administration revealed reduced expression levels of proteins mediating synaptic plasticity CamKIIβ, as well as the AMPA receptor subunits GluA2 (Fig. 6d–i). In agreement with the results from the behavioral test, microglial activation, and IDO expression, ASA VI reversed the LPS-induced decrease in the expression of synaptic proteins (GluA2 and CamKIIβ) in a dose-dependent manner (Fig. 6d–i). The 40 mg/kg ASA VI consistently normalized the expression of GluA2 and CamKIIβ both in the hippocampus and the PFC of LPS-treated mice. Although both 10 mg/kg and 20 mg/kg of ASA VI increased the expression of GluA2 or/and CamKIIβ, the synaptic protein expression levels did not return to normal. The expressions of GluA2 in the hippocampus and PFC of LPS-treated mice were normalized by 80 mg/kg ASA VI. But the expression of CamKIIβ was only normalized in the hippocampus but not in PFC of 80 mg/kg ASA VI–treated mice (Fig. 6d–i).

Effects of ASA VI on GluA2 and CamKIIβ expression in the hippocampus and prefrontal cortex of LPS-treated mice. a Immunohistochemistry detects the changes of mature neurons (NeuN, green) in prefrontal cortex of mice following ASA VI pretreatment before LPS or saline administration. Scale bar, 20 μm. b, c Quantification of the area of NeuN+ cells in the hippocampus and prefrontal cortex of mice. d, e Western blotting detects GluA2 and CamKIIβ in the hippocampus and prefrontal cortex of mice following ASA VI pretreatment before LPS administration. f–i Quantification of the protein level of GluA2 and CamKIIβ in the hippocampus and prefrontal cortex. GluA2 and CamKIIβ were normalized β-actin. Data were displayed individually (n = 3–5 per group); each sample was repeated 3 times for western blotting. Five immunofluorescence images of each sample were used to analysis. *P < 0.05, **P < 0.01, ***P < 0.005 when compared with control group, #P < 0.05, ##P < 0.01, ###P < 0.005 when compared with LPS group (one-way ANOVA with LSD test)
Discussion
Based on our results, ASA VI showed novel promising antidepressant effects on a validated mouse model of inflammatory depression. Improvements in mice depressive-like behaviors were reflected by the reduction of immobility times in the FST test. Furthermore, the underlying mechanisms were investigated by assessing microglial activation, inflammatory expression, NF-κB activation, IDO expression, and expression of synaptic proteins. Our results were consistent with previous studies which had demonstrated that LPS induces microglial activation and IDO expression through upregulation of active pNF-κB. However, the suppressant effects of ASA VI on microglia-mediated neuroinflammatory response, NF-κB activation, and the kynurenine pathway are investigated for the first time.
ASA VI is the major active ingredient in Radix Dipsaci. Although Radix Dipsaci is a natural, stable and widely distributed medicinal herb, there are few reports on ASA VI in the literature. There is a small body of literature showing that ASA VI can cross the blood-brain barrier and has anti-inflammatory and antioxidant effects (Song et al. 2014; Wang et al. 2018; Yang et al. 2019). Based on these, we hypothesized that ASA VI protects the brain from neuroinflammatory damage by inhibiting the expression of microglia-mediated pro-inflammatory cytokines. In this study, the model animals of inflammation-induced depression were successfully established by injection of LPS, which was represented by the increased in center time in OFT and decreased immobility times in FST. OFT is often used to evaluate the motor ability and anxiety-like behavior of animals (Rajabi et al. 2018). And the latency time and immobility times in FST are commonly used to assess the behavioral despair (Yan et al. 2014). We conducted a dose screening, from 10 to 80 mg/kg, for antidepressant effects of ASA VI. The results showed that the mobility and stationary time and moved distance both in control mice and LPS-treated mice were not significantly affected by the ASA VI application regardless of the dose. This means that ASA VI does not promote excitement or sedation. However, all the ASA VI treatments except for the 10 mg/kg treatment significantly increased the time in center in LPS-treated mice, suggesting ASA VI ameliorated the LPS-induced anxiety-like behavior. In addition, 40 mg/kg and 80 mg/kg of ASA VI treatments significantly reversed LPS-induced increase in immobility times and decrease in latency time in FST, which means ASA VI can ameliorate the LPS-induced depressive-like behavior. Surprisingly, ASA VI did not reserve the LPS-induced decrease in food consumption and body weight. These results suggest that in despite of the improvement in depression-like behavior, the ASA VI cannot completely eliminate the other effects of LPS injection on animals. Weight and appetite are influenced by many factors, just as LPS affects animals in many ways (Martinez et al. 2014; Subramaniapillai and McIntyre 2017). It is worth noting that 80 mg/kg of ASA VI generally reduced the weight of control mice. The dose may be too high and cause minor damage to the animals.
In the brain, glia especially microglia could respond positively to LPS stimulation (Zhang et al. 2017). In this study, we found that LPS did not affect GFAP mRNA expression and the GFAP+ staining area. Significantly, LPS administration resulted in an increase in microglial marker (Iba1 and CD11b) expression and changes in the microglial morphology, including the increased area of microglia and decreased number and length of branches in the hippocampus and PFC. These results suggest that microglia are more sensitive to immune stimulation than astrocytes (Zhang et al. 2017). When the mice were pretreated with ASA VI before LPS administration, the microglial activation and neuroinflammatory response in the hippocampus and PFC were inhibited by ASA VI, which exhibited a dose-dependent pattern. This result supports our previous hypothesis that the antidepressant effect of ASA VI is potentially mediated by inhibiting LPS-induced activation of microglia and neuroinflammation. Microglia activation plays an important role in the development of depression (Zhang et al. 2018). Neuroinflammatory injury is considered one of the main reasons that microglia cells aggravate the pathological process of depression (Zhang et al. 2018). In particular, pro-inflammatory cytokines (such as IL-1β and TNF-α) not only affect the neural function but also induce secondary activation of microglia (Jung et al. 2016; Zhang et al. 2016). ASA VI also inhibited the LPS-induced increase in expression of IL-1β, TNF-α, iNOS, and IL-6 both in the hippocampus and prefrontal cortex.
NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) is a protein complex that controls the transcription of DNA (Zhu et al. 2018). NF-κB has been found in most types of animal cells and is involved in cellular responses to stimuli such as stress, cytokines, and bacterial or viral antigens (Pan et al. 2017). NF-κB plays a key role in regulating the immune response to infection (Guo et al. 2019). LPS has been shown to promote microglial activation and neuroinflammatory response by activating NF-κB through their specific receptors (TLR4) (Guo et al. 2019). Our data showed that ASA VI inhibited the LPS-induced phosphorylation of NF-κB, and the effect varies with different doses. ASA VI also inhibited TLR4 expression and the p65 transposition to nucleus in LPS-treated microglia. These results suggest that ASA VI can suppress microglia-mediated neuroinflammation by inhibiting the TLR4/NF-κB signaling pathway (Yu et al. 2012).
Microglial IDO is activated by inflammatory cytokines (including IFN-γ, IL-1β, IL-6, and TNF-α) and psychological stress. It facilitates the microglial kynurenine pathway (KP) and results in depressive-like symptoms (Agudelo et al. 2014; Du et al. 2019). The KP was proposed to serve as the switch from acute (sickness-like) effects of inflammatory challenges and stress to the development of depression (Corona et al. 2013). The pro-inflammatory cytokines induced by LPS stimulation can promote the expression of IDO in microglia (O’Connor et al. 2009a). TLR4/NF-κB signaling appears to be the most promising route for IDO elevation in the brain of mice undergoing systemic LPS administration and consequent tryptophan catabolite formations (Hemmati et al. 2019). In this study, we found that ASA VI also inhibited the LPS-induced increase of IDO expression both in the hippocampus and PFC. These results suggest that the antidepressant effects of ASA VI are mediated by the suppression of microglial activation and reduction of TLR4/NF-κB-induced IDO expression.
Studies in rodents provide even more specific evidence for implicating the activation of the enzyme’s IDO and signaling via the kynurenine pathway in inflammation-induced depression (Wang et al. 2010a; Wang et al. 2010b). The blockade of the KP activation and the neurobehavioral consequences of this activation can be achieved by the IDO inhibitor (Yirmiya et al. 2015). Levels of kynurenine (KYN) and its metabolites 3-hydroxykynurenine (3HK) and kynurenic acid (KYNA) in patients are strongly correlated to depression (Agudelo et al. 2014). The elevated QUIN levels intensely stimulate NMDA receptor signaling, leading to depression (Agudelo et al. 2014). The depressive-like symptoms induced by LPS and other immune challenges were attenuated by NMDA receptor antagonists such as ketamine (Jiang et al. 2019; Walker et al. 2013). Imbalances in glutamate transmission and decreased synaptic plasticity have been suggested as possible mechanisms of depression (Leal and Lopes 2018). Given that the effect of IDO and inflammatory cytokines on glutamate transmission may be the main cause of depression-like behavior, we further examined the expression of synaptic proteins (GluA2 and CamKIIβ). Our analysis of synaptic proteins in mice revealed that expression level of proteins mediating synaptic plasticity CamKIIβ and the AMPA receptor subunit GluA2 decreased after LPS administration. In agreement with the behavioral test results, ASA VI consistently normalized the expression of GluA2 and CamKIIβ both in the hippocampus and the PFC of LPS-treated mice. The results suggest that ASA VI normalized the aberrant glutamate transmission in the hippocampus and prefrontal cortex of LPS-treated mice.
In conclusion, our results propose a promising antidepressant effect for ASA VI possibly through the downregulation of IDO expression and normalization of the aberrant glutamate transmission. This remedying effect of ASA VI is attributed to suppress microglia-mediated neuroinflammatory response via inhibiting the TLR4/NF-κB signaling pathway (Fig. 7).

Schematic representation of the mechanism underlying the antidepressant effects of ASA VI. LPS induces the activation of NF-κB pathway by TLR4/MD2 in microglia. Activation of NF-κB results in the activation of microglia and transcriptional expression of pro-inflammatory cytokines including IL-1β, TNF-α, and IL-6. These cytokines induce the increase of indoleamine 2,3-dioxygenase (IDO) which catalyze the tryptophan (Trp) into kynurenine (KYN) in microglia. Kynurenine is metabolized to 3-hydroxykynurenine (3HK) in microglia and the 3HK is further metabolized into quinolinic acid (QUIN) which is the agonist of the N-methyl-D-aspartate (NMDA) receptor that leads to neuronal dysfunction and aberrant glutamate transmission. On the other hand, the pro-inflammatory cytokines also directly leads to neuronal dysfunction and aberrant glutamate transmission. This series of changes eventually leads to depression-like symptoms. Antidepressant effect of ASA VI is potentially mediated by inhibiting LPS-induced activation of microglia and neuroinflammation via inhibiting TLR4/NF-κB
References
Agudelo LZ, Femenia T, Orhan F, Porsmyr-Palmertz M, Goiny M, Martinez-Redondo V, Correia JC, Izadi M, Bhat M, Schuppe-Koistinen I, Pettersson AT, Ferreira DMS, Krook A, Barres R, Zierath JR, Erhardt S, Lindskog M, Ruas JL (2014) Skeletal muscle PGC-1alpha1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 159:33–45
Al-Harbi KS (2012) Treatment-resistant depression: therapeutic trends, challenges, and future directions. Patient Prefer Adherence 6:369–388
Brites D, Fernandes A (2015) Neuroinflammation and depression: microglia activation, extracellular microvesicles and microRNA dysregulation. Front Cell Neurosci 9:476
Corona AW, Norden DM, Skendelas JP, Huang Y, O’Connor JC, Lawson M, Dantzer R, Kelley KW, Godbout JP (2013) Indoleamine 2,3-dioxygenase inhibition attenuates lipopolysaccharide induced persistent microglial activation and depressive-like complications in fractalkine receptor (CX(3)CR1)-deficient mice. Brain Behav Immun 31:134–142
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56
Du HX, Chen XG, Zhang L, Liu Y, Zhan CS, Chen J, Zhang Y, Yu ZQ, Zhang J, Yang HY, Zhong K, Liang CZ (2019) Microglial activation and neurobiological alterations in experimental autoimmune prostatitis-induced depressive-like behavior in mice. Neuropsychiatr Dis Treat 15:2231–2245
Duman RS, Aghajanian GK (2012) Synaptic dysfunction in depression: potential therapeutic targets. Science 338:68–72
Frick LR, Williams K, Pittenger C (2013) Microglial dysregulation in psychiatric disease. Clin Dev Immunol 2013:608654
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845
Guo DK, Zhu Y, Sun HY, Xu XY, Zhang S, Hao ZB, Wang GH, Mu CC, Ren HG (2019) Pharmacological activation of REV-ERBalpha represses LPS-induced microglial activation through the NF-kappaB pathway. Acta Pharmacol Sin 40:26–34
Hanisch UK, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10:1387–1394
Hashmi AM, Butt Z, Umair M (2013) Is depression an inflammatory condition? A review of available evidence. J Pak Med Assoc 63:899–906
Hemmati S, Sadeghi MA, Mohammad Jafari R, Yousefi-Manesh H, Dehpour AR (2019) The antidepressant effects of GM-CSF are mediated by the reduction of TLR4/NF-kB-induced IDO expression. 16, 117
Jiang J, Jian Q, Jing M, Zhang Z, Zhang G, Shan L, Yu P, Wang Y, Xu L (2019) The novel N-methyl-d-aspartate receptor antagonist MN-08 ameliorates lipopolysaccharide-induced acute lung injury in mice. Int Immunopharmacol 66:109–118
Jung YS, Park JH, Kim H, Kim SY, Hwang JY, Hong KW, Bae SS, Choi BT, Lee SW, Shin HK (2016) Probucol inhibits LPS-induced microglia activation and ameliorates brain ischemic injury in normal and hyperlipidemic mice. Acta Pharmacol Sin 37:1031–1044
Ke K, Li Q, Yang X, Xie Z, Wang Y, Shi J, Chi L, Xu W, Hu L, Shi H (2016) Asperosaponin VI promotes bone marrow stromal cell osteogenic differentiation through the PI3K/AKT signaling pathway in an osteoporosis model. Sci Rep 6:35233
Kohler O, Krogh J, Mors O, Benros ME (2016) Inflammation in depression and the potential for anti-inflammatory treatment. Curr Neuropharmacol 14:732–742
Kubicova L, Hadacek F, Chobot V (2013) Quinolinic acid: neurotoxin or oxidative stress modulator? Int J Mol Sci 14:21328–21338
Leal RB, Lopes MW (2018) Amygdala levels of the GluA1 subunit of glutamate receptors and its phosphorylation state at serine 845 in the anterior hippocampus are biomarkers of ictal fear but not anxiety
Lee JS, Song JH, Sohn NW, Shin JW (2013) Inhibitory effects of ginsenoside Rb1 on neuroinflammation following systemic lipopolysaccharide treatment in mice. Phytother Res 27:1270–1276
Martinez JA, Milagro FI, Claycombe KJ, Schalinske KL (2014) Epigenetics in adipose tissue, obesity, weight loss, and diabetes. Adv Nutr 5:71–81
O’Connor JC, Andre C, Wang Y, Lawson MA, Szegedi SS, Lestage J, Castanon N, Kelley KW, Dantzer R (2009a) Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J Neurosci 29:4200–4209
O’Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, Kelley KW, Dantzer R (2009b) Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry 14:511–522
Ogyu K, Kubo K, Noda Y, Iwata Y, Tsugawa S, Omura Y, Wada M, Tarumi R, Plitman E, Moriguchi S, Miyazaki T, Uchida H, Graff-Guerrero A, Mimura M, Nakajima S (2018) Kynurenine pathway in depression: a systematic review and meta-analysis. Neurosci Biobehav Rev 90:16–25
Oxenkrug GF (2010) Tryptophan kynurenine metabolism as a common mediator of genetic and environmental impacts in major depressive disorder: the serotonin hypothesis revisited 40 years later. Isr J Psychiatry Relat Sci 47:56–63
Pan N, Lu LY, Li M, Wang GH, Sun FY, Sun HS, Wen XJ, Cheng JD, Chen JW, Pang JY, Liu J, Guan YY, Zhao LY, Chen WL, Wang GL (2017) Xyloketal B alleviates cerebral infarction and neurologic deficits in a mouse stroke model by suppressing the ROS/TLR4/NF-kappaB inflammatory signaling pathway. Acta Pharmacol Sin 38:1236–1247
Rajabi M, Mohaddes G, Farajdokht F, Nayebi Rad S, Mesgari M, Babri S (2018) Impact of loganin on pro-inflammatory cytokines and depression- and anxiety-like behaviors in male diabetic rats. Physiol Int 105:199–209
Savitz J (2017) Role of kynurenine metabolism pathway activation in major depressive disorders. Curr Top Behav Neurosci 31:249–267
Schwarcz R, Stone TW (2017) The kynurenine pathway and the brain: challenges, controversies and promises. Neuropharmacology 112:237–247
Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ (2012) Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci 13:465–477
Song L, Wang S, Duan X, Liu X, Li Z, Nie L, Chu G (2014) Chromatographic fingerprint combined with content of asperosaponin VI and antioxidant activity for quality evaluation of wine-fried Dipsaci Radix. Nat Prod Commun 9:773–778
Subramaniapillai M, McIntyre RS (2017) A review of the neurobiology of obesity and the available pharmacotherapies. CNS Spectr 22:29–38
Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B, Kelley KW, Dantzer R (2013) NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology 38:1609–1616
Wang Y, Lawson MA, Dantzer R, Kelley KW (2010a) LPS-induced indoleamine 2,3-dioxygenase is regulated in an interferon-gamma-independent manner by a JNK signaling pathway in primary murine microglia. Brain Behav Immun 24:201–209
Wang Y, Lawson MA, Kelley KW, Dantzer R (2010b) Primary murine microglia are resistant to nitric oxide inhibition of indoleamine 2,3-dioxygenase. Brain Behav Immun 24:1249–1253
Wang Y, Shen J, Yang X, Jin Y, Yang Z, Wang R, Zhang F, Linhardt RJ (2018) Akebia saponin D reverses corticosterone hypersecretion in an Alzheimer’s disease rat model. Biomed Pharmacother 107:219–225
Yan Y, Wang YL, Su Z, Zhang Y, Guo SX, Liu AJ, Wang CH, Sun FJ, Yang J (2014) Effect of oxytocin on the behavioral activity in the behavioral despair depression rat model. Neuropeptides 48:83–89
Yang S, Zhang W, Xuan LL, Han FF, Lv YL, Wan ZR, Liu H, Ren LL, Gong LL, Liu LH (2019) Akebia Saponin D inhibits the formation of atherosclerosis in ApoE(-/-) mice by attenuating oxidative stress-induced apoptosis in endothelial cells. Atherosclerosis 285:23–30
Yirmiya R, Rimmerman N, Reshef R (2015) Depression as a microglial disease. Trends Neurosci 38:637–658
Yu X, Wang LN, Du QM, Ma L, Chen L, You R, Liu L, Ling JJ, Yang ZL, Ji H (2012) Akebia Saponin D attenuates amyloid beta-induced cognitive deficits and inflammatory response in rats: involvement of Akt/NF-kappaB pathway. Behav Brain Res 235:200–209
Zhang JQ, Wu XH, Feng Y, Xie XF, Fan YH, Yan S, Zhao QY, Peng C, You ZL (2016) Salvianolic acid B ameliorates depressive-like behaviors in chronic mild stress-treated mice: involvement of the neuroinflammatory pathway. Acta Pharmacol Sin 37:1141–1153
Zhang J, Xie X, Tang M, Zhang J, Zhang B, Zhao Q, Han Y, Yan W, Peng C, You Z (2017) Salvianolic acid B promotes microglial M2-polarization and rescues neurogenesis in stress-exposed mice. Brain Behav Immun 66:111–124
Zhang L, Zhang J, You Z (2018) Switching of the microglial activation phenotype is a possible treatment for depression disorder. Front Cell Neurosci 12:306
Zhang W, Xue K, Gao Y, Huai Y, Wang W, Miao Z, Dang K, Jiang S, Qian A (2019) Systems pharmacology dissection of action mechanisms of Dipsaci Radix for osteoporosis. Life Sci 235:116820
Zhu M, Liu M, Guo QL, Zhu CQ, Guo JC (2018) Prolonged DADLE exposure epigenetically promotes Bcl-2 expression and elicits neuroprotection in primary rat cortical neurons via the PI3K/Akt/NF-kappaB pathway. Acta Pharmacol Sin 39:1582–1589
Acknowledgments
We thank Professor Lanping Guo at the Resource Center of the Chinese Academy of Traditional Chinese Medicine for giving us guidance on the experiment and writing. We are grateful to Jun Yu for help in revising the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81860675), Guizhou Science and Technology Plan Project ([2019]5611), and Department of Science and Technology of Guizhou High-level Innovative Talents ([2018]5638).
Author information
Authors and Affiliations
Contributions
JQZ, SNY, and TZ designed the conceptual idea for this study and wrote the manuscript. SNY, YHL, CHX, and CL performed the experiments and analyzed these data. CGY and WKJ contributed new analytical tools and reagents. All the authors participated in the discussion and approved the manuscript as submitted.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Table S1
(DOCX 16 kb)
Rights and permissions
About this article

Cite this article
Zhang, J., Yi, S., Li, Y. et al. The antidepressant effects of asperosaponin VI are mediated by the suppression of microglial activation and reduction of TLR4/NF-κB-induced IDO expression. Psychopharmacology 237, 2531–2545 (2020). https://doi.org/10.1007/s00213-020-05553-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-020-05553-5