
Abstract
Rationale
Dysregulation of the one carbon cycle is documented in depression. Thereby, S-adenosylmethionine (SAMe), a one–carbon cycle nutraceutical compound with a favourable side effect profile, has a theoretical rationale for efficacy. However, further controlled studies are required to confirm SAMe’s efficacy.
Objectives
To test the efficacy of SAMe versus placebo in unmedicated DSM-5 diagnosed (major depressive disorder) (MDD) patients with mild-to-moderate levels of depressive symptoms.
Methods
We conducted an 8-week, double-blind, randomised controlled trial testing 800 mg/day of SAMe monotherapy versus placebo in 49 patients with MDD (Montgomery-Åsberg Depression Rating Scale [MADRS] score 14–25) who were not currently taking antidepressants. One–carbon cycle biomarkers, brain-derived neurotropic factor (BDNF), and relevant single nucleotide polymorphisms (SNPs) were analysed as potential treatment moderators.
Results
A clinically relevant differential reduction from baseline to week 8 of 3.76 points occurred on the primary outcome (MADRS) in favour of SAMe. This however was not significant (p = 0.13) on an adjusted linear mixed model, notwithstanding a medium to large effect size of 0.72. A high placebo response rate of 53% occurred (> 50% reduction on MADRS). Exploratory analyses showed that SAMe was however effective in reducing depression amongst participants with milder depression severity (MADRS ≤ 22, p = 0.045). Response was not moderated by BDNF, SNPs, or one–carbon cycle biomarkers, although increased folate concentrations were correlated with improved symptoms in the SAMe group (r = − 0.57, p = 0.026). The treatment was safe and well tolerated.
Conclusions
Although a differential reduction in depression symptoms between groups was observed in favour of SAMe, the results of this pilot study were not statistically significant.
Trial registration
ANZCTR—Australian New Zealand Clinical Trials Registry; No.: ACTRN12613001299796; URL: https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=364900
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Major depressive disorder (MDD) is a mental disorder that remains difficult-to-treat. Reports suggest that only 50 to 60% of patients respond to the first antidepressant prescribed (Frodl 2017). In addition, for many patients, antidepressants are not well tolerated and may elicit a range of side effects. Novel compounds (e.g. nutraceuticals, amino-acids, and herbal medicines) are potential treatments for depression (Sarris et al. 2016). These compounds generally have favourable side effect profiles and have the potential to act on brain pathways associated with depression (Sarris et al. 2011; De Sousa et al. 2017).
S-Adenosylmethonine (SAMe) is a nutraceutical that occurs naturally in the body (although is not readily available in the diet) and is an active cofactor in the one carbon cycle and the process of methylation (Sharma et al. 2017). As a methyl donor, SAMe is involved in many cell membrane functions and in the synthesis of monoamines, particularly serotonin, noradrenaline, and dopamine (cited in Arnold et al. 2005; Sharma et al. 2017). Methylfolate is another important one–carbon cycle component, having some evidence for antidepressant activity (Sarris et al. 2016), and may be potentially prescribed alongside SAMe.
Several RCTs have investigated the safety and efficacy of SAMe as monotherapy in depression (Galizia et al. 2016; Sharma et al. 2017). In two recent reviews, SAMe showed a similar efficacy to antidepressants (with very few adverse effects) (Galizia et al. 2016; Sharma 2017). However, the reviews have concluded that the evidence quality was low, owing to the heterogeneity of the studies, including formulation of SAMe used (e.g. if tosylated, kept in blister packs, or refrigerated, dose, length of study, and patient population (e.g. primary MDD diagnosis vs. general depressive symptoms) (Galizia et al. 2016).
In a recent research report, it should be noted that SAMe (1600 mg/day) and escitalopram (20 mg) both failed to outperform placebo in a 12-week, 3-arm, double-blind RCT (Mischoulon et al. 2014). There was however a suggestion of a gender effect, with males more in this study likely to respond to SAMe than females (Sarris et al. 2015). However, our sub-analysis of that study, which involved 144 adults with MDD (who had biomarker data), found a significant difference on Hamilton Depression Rating Scale (HAM-D) depression scores in favour of SAMe versus placebo from baseline to week 12 (p = 0.039) (Sarris et al. 2014). Regardless, it should be noted that this result was based on post hoc analyses (including only participants with available biomarker data); thus, the findings there cannot be reliably attributed to the intervention. Further, no correlations between biomarkers and outcomes were observed.
Due to conflicting findings, we thereby assessed the efficacy of SAMe versus placebo in a double-blind RCT. Since SAMe is commercially expensive, we used a dose commonly recommended in over the counter products for this RCT. Further, concordant with the RDoC philosophy (Cuthbert and Insel 2013), we also investigated one–carbon cycle nutrients, homocysteine, and associated single nucleotide polymorphisms (SNPs) as well as brain-derived neurotrophic factor (BDNF), as potential moderators of response.
Methods
Trial design
The study was a phase II, multicentre, 8-week, double-blind RCT testing SAMe monotherapy vs. placebo (Sarris et al. 2015). Participants were recruited from September 2013 to July 2017 at The Melbourne Clinic (The University of Melbourne), Richmond, Melbourne, Australia, and The Royal Brisbane and Women’s Hospital (The University of Queensland) Herston, Brisbane, Australia. The trial was funded by the Australian National Health and Medical Research Council (APP1048222) and was co-sponsored by FIT-BioCeuticals (who provided the study product but were uninvolved in all pre/post study aspects). The trial had ethics approval (TMC REC: 232; UQ MREC: 2014000702) and is registered with ANZCTR (protocol number: 12613001299796).
Inclusion/exclusion criteria
Participants who were currently depressed and not taking antidepressants for their depression were included. Specifically, eligible participants were aged between 18 and 75 years old, fulfilled the DSM-5 diagnostic criteria for current MDD, presented with mild-to-moderate depression (Montgomery-Åsberg Depression Rating Scale; MADRS, between 14 and 25) (Montgomery and Asberg 1979); met SAFER 2.0 criteria for participation in a clinical trial (testing pervasiveness, persistence, and pathology of MDD to ensure depression is valid and assessable) (Desseilles et al. 2013); and fluent in spoken and written English with the capacity to provide informed consent and follow the trial procedures.
Participants were excluded if they were currently taking an antidepressant, mood stabiliser or antipsychotic, or any mood-modulating nutraceuticals (e.g. St John’s wort or fish oil; two participants underwent a 1- to 2-week washout of fish oil and B vitamins before study commencement); presented with any suicidal ideation (> 1 on suicidal thoughts domain of the MADRS); had severe depression symptomatology (MADRS total score over 25) at time of study entry (an ethics committee requirement); had failed three or more trials of pharmacotherapy or somatic therapy for the current major depressive episode; met DSM-5 criteria for Bipolar I/II or Schizophrenia; met criteria for a primary diagnosis of a DSM-5 substance/alcohol use disorder within the past 12 months; recently commenced psychotherapy (stable treatment was acceptable; > 4 weeks); taking warfarin or phenytoin, had a known or suspected clinically unstable systemic medical disorder; were breastfeeding or pregnant; or were not using medically approved contraception if female and of childbearing age.
Outcome measures
Participants were screened for MDD and psychiatric comorbidities with the MINI 6.0 at the baseline visit (Sheehan et al. 1998). The primary outcome was change in MADRS depressive symptom severity score from baseline to follow-up. The secondary outcome measures were as follows: response (MADRS > 50% improvement) and remission (< 10 MADRS week-8 score), Beck depression inventory (BDI-II) (Beck et al. 1961); Hamilton Anxiety Rating Scale (HAMA) (Hamilton 1959); Short Form Survey-12 (SF-12) (Ware et al. 1996); Leeds Sleep Evaluation Questionnaire (LSEQ) (Parrott and Hindmarch 1980); the Systematic Assessment for Treatment Emergent Effects (SAFTEE) (Levine and Schooler 1986); and the Clinical Global Impression—Improvement (CGI-I) and Severity (CGI-S) (Guy 1976). The Sternbach (Sternbach 1991) and Hunter (Dunkley et al. 2003) Serotonin Toxicity Criteria were used to assess for potential (but unlikely) serotonin syndrome (assessing signs such as hyperthermia, myoclonus, tremor, hyperflexia, diaphoresis, agitation). Blinding was assessed via a blinding questionnaire at study endpoint asking participants whether they thought they received Active, Placebo, or were Unsure.
Biochemical analysis
A range of biomarkers were studied in exploratory analyses. Serum nutrient levels, serum BDNF protein levels, and targeted genotyping of one–carbon cycle genes were processed and analysed by Australian Clinical Labs Pty Ltd. in Melbourne, Australia. Serum BDNF was extracted from whole blood and analysed in triplicate using ELISA kits (anti-human BDNF-antibodies) at week 0 and 8. The mean of the three results for each participant was used. Eleven polymorphisms in 10 genes (Table 3) were assayed. DNA was extracted from whole blood using Qiagen, QIAmp mini-columns according to the manufacturer’s instruction and genotyping was performed by single-base extension assays and analysed on the Sequenom Massarray.
Randomisation and intervention
The randomisation schedule (computerised two-block randomisation, e.g. AABABBABABAA) and blinding were prepared by an independent researcher. Participants were randomly assigned to either SAMe or placebo and were blinded to group assignment. Trial researchers and investigators were also blinded to the randomisation. Participants were required to take two tablets, twice per day, with a daily dose of 800 mg/day of SAMe and pertinent one–carbon cycle cofactors—500 mcg/day of folinic acid and 200 mcg/day of vitamin B12. Placebos were matched externally and internally to the active tablets in colour, size, odour, and shape.
Procedure
Participants were recruited via social media, radio, television, internet advertising, medical referrals, and flyers in the community. Potential participants were screened over the phone to determine whether they might be eligible for the trial. They were asked to attend the trial site to confirm their eligibility. All participants provided informed consent to participate and were assessed for MDD using the MINI 6.0 and MADRS. Other health domain data collected included sleep, anxiety, general health, and perceived wellbeing (via self-report questionnaires and/or clinician administrated measures). If eligible, participants were randomly allocated to a treatment group and were asked to attend their local Australian Clinical Laboratories collection site to have a pre-treatment blood sample taken for nutrient biomarkers, BDNF, and SNP analyses. Participants attended a week-1 safety visit, and then fortnightly visits at the trial site (weeks 2, 4, 6, and 8), repeating the same self-report and clinician-administered measures. Participants were also asked about compliance and adverse effects at every visit. At the final visit (week 8), participants were asked to provide a post-treatment blood sample. A total of AU$100 was provided to participants to cover travel expenses to and from the trial sites over the 8 weeks. All participants who completed the study received a 2-month supply of SAMe tablets (approved by the ethics committee).
Statistical analysis
Initial analysis was conducted blind to group assignment. Sociodemographic and clinical characteristics were assessed for any baseline differences between groups using chi-squared tests for categorical variables and t tests for continuous variables. ANOVA models were utilised to test sociodemographic and clinical characteristics for associations with change in MADRS score (baseline to week 8). Employment status, general health, and baseline serum B12 concentrations each significantly predicted change in MADRS score. These variables were retained for use as covariates in the primary adjusted linear mixed-effects model (LMM). An a priori power calculation was not undertaken as this was specifically designed as a phase II pilot study seeking to recruit as many people as possible within our allocated budget. A target of 50 participants (SAMe arm vs placebo arm) was set in our protocol.
Analysis of primary and secondary outcomes was undertaken using LMMs. Models included main effects of time, group, and a group × time interaction (the latter indicating treatment effect). Covariates were included only in the primary model (MADRS). Random effects of subject (participant × site) as well as random intercepts and random slopes were utilised in each model. An autoregressive covariance structure suited the data based on visual inspection. Pertinent secondary analyses were also undertaken utilising LMMs. Due to the high prevalence of comorbid generalised anxiety disorder (GAD) in the sample, three-way interactions were tested to determine if comorbid GAD diagnosis moderated treatment response. Further exploratory secondary analyses were undertaken to determine if depression severity, one–carbon cycle biomarkers, BDNF, or SNPs moderated treatment response. In the case of depression severity, the sample was split at the median baseline MADRS score (23 points) rather than at defined severity thresholds (owing to clustering at higher scores), and sub-analyses were performed. Pearson and Spearman’s correlations were performed to assess the relationship between change in biomarker concentration over time and treatment response. A two-sided alpha level of less than 0.05 was regarded as being statistically significant. Statistical analysis was performed using the Statistical Package for Social Sciences software (SPSS, version 25.0, IBM, Chicago).
Results
Participant characteristics
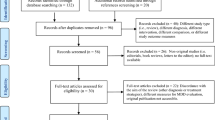
After screening and baseline visit exclusions, 49 participants were randomised to either SAMe (n = 25) or placebo (n = 24) (see CONSORT; Fig. 1). All but four participants attended at least one follow-up visit (n = 21 placebo, n = 24 SAMe). There were 22 study completers in the SAMe group (91.7%) and 19 in the placebo group (90.5%), χ2(1,49) = 0.699, p = 0.40. Sociodemographic and clinical characteristics of each group are displayed in Table 1. No significant between-group differences in any of these characteristics were noted. However, there was a greater frequency of comorbid GAD in the placebo group and a trend towards a greater number of lifetime episodes of depression in the SAMe group (albeit marginally non-significantly), X2(1,49) = 3.47, p = 0.062 and t(30) = − 0.199, p = 0.056 (Welch t test), respectively.

CONSORT flow diagram displaying participant flow from enrolment to trial completion
Primary outcome
Mean scores at week 8 and change scores from baseline for primary and secondary measures are displayed in Table 2. On the primary outcome, the MADRS, an adjusted LMM revealed a greater reduction, from baseline to week 8, of 3.76 points (and a medium to large Cohen’s d effect size of 0.72) for the SAMe group (see Fig. 2). This equated to a standardised mean difference of 0.63. However, the primary group × time interaction was found to be non-significant F(1,81) = 2.36, p = 0.13. This result was weakened when applying an unadjusted model F(1,101) = 1.11, p = 0.30. Results remained unchanged when including only participants whose compliance was rated as high.

MADRS, Montgomery-Asberg Depression Rating Scale; values are estimated marginal means derived from unadjusted linear mixed model
Secondary analyses
The sample was dichotomised at the median baseline MADRS score (< 23 points classed as mild; 23–25 points classed as severe) and sub-analyses were performed in each group to investigate whether depression severity moderated response. An unadjusted LMM revealed a marginally significant treatment effect in those with mild depression severity (F(1,36) = 4.29, p = 0.045), but no separation in those whose depression was more severe, F(1,46) = 0.005, p = 0.95. Furthermore, a comorbid GAD × group × time interaction was revealed on the MADRS, indicating that GAD moderated the relationship between treatment arm and response, with GAD participants (n = 19) more responsive to SAMe than placebo, F(1,81) = 4.59, p = 0.035. However, no interaction was present for baseline HAMA score, indicating that anxiety severity did not moderate response (p = 0.54). No gender effect was found with respect to males outperforming females, although the number of males in the study was low (n = 10).
Response (> 50% reduction in MADRS score) was achieved by 12 (54.5%) study completers in the SAMe group and 10 (52.6%) in the placebo group, Χ2 = 0.015, p = 0.90. Similarly, eight in the SAMe group (42.1%) and seven (31.8%) participants in the placebo group reached remission of depressive symptoms (MADRS score < 10) at study completion, Χ2 = 0.465, p = 0.50. There were no significant group × time interactions noted on the HAMA (F[1,105] = 0.388, p = 0.54), BDI (F[1,111] = 0.041, p = 0.64), CGI-S (F[1,87] = 0.885, p = 0.35), CGI-I (F(1,100) = 2.42, p = 0.12), or LSEQ (F[1,107] = 0.48,8, p = 0.51).
Genetic biomarkers, folate, B12, homocysteine, and BDNF
Baseline concentrations of folate, B12, homocysteine, and BDNF are displayed in Table 1. Homocysteine was higher in the active group at baseline, albeit non-significantly, t(31) = − 1.96, p = 0.059 (Welch’s t test). After treatment, B12 concentrations increased significantly more in the SAMe group (55.0% ± 37.1), than the placebo group (2.51% ± 17.2), t(28) = − 4.58, p < 0.001. Folate concentrations increased in the SAMe group (9.14% ± 22.1) and decreased in the placebo group (− 5.07% ± 32.4), although the difference was non-significant t(26) = − 1.42, p = 0.17. Conversely, homocysteine concentrations decreased in the SAMe group (− 5.67% ± 24.9) and increased in the placebo group (3.79% ± 20.4), which was also non-significant between groups, t(29) = 1.69, p = 0.10. Group × time × baseline biomarker interactions revealed that none of these indices moderated treatment response (all p > 0.05).
In the SAMe group, a significant correlation was found between change in folate concentration and response on the MADRS, r = − 0.57, p = 0.026. Specifically, increased folate concentrations were associated with greater treatment response. No correlations were noted between change in B12, BDNF, or HCY concentrations and response on the MADRS in the SAMe group (all p > 0.05). There were similarly no correlations for any markers in the placebo group (all p > 0.05). No genetic marker investigated in this study demonstrated any interaction with group and/or time (see Table 3 for list of SNPs).
Adverse events and blinding assessment
The mean total number of adverse events recorded on the SAFTEE across the trial was 19.2 ± 11.8 in the SAMe group and 17.0 ± 10.0 in the placebo group, which was not significantly different between groups, t(43) = − 0.630, p = 0.53. The most common adverse events recorded in both arms of the trial were as follows: drowsiness (SAMe = 21, placebo = 17), irritability (SAMe = 19, placebo = 17), weakness/fatigue (SAMe = 17, placebo = 18), trouble sleeping (SAMe = 15, placebo = 19), poor concentration (SAMe = 15, placebo = 13), headache (SAMe = 17, placebo = 10), feeling nervous or hyper (SAMe = 14, placebo = 12), difficulty finding words (SAMe = 14, placebo = 12), poor memory (SAMe = 14, placebo = 12), apathy (SAMe = 12, placebo = 13), abdominal discomfort (SAMe = 16, placebo = 8), and loss of sexual interest (SAMe = 14, placebo = 8). Although a trend towards significance was noted for abdominal discomfort being more common in the SAMe group (p = 0.055), a significantly different adverse event frequency was only noted for one adverse event—strange taste in the mouth (SAMe = 7, placebo = 0), which was significantly more common in the SAMe group, p = 0.010 (Fisher’s exact test). One serious adverse event (SAE) was recorded in the trial, which occurred in the placebo group. This SAE, involving hypertension which required a clinician visit, was recovered without sequelae. Due to the potential symptoms of serotonin syndrome not presenting, this was not formally assessed or diagnosed in any participant.
In respect to blinding assessment, at the end of the trial, 13 participants within the SAMe group (62%) believed they had received the active treatment, five (24%) believed they had received placebo, and three (14%) were unsure. In the placebo group, nine participants (47%) believed they had received the active treatment, five (26%) believed they had received placebo, and five (26%) were unsure. Perceived group allocation was therefore independent of actual group allocation χ2(2) = 1.13, p = 0.57.
Discussion
In this double-blind RCT, SAMe did not show a statistically significant reduction in MADRS score between groups, notwithstanding a medium to large effect size occurring in the adjusted model. To illustrate, the standardised mean difference between groups was greater than 0.50, which is generally considered to be a ‘clinically significant’ effect size in the literature (cf. Jakobsen et al. 2017). Despite this, the threshold of significance was not met in this study, potentially related to the high placebo response rate of 53% and the limited statistical power. As a preliminary phase II trial, however, a definitive determination of efficacy was not the purpose of this investigation, but rather the detection of a ‘signal’ which may warrant further investigation in a subsequent expanded trial. Although supported by the clinically significant effect sizes on the primary adjusted model, a similar signal of efficacy was not similarly found on secondary outcome measures. For example, no clear inter-group separation was noted in response or remission rates on the MADRS, nor was any effect evident on the BDI-II (clinician-rated scales tend to have stronger effect sizes).
Interestingly, when the sample was split at the median baseline MADRS score (23 points), sub-group analyses revealed a marginally significant treatment effect in those with milder depression, while no benefit was noted in those with more severe depression. This may suggest that SAMe is efficacious in cases of milder levels of depression, a potentially counter intuitive finding as most biological therapies are more efficacious in more unwell individuals (Khan et al. 2002). An interaction with GAD diagnosis was also noted, in which participants with GAD were more responsive to treatment than those without GAD (although baseline anxiety levels on the HAMA did not moderate treatment response). This is also a surprising result given that SAMe treatment has been associated with anxiety in those sensitive to its potential stimulatory effects (Sharma et al. 2017). It must be noted that statistical power in this analysis was restricted, and these latter findings which relied on sub-group analysis and three-way interactions may represent type I error, and thus should be interpreted with caution. No significantly beneficial effect was found preferentially for males (as found in our previous study; Sarris et al. 2015); however, there was a small sample of males in this present study, and thus, it was not powered properly for this sub-analysis.
As detailed in the “Introduction” section, recent reviews of SAMe in neuropsychiatric disorders have found overall encouraging (but low quality) evidence for the efficacy of SAMe for use in depression. Many of these studies were performed before the 1990s with heterogeneous dosing and administration (intravenous, intramuscular, and oral were all common) and consisted of samples in which depression may not have been the primary diagnosis. Our RCT, utilising a therapeutic dose of stable oral SAMe in participants with primary MDD, does not support the positivity endorsed in these previous reviews. Interestingly, results mirrored those reported in a large (n = 189) recent study which tested monotherapy SAMe against placebo and escitalopram (Mischoulon et al. 2014). In this study, remission rates on the HAM-D were notably higher in the treatment group (28%) than the placebo group (17%), but this difference similarly did not reach significance. The marked placebo response rate was also found in our recent RCT testing SAMe adjunctively with antidepressants in MDD (Sarris et al. 2018). It is possibly due to a high rate of expectancy bias driven by specific recruitment targeting from online recruitment measures (such as Facebook), and potentially the use of six time point visits (which may increase the therapeutic benefits of researcher interaction).
Strengths of the study include its rigorous design and strict inclusion and exclusion criteria. The study also assessed the nutraceutical in a potentially appropriate formulation (cofactors added with the SAMe, and being enteric coated, blister-packed, and refrigerated) in a sample of participants who were determined to possess primary MDD by clinically trained researchers. Further, there were various novel elements in the study design, including the assessment of pertinent nutrient and genetic markers to investigate whether treatment response may be moderated or correlated with these biomarkers. It is however recognised that a higher dose and longer length of intervention may have been of further benefit, and that the sample size was modest and limited to subjects with mild-to-moderate levels of depression. Further, between-group differences were weaker in the unadjusted model than in the adjusted model. Finally, it must be recognised that the operative biological pathways of SAMe action in depression are not singular nor completely defined; thereby, the biomarkers explored in this analysis may not truly reflect the underlying mechanisms of action or modifying biological factors.
In summation, although the differential reduction in depression symptoms observed between groups may be clinically relevant, this study did not find SAMe to be statistically significant in treating depression. This could potentially be due to the modest sample size and pronounced placebo response, with a larger study aiming also to minimise expectancy bias, being an important next step.
References
Arnold O, Saletu B, Anderer P et al (2005) Double-blind, placebo-controlled pharmacodynamic studies with a nutraceutical and a pharmaceutical dose of ademetionine (SAMe) in elderly subjects, utilizing EEG mapping and psychometry. Eur Neuropsychopharmacol 15:533–543. https://doi.org/10.1016/j.euroneuro.2005.01.004
Beck AT, Ward C, Mendelson M (1961) Beck depression inventory (BDI). Arch Gen Psychiatry 4:561–571
Cuthbert BN, Insel TR (2013) Toward the future of psychiatric diagnosis: the seven pillars of RDoC. BMC Med 11. https://doi.org/10.1186/1741-7015-11-126
De Sousa DP, Silva RHN, Da Silva EF, Gavioli EC (2017) Essential oils and their constituents: an alternative source for novel antidepressants. Molecules. 22. https://doi.org/10.3390/molecules22081290
Desseilles M, Witte J, Chang TE, Iovieno N, Dording C, Ashih H, Nyer M, Freeman MP, Fava M, Mischoulon D (2013) Massachusetts general hospital SAFER criteria for clinical trials and research. Harv Rev Psychiatry 21:269–274. https://doi.org/10.1097/HRP.0b013e3182a75cc7
Dunkley EJ, Isbister GK, Sibbritt D et al (2003) The hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 96:635–642
Duru G, Fantino B (2008) The clinical relevance of changes in the Montgomery–Asberg depression rating scale using the minimum clinically important difference approach. Curr Med Res Opin 24:1329–1335. https://doi.org/10.1185/030079908X291958
Frodl T (2017) Recent advances in predicting responses to antidepressant treatment. F1000Research. https://doi.org/10.12688/f1000research.10300.1
Galizia I, Oldani L, Macritchie K, Amari E, Dougall D, Jones TN, Lam RW, Massei GJ, Yatham LN, Young AH, Cochrane Common Mental Disorders Group (2016) S-adenosyl methionine (SAMe) for depression in adults. Cochrane Database Syst Rev
Guy W (1976) Clinical global impression scale. ECDEU assess man psychopharmacol Vol DHEW Publ No ADM 76 338:218–222
Hamilton M (1959) Hamilton anxiety rating scale (HAM-A). J Med 61:81–82. https://doi.org/10.1145/363332.363339
Jakobsen JC, Katakam KK, Schou A, Hellmuth SG, Stallknecht SE, Leth-Møller K, Iversen M, Banke MB, Petersen IJ, Klingenberg SL, Krogh J, Ebert SE, Timm A, Lindschou J, Gluud C (2017) Selective serotonin reuptake inhibitors versus placebo in patients with major depressive disorder. A systematic review with meta-analysis and Trial Sequential Analysis. BMC Psychiatry 17. https://doi.org/10.1186/s12888-016-1173-2
Khan A, Leventhal RM, Khan SR, Brown WA (2002) Severity of depression and response to antidepressants and placebo: an analysis of the Food and Drug Administration database. J Clin Psychopharmacol 22:40–45
Levine J, Schooler NR (1986) SAFTEE: a technique for the systematic assessment of side effects in clinical trials. Psychopharmacol Bull 22:343–381. https://doi.org/10.1093/jnci/djm189
Mischoulon D, Price LH, Carpenter LL, Tyrka AR, Papakostas GI, Baer L, Dording CM, Clain AJ, Durham K, Walker R, Ludington E, Fava M (2014) A double-blind, randomized, placebo-controlled clinical trial of S-adenosyl-L-methionine (SAMe) versus escitalopram in major depressive disorder. J Clin Psychiatry 75:370–376. https://doi.org/10.4088/JCP.13m08591
Montgomery SA, Asberg M (1979) A new depression scale designed to be sensitive to change. Br J Psychiatry 134:382–389
Parrott AC, Hindmarch I (1980) The leeds sleep evaluation questionnaire in psychopharmacological investigations-a review. Psychopharmacology 71:173–179. https://doi.org/10.1007/BF00434408
Sarris J, Panossian A, Schweitzer I, Stough C, Scholey A (2011) Herbal medicine for depression, anxiety and insomnia: a review of psychopharmacology and clinical evidence. Eur Neuropsychopharmacol 21:841–860. https://doi.org/10.1016/j.euroneuro.2011.04.002
Sarris J, Papakostas GI, Vitolo O et al (2014) S-Adenosyl methionine (SAMe) versus escitalopram and placebo in major depression: efficacy and effects of histamine and carnitine as moderators of response. J Affect Disord 164:76–81
Sarris J, Price LH, Carpenter LL, Tyrka A, Ng C, Papakostas G, Jaeger A, Fava M, Mischoulon D (2015) Is S-adenosyl methionine (SAMe) for depression only effective in males? A re-analysis of data from a randomized clinical trial. Pharmacopsychiatry 48:141–144. https://doi.org/10.1055/s-0035-1549928
Sarris J, Murphy J, Mischoulon D, Papakostas GI, Fava M, Berk M, Ng CH (2016) Adjunctive nutrient nutraceuticals for depression: a systematic review and meta-analyses. Am J Psychiatry 173:575–587
Sarris J, Byrne GJ, Bousman C, Stough C, Murphy J, MacDonald P, Adams L, Nazareth S, Oliver G, Cribb L, Savage K, Menon R, Chamoli S, Berk M, Ng C, Mischoulon D (2018) Adjunctive S-adenosylmethionine (SAMe) in treating non-remittent major depressive disorder: an 8-week double-blind, randomized, controlled trial. Eur Neuropsychopharmacol 28:1126–1136. https://doi.org/10.1016/j.euroneuro.2018.07.098
Sharma A, Gerbarg P, Bottiglieri T, Massoumi L, Carpenter LL, Lavretsky H, Muskin PR, Brown RP, Mischoulon D, as Work Group of the American Psychiatric Association Council on Research (2017) S-adenosylmethionine (SAMe) for neuropsychiatric disorders: a clinician-oriented review of research. J Clin Psychiatry 78:e656–e667
Sheehan DV, Lecrubier Y, Sheehan KH et al (1998) The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry 59:34–57
Sternbach H (1991) The serotonin syndrome. Am J Psychiatry 148:705–713. https://doi.org/10.1176/ajp.148.6.705
Ware JE, Kosinski M, Keller SD (1996) A 12-item short-form health survey: construction of scales and preliminary tests of reliability and validity. Med Care 34:220–233. https://doi.org/10.1097/00005650-199603000-00003
Acknowledgments
We would like to thank the study participants who contributed to this project.
Funding
This study is funded by a National Health and Medical Research Council (NHMRC) project grant (APP1048222), and is co-sponsored by FIT-BioCeuticals. Jerome Sarris is supported by an NHMRC Fellowship (APP1125000). Michael Berk is supported by a National Health and Medical Research Council (NHMRC) Senior Principal Research Fellowship (APP1059660 and APP1156072). Study sponsors had no role in study design, in the collection and analysis of data, manuscript preparation, nor the decision to submit the paper for publication.
Author information
Authors and Affiliations
Contributions
Author Sarris designed the study. Authors Sarris, Byrne, Bousman, Stough, Berk, Ng, Chamoli, Menon, and Mischoulon contributed to the design and write-up of the protocol. Authors Sarris, Bousman, and Cribb designed and implemented the statistical analysis. Authors Murphy, Cribb, MacDonald, Adams, Nazareth, Oliver, and Savage were involved in the data collection and entry. All authors have contributed to and approved the final manuscript.
Corresponding author
Ethics declarations
The trial had ethics approval (TMC REC: 232; UQ MREC: 2014000702) and is registered with ANZCTR (protocol number: 12613001299796).
Conflict of interest
JS has received honoraria, research support, royalties, or consultancy or travel grant funding from Integria Healthcare & MediHerb, Pfizer, Scius Health, Key Pharmaceuticals, Taki Mai, FIT-BioCeuticals, Blackmores, Soho-Flordis, Australian Natural Therapeutics Group, Grünbiotics, Healthworld, HealthEd, HealthMasters, SPRIM, Elsevier, Chaminade University, International Society for Affective Disorders, Complementary Medicines Australia, Terry White Chemists, ANS, Society for Medicinal Plant and Natural Product Research, Sanofi-Aventis, Omega-3 Centre, the National Health and Medical Research Council, and CR Roper Fellowship. DM has received research support from Nordic Naturals. He has received honoraria for consulting, speaking, and writing from the Massachusetts General Hospital Psychiatry Academy, Blackmores, and PeerPoint Medical Education Institute, LLC. He has provided unpaid consulting for Pharmavite LLC and Gnosis USA, Inc. He has received royalties from Lippincott Williams & Wilkins for published book ‘Natural Medications for Psychiatric Disorders: Considering the Alternatives.’ MB has received Grant/Research Support from the NIH, Cooperative Research Centre, Simons Autism Foundation, Cancer Council of Victoria, Stanley Medical Research Foundation, MBF, NHMRC, Beyond Blue, Rotary Health, Geelong Medical Research Foundation, Bristol-Myers Squibb, Eli Lilly, Glaxo SmithKline, Meat and Livestock Board, Organon, Novartis, Mayne Pharma, Servier, Woolworths, Avant, and the Harry Windsor Foundation; has been a speaker for Astra Zeneca, Bristol-Myers Squibb, Eli Lilly, Glaxo SmithKline, Janssen-Cilag, Lundbeck, Merck, Pfizer, Sanofi Synthelabo, Servier, Solvay, and Wyeth; and served as a consultant to Allergan, Astra Zeneca, Bioadvantex, Bionomics, Collaborative Medicinal Development, Eli Lilly, Grunbiotics, Glaxo SmithKline, Janssen-Cilag, LivaNova, Lundbeck, Merck, Mylan, Otsuka, Pfizer, and Servier. CS has received research grants from Government: Australian Research Council; NHMRC; NDLERF; NIDA; VDLERF; ARC; Vicpolice; Vicroads; Nestle; SFI; Barry Callebaut; Blackmores; Flordis; Zeller; Clover Corporation; Horphag; GSK; Bayer; Pharmalink; Siemens; Securatek; Efamol; Cognis; Integria; Medvet, Cooper Nutrition, and paid strategic consulting advice for SFI, Blackmores, Bayer; ARU, Carlton FC, GSK India. DM has received research support from Nordic Naturals. He has provided unpaid consulting for Pharmavite LLC and Gnosis USA, Inc. He has received honoraria for speaking from the Massachusetts General Hospital Psychiatry Academy, from PeerPoint Medical Education Institute LLC, and from Blackmores. He has received royalties from Lippincott Williams & Wilkins for published book ‘Natural Medications for Psychiatric Disorders: Considering the Alternatives.’ GJB receives research support from Janssen-Cilag, Eli Lilly, Biogen, NHMRC (APP1104460, APP1095227, APP1063383, APP1048222) and the RBWH Foundation. He owns shares in CSL. He receives royalties from the sale of his books Community Mental Health for Older People and Psychosocial Dimensions of Medicine. He receives licensing fees from the use of his rating scale, the Geriatric Anxiety Inventory. He is employed by the University of Queensland, the Royal Brisbane & Women’s Hospital, and the Repatriation Medical Authority. CN had served as a consultant for Lundbeck, Grunbiotics, Servier, Janssen-Cilag, Wyeth and Eli Lilly, received research grant support from Wyeth and Lundbeck, and speaker honoraria from Servier, Lundbeck, Bristol-Myers Squibb, Organon, Eli Lilly, GlaxoSmithKline, Janssen-Cilag, Astra-Zenaca, Wyeth, and Pfizer.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sarris, J., Murphy, J., Stough, C. et al. S-Adenosylmethionine (SAMe) monotherapy for depression: an 8-week double-blind, randomised, controlled trial. Psychopharmacology 237, 209–218 (2020). https://doi.org/10.1007/s00213-019-05358-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-019-05358-1