
Abstract
Background
Adolescent alcohol exposure may increase depression vulnerability in adulthood by increasing the anhedonic response to stress.
Methods
Male Wistar rats (postnatal days 28–53) were exposed to binge-like adolescent intermittent ethanol (AIE) or water. In adulthood, rats were exposed to social defeat, consisting of daily confrontations with an aggressive conspecific, followed by testing of brain reward function in a discrete-trial current-intensity intracranial self-stimulation procedure for 10 consecutive days. Neurochemistry and corticotropin-releasing factor (CRF) and CRF receptor 1 (CRFR1) mRNA levels were assessed in corticolimbic brain areas on day 11 of social defeat stress.
Results
Social defeat elevated reward thresholds in both AIE- and water-exposed rats indicating stress-induced anhedonia. However, AIE-exposed rats were more likely to show threshold elevations after repeated stress compared to water-exposed rats. AIE exposure decreased CRF mRNA levels in the nucleus accumbens and increased CRFR1 mRNA levels in the prefrontal cortex, while stress increased CRF mRNA levels in the central amygdala. In the caudate putamen, AIE exposure decreased dopamine turnover, while stress increased glutamate and serotonin metabolism and turnover.
Conclusions
These results demonstrate increased risk of repeated stress-induced anhedonia after AIE exposure, an effect that may be due to alterations in brain CRF and dopamine systems. These results suggest that the increased rates of depression reported in people with a history of adolescent alcohol exposure may be related to alterations in brain reward and stress systems that may contribute to increased stress-induced anhedonia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The high prevalence of alcohol binge drinking at very high intoxication levels among adolescents is an important public health concern (Johnston et al. 2014; Schuckit et al. 2014; Windle et al. 2008). Ethanol consumption at neurotoxic doses during adolescence causes damage to frontal cortical brain areas undergoing maturation (Bava and Tapert 2010; Jacobus and Tapert 2013; Squeglia et al. 2014). Considering that corticolimbic brain areas mediate reward processing and depression (Koob and Volkow 2010; Russo and Nestler 2013), underage drinking may lead to altered responses to reward (Migliorini et al. 2013) and an increased risk of major depression (Boden and Fergusson 2011; Briere et al. 2014) later in life. A few longitudinal studies have found causal links between heavy ethanol use during adolescence and the onset of depressive symptoms (Fergusson et al. 2009), as well as increased frequency of depressive symptoms among adolescents and young adults (Paschall et al. 2005). However, the long-term effects of adolescent exposure to ethanol on susceptibility to depression and the underlying neurobiological mechanisms remain largely unknown.
Our previous work in rats demonstrated that adolescent intermittent ethanol (AIE) exposure produced long-term alterations in brain reward function manifested behaviorally as diminished reward deficits or anhedonia, a core symptom of depression (American Psychiatric Association, 2013), during ethanol withdrawal in adult male rats (Boutros et al. 2014a). In rodents, AIE-induced alterations in reward processes can be attributed to dysregulated neurotransmission in corticolimbic reward circuits. For example, levels of dopamine and norepinephrine were decreased in cortical regions and increased in subcortical brain structures following adolescent ethanol exposure (Badanich et al. 2007; Boutros et al. 2014b; Pascual et al. 2009; Philpot et al. 2009). Serotonin levels in the dorsal raphe nucleus were increased (Evrard et al. 2006), and glutamate/GABA transmission balance was altered after exposure to ethanol during adolescence (Fleming et al. 2012; Fleming et al. 2013; Gomez et al. 2012; Silveri et al. 2014).
Stress can precipitate the onset and increase the severity of depression in humans (Kessler 1997). In rodents, AIE exposure produced persistent adaptations in stress systems including corticotropin-releasing factor (CRF), which plays a pivotal role in the stress response, alcohol dependence, and depression (Binder and Nemeroff 2010; George et al. 2012a; Paez-Pereda et al. 2011; Zorrilla et al. 2014). Specifically, AIE exposure led to blunted basal CRF levels in the hypothalamic-pituitary-adrenal (HPA) axis (Allen et al. 2011), reduced CRF cell counts in the central amygdala (Gilpin et al. 2012), as well as a blunted CRF response following an acute physiological stressor (ethanol challenge) (Logrip et al. 2013). Considering that CRF and dopaminergic neurotransmission are interconnected in corticostriatal reward pathways (George et al. 2012a; Wise and Morales 2010), we hypothesized that AIE exposure may alter susceptibility to stress-induced anhedonia during adulthood.
In the present studies, we investigated the long-term effects of AIE exposure on brain reward function in response to acute and repeated stress induced by social defeat in adult Wistar male rats. There are two key alcohol use patterns among human adolescents: (1) early initiation of use during adolescence and (2) high rates of binge drinking that are particularly prevalent late in adolescence. Recent work has begun to dissect effects of AIE exposure that are specific to age of exposure (e.g., early vs late adolescence; for review, see Spear 2015). In the present studies, rats were exposed to AIE throughout adolescence (postnatal day (PND) 28–53) starting at a particularly young age to model extreme binge drinking in early adolescence (e.g., > 20% of 12th graders report consumption of 5+ drinks/occasion within the past 2 weeks, 10.5% report consumption of 10+ drinks, and 5.6% report consuming 15+ drinks (Patrick et al. 2013)). The social defeat procedure has been used as a stressor to produce depression-like behavior in rodents. Social stress-induced reward deficits, as reflected by elevated reward thresholds (Der-Avakian et al. 2014; Donahue et al. 2014), can be reversed by administration of antidepressant medications (Der-Avakian et al. 2014), suggesting that the anhedonic state after social defeat may model key features of stress-induced depression in humans. Considering that interaction between stressful life events and the CRF gene predicted both adolescent heavy alcohol use (Blomeyer et al. 2008; Schmid et al. 2010) and major depression (Polanczyk et al. 2009; Wasserman et al. 2008; Wasserman et al. 2009), we assessed both CRF and CRF1 receptor (CRFR1) mRNA levels in the nucleus accumbens (NAc), prefrontal cortex (PFC), and central amygdala (CeA) after AIE exposure and social defeat. To assess the broad effects of AIE exposure and social defeat on brain neurochemistry, we measured monoamine, glutamate, and GABA levels in the caudate putamen (CPu) using high-performance liquid chromatography (HPLC) that allows for assessments of changes in multiple neurotransmitter systems from the same sample.
Methods
Subjects
Wistar male rats (Charles River, Raleigh, NC) arrived in the vivarium on PND 9 in groups of four males together with a nursing female. Pups were weaned on PND 21 and assigned to four experimental groups (water-no stress, water-stress, AIE-no stress and AIE-stress) with one rat from each litter assigned to each experimental group. The rats were pair-housed in a humidity- and temperature-controlled vivarium on a 12 h/12 h reverse light/dark cycle. Food and water were available ad libitum. All of the procedures were conducted in accordance with the guidelines of the American Association for the Accreditation of Laboratory Animal Care and the National Research Council’s Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of California, San Diego.
AIE exposure
A timeline of the experimental events, including AIE exposure, behavioral testing, repeated social defeat exposure and brain sample collection, is presented in Fig. 1.

Timeline of experimental design showing the sequence of AIE exposure, training and testing in the intracranial self-stimulation (ICSS) procedure, exposure to social defeat and brain sample collection across the lifespan of the rats. See text for details
The AIE exposure regimen has been described in detail in our previously published work (Boutros et al. 2014b). Briefly, rats were administered either water (control rats) or 5 g/kg (v/v) 25% ethanol intragastrically via oral gavage three times a day in a 2 days on, 2 days off exposure regimen throughout adolescence (PND 28–53). During AIE exposure, rats were observed for behavioral intoxication signs (BIS) before each ethanol administration, and doses were adjusted according to BIS as previously described (Boutros et al. 2014a; Majchrowicz 1975; Mejia-Toiber et al. 2014). This AIE exposure regimen resulted in high blood ethanol concentrations (215.07 mg/dl) but low levels of behavioral intoxication (See Supplementary Methods and Results).
ICSS procedure
A discrete-trial current-intensity ICSS procedure was used. Details of the surgery, ICSS chambers, training and testing procedures have been described elsewhere (Boutros et al. 2014a; Der-Avakian et al. 2014). Briefly, rats were prepared with bilateral electrodes directed at the lateral hypothalamus and trained to respond for rewarding electrical stimulation. During each session, the electrical current was varied in descending and ascending series to determine a minimal electrical current that elicited a positive operant response, termed the reward threshold. Elevations in reward thresholds are operationally defined as an anhedonic response.
Social defeat procedure
Details of the social defeat procedure have been described elsewhere (Der-Avakian et al. 2014). Briefly, after stable reward thresholds were established, both AIE-exposed and water-exposed rats were subjected to either stress (11 days of social defeat) or no stress. Using a resident/intruder procedure, the stress-exposed rats (i.e., intruders) were housed in a separate compartment within the cage of a reproductive pair of rats and any pre-weanling pups (i.e., residents). Resident Long-Evans male rats were previously selected for highly aggressive behavior based on the presence of attacks against a novel male conspecific during three prescreening tests (i.e., direct exposure to a new rat once a day for three consecutive days). Each day, the female resident and pups were removed from the cage and the barrier separating the intruder rat from the male resident rat was removed. The interaction between the rats lasted for 3 min, or until the intruder rat adopted a supine defeat position. The rats were immediately tested in the ICSS procedure after each of the first 10 daily social defeat sessions and returned to the cage of another resident pair afterward. Resident/intruder pairings were alternated daily. Non-stressed control rats were briefly handled daily before ICSS testing.
Corticosterone levels
Plasma corticosterone levels were determined on day 1 (tail-tip blood samples collected immediately after ICSS testing) and day 11 (trunk blood samples) of stress exposure. Plasma corticosterone levels were measured according to manufacturer’s instructions using a widely applied commercial double antibody RIA kit for rat and mouse samples (MPBiomedicals, Diagnostic Division, Orangeburg, NY, USA, catalog # 07120103).
Brain sample collection
On day 11, rats were exposed to social defeat only (i.e., without ICSS testing). All rats (except resident males and females which were maintained in the colony) were humanely euthanized by decapitation without anesthesia, and brains were snap frozen 150 min after stress exposure to avoid immediate physiological effects of stress. Based on literature findings, significant levels of CRF mRNA were reliably observed at 1 h (e.g., (Funk et al. 2006)) and 6 h (e.g., (Panksepp et al. 2007)) after exposure to social defeat. The PFC, NAc and CeA were dissected for analysis of CRF and CRFR1 mRNA levels. The CPu was dissected for analysis of monoamines, glutamate, and GABA. All brain areas were dissected with Harris micro-punch (1 mm) from coronal cryostat sections of 200-um slices for analysis.
Quantitative real-time PCR (qPCR)
DNase-treated RNA was isolated by “RNeasy Mini Kit” (Qiagen, Hilden, Germany) from the dissected PFC, NAc, and CeA regions. cDNA was synthesized using “High Capacity cDNA Reverse Transcription” kit (Life Technologies, CA, USA). For this study, actb (ID: Rn00667869_m1) was used as house-keeping gene (average % control ΔΔct calculation for CRF and CRFR1 for each treatment group). To examine the expression of CRF and CRFR1 mRNA levels, TaqMan qPCR was performed (Applied Biosystems, CA, USA). The TaqMan probes and primers used were actb (ID: Rn00667869_m1), crh (ID: Rn01462137_m1), and crhr1 (ID: Rn00578611_m1). Threshold cycle (Ct) values were measured for each primer and were compared using statistical analysis.
HPLC and analysis
Catecholamines and amino acids from brain tissue were measured by HPLC with electrochemical detection for catecholamines and fluorescence detection for amino acids (Groves et al. 2013; Kesby et al. 2016a; Kesby et al. 2016b; Kesby et al. 2009). Brain tissues were homogenized in 0.1 M perchloric acid with 50 ng/mL deoxyepinephrine (catecholamine internal standard) using probe sonication (Vibra-Cell, Sonics & Materials, CT, USA) and centrifuged at 13,000 rpm for 5 min. The supernatant was filtered by a 4-mm 0.22-μM nylon syringe filter (MicroSolv Technology Corporation, NJ, USA). For catecholamines, 10 μL of sample was injected into the HPLC system, which consisted of an autosampler (Dionex UltiMate 3000, Thermo Scientific, CA, USA), an isocratic HPLC pump (Model 584, ESA Laboratories, MA, USA), a Sunfire C18 column, (4.6 mm × 100 mm, 3 um; Waters Corporation, MA, USA), and a Coulochem III (ESA Laboratories) electrochemical detector. The mobile phase consisted of a 12% acetonitrile/50 mM citric acid and 25 mM potassium dihydrogen phosphate buffer containing 1 mM EDTA and 1.4 mM octane sulfonic acid adjusted to pH 4.3 with phosphoric acid. Flow rate was 0.5 ml/min. An analytical cell (Model 5014B, ESA Laboratories) with the first and second electrodes maintained at − 150 and + 300 mV, respectively, was used for detection. Amino acids were analyzed by HPLC using pre-column derivatization at 4 °C and fluorescence detection. The derivatization protocol was conducted by the autosampler as follows: 10 μL of 1 nM/μL homoserine (amino acid internal standard) was mixed with 10 μL of sample; then, 20 μL of borate buffer (0.4 M at pH 10) was added and mixed; then, 5 μL of OPA reagent (150 mg o-phthalaldehyde in 9 ml methanol with 1 ml borate buffer and 100 μl mercaptoethanol) was added and mixed; 1 μl of the final solution was injected into the system after a 45 s wait. The system consisted of an isocratic pump and autosampler (Dionex UltiMate 3000, Thermo Scientific), and fluorescence detector (Model 2475, Waters Corporation) equipped with a Phenomenex Gemini C18 column (4.6 mm × 150 mm, 3 um; Phenomenex, CA, USA). The mobile phase consisted of 0.05 M sodium acetate, tetrahydrofuran, and acetonitrile (74:1:25, v/v) adjusted to pH 4.0 using 100% acetic acid. Flow rate was 1 ml/min, and the fluorescence detector was set to an excitation wavelength of 337 nm and an emission wavelength of 454 nm. All data were stored and processed with Dionex Chromeleon software (version 7.2, Thermo Scientific). Data were quantified by calculating peak-area ratios of each compound compared to the relevant internal standard.
Statistical methods
All data were subjected to analysis of variance (ANOVA) using SPSS 18 (SPSS, Chicago, IL, USA). For ICSS data, thresholds were expressed as a percent of the mean absolute thresholds over the last 5 days of baseline testing to normalize variability of absolute thresholds that is commonly due to factors like electrode preparation and placement (Der-Avakian and Markou 2010; Der-Avakian et al. 2014). Rats were randomly assigned to stress and no stress groups such that both groups had similar average absolute thresholds before initiation of social defeat (see Results). Behavioral data were analyzed using three-way ANOVAs with AIE and Stress as the between-subject factors and ICSS Session as a within-subject factor. Due to large variability in the stress response, we performed chi-square analyses on the number of rats from each experimental group showing anhedonia, defined as threshold elevations greater than two standard deviations above pre-stress baseline thresholds averaged across all animals in both stress groups (i.e., AIE- and water-exposed) (Der-Avakian et al. 2014). Neurochemical data were analyzed using two-way ANOVAs with AIE and Stress as the between-subject factors. Significant main and interaction effects were followed by t tests using a Šidák adjustment for multiple comparisons. For repeated-measure analyses, Mauchly’s test of sphericity of the covariance matrix was applied. When the sphericity assumption was violated, the degrees of freedom for any term that involved that factor were adjusted to more conservative values by applying the Huynh-Feldt correction. We report the uncorrected degrees of freedom. The level of significance was set to 0.05.
For the neurochemical data, some brain samples were not viable in some regions, leading to varying sample sizes across the brain regions examined. In addition, outliers that differed from the group mean by more than two standard deviations were excluded from the neurochemical analyses (i.e., one outlier from each experimental group; total four outliers). Final sample sizes for each analysis are presented in the figures.
Results
During AIE exposure, the average blood alcohol concentration, ethanol dose, and behavioral intoxication were 215.17 mg/dl, 14.5 g/kg/day and 0.25, respectively (see Supplementary Results for details). There were no differences in baseline reward thresholds between AIE-exposed (140.98 ± 6.68 μA) and water-exposed (135.49 ± 6.45 μA) rats before social defeat was initiated.
Reward thresholds
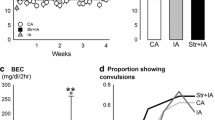
Rats exposed to social defeat had elevated reward thresholds compared to non-stressed rats (Fig. 2; main effect of Stress, F 1,40 = 16.87, p < .001; Stress × Session interaction, F 9,360 = 2.69, p < .05), with no effect of AIE exposure and no AIE exposure × Stress interaction.

Intracranial self-stimulation reward thresholds (expressed as a percent of baseline thresholds) during the 5 baseline days before social defeat and throughout the 10-day stress exposure period for AIE- and water-exposed rats. Data are presented as group mean ± SEM. Carat sign denotes a significant main effect of stress (^, p < .001). Asterisks denote a significant Stress × Session interaction (*, p < .05). bl baseline, d day
After acute stress (Fig. 3a), the proportion of AIE-stress rats with anhedonia (23%) did not differ from AIE-no stress rats (23%).Threshold elevations in this small proportion of non-stressed rats may have resulted from handling prior to the ICSS session. Similarly, there was no significant difference in the proportion of water-stress rats with anhedonia (67%) compared to water-no stress rats (22%). After 10 days of social defeat (Fig. 3b), a significantly greater proportion of AIE-stress rats exhibited anhedonia (85%) compared to AIE-no stress rats (0%; χ 2 3 = 28.02, p < .0001) and compared to AIE-stress rats after 1 day of social defeat (23%, χ 2 = 3.9, p < .05). The proportion of water-stress rats with anhedonia (67%) and water-no stress rats with anhedonia (22%) did not differ significantly and were identical to the proportions of water-exposed rats showing anhedonia after the first day of social defeat.

Percent of rats in each group showing anhedonia on the first day (a) and on the 10th day of social defeat stress (b). Carat sign depicts a difference between stress and non-stress groups based on a chi-square test (^, p < 0.0001)
CRF and CRFR1 analyses
Main effects of AIE exposure on CRF and CRFR1 mRNA levels were revealed in the ANOVAs (Fig. 4). Compared to water-exposed rats, AIE-exposed rats had significantly decreased levels of CRF mRNA in the NAc (Fig. 4a; F 1,34 = 5.20, p < .05) and significantly higher CRFR1 mRNA levels in the PFC (Fig. 4d; F 1,36 = 10.41, p < .005). There was no effect of AIE on CRF mRNA levels in the PFC (Fig. 4c) and CeA (Fig. 3e) or CRFR1 mRNA levels in the NAc (Fig. 4b) and CeA (Fig. 4f).

CRF and CRFR1 mRNA levels in the nucleus accumbens, prefrontal cortex, and central amygdala for each group. Data are presented as percent of water-no stress rats. a CRF in NAC. b CRFR1 in NAC. c CRF in PFC. d CRFR1 in PFC. e CRF in CeA. f CRFR1 in CeA. Asterisks denote significant main effects of AIE exposure, and carat denotes a significant main effect of stress
A main effect of Stress was detected for CRF mRNA levels in the CeA, with stress-exposed rats showing increased CRF mRNA levels compared to no stress rats (Fig. 4e; F 1,32 = 14.97, p < .001). There were no other significant effects of Stress on CRF mRNA levels or CRFR1 mRNA levels in other brain areas (Fig. 4). There were no AIE × Stress interactions detected in the ANOVAs.
Dopamine, norepinephrine, and serotonin in the caudate putamen
There were no significant differences between groups on any of the measures of norepinephrine, dopamine or their metabolites in the CPu (Table S2). However, independent of stress exposure, AIE-exposed rats exhibited decreased dopamine turnover, as demonstrated by a decreased DOPAC/DA ratio (Fig. 5a; main effect of AIE, F 1,35 = 4.31, p < .05) and a decreased DOPAC/HVA ratio (Fig. 5b; main effect of AIE, F 1,35 = 6.19, p < .05). Independent of AIE exposure, stress-exposed rats showed strong trends for higher levels of 5HIAA (main effect of Stress, F 1,35 = 3.73, p < .06; Table S2) and higher serotonin turnover as demonstrated by an increased 5HIAA/5HT ratio (main effect of Stress, F 1,35 = 3.97, p < .054).

Measures of dopamine turnover (a, b), glutamate levels (c), and glutamine/glutamate ratio (d) in the caudate putamen for each group of rats. All data are presented as mean ± SEM. * p < .05, ** p < .01, *** p < .001
Glutamate and GABA in the caudate putamen
Independent of AIE exposure, stress-exposed rats had higher levels of glutamate (main effect of Stress, F 1,35 = 6.00, p < .05; Fig. 5c) and decreased glutamate buffering as demonstrated by a decreased glutamine/glutamate ratio (main effect of Stress, F 1,35 = 18.07, p < .001; Fig. 5d) in the CPu. There was also a significant AIE × Stress interaction on the glutamine/glutamate ratio (F 1,35 = 6.61, p < .05), but no significant AIE × Stress interactions on the levels of glutamine, glutamate or GABA in the CPu (Table S3).
Plasma corticosterone analyses
Exposure to social defeat increased plasma corticosterone (Fig. S1) on day 1 (F 1,40 = 4.11, p < .05) and on day 11 (F 1,40 = 15.48, p < .001), with no differences between AIE-exposed and water-exposed rats.
Discussion
Exposure to social defeat increased reward thresholds in both AIE-exposed and water-exposed rats, indicating stress-induced reward deficits or anhedonia. Repeated stress was more likely to increase reward thresholds in AIE-exposed rats, while in water-exposed rats, the number of rats showing anhedonia was similar after acute and repeated stress exposures. Independent of stress exposure, AIE exposure decreased CRF mRNA levels in the NAc, increased CRFR1 mRNA levels in the PFC, and decreased dopamine turnover in the CPu. Independent of AIE exposure, repeated stress increased CRF mRNA in the CeA, increased glutamate levels, decreased glutamate buffering in the CPu and tended to increase serotonin turnover in the CPu. Furthermore, the effects of stress on glutamate function were more severe in water-exposed rats compared with AIE-exposed rats.
Repeated social defeat elevated reward thresholds in all rats indicating that stress-induced anhedonia was not more severe in AIE-exposed than in water-exposed rats. Consistent with these findings, plasma corticosterone levels were increased in all stressed rats with no difference between AIE- and water-exposed rats. Exposure to acute stress tended to increase the proportion of water-exposed rats that showed anhedonia but did not affect the proportion of AIE-exposed rats that showed anhedonia. These findings suggest that AIE exposure produced a blunted response to acute stress in adulthood. Repeated stress, however, significantly increased the proportion of AIE-exposed rats that showed anhedonia, while the proportion of water-exposed rats remained at the level observed during exposure to acute stress. Similar to these findings, rats that experienced maternal separation in the early postnatal period did not show acute stress-induced reward threshold elevations, but did show increased reward deficits after chronic stress (Der-Avakian and Markou 2010). Alternatively, AIE may have produced a late and persistent anhedonic response to acute social defeat that emerged only after several days of testing, suggesting that AIE may indeed produce vulnerability to acute stress in adulthood. It is unlikely that water-exposed rats would show such a protracted ICSS response to acute social defeat; however, this hypothesis must be tested in a separate cohort of rats exposed to AIE or water and a single social defeat session during adulthood. In addition, it is also possible that prior early life stressors (e.g., exposure to shipment/transport stress during the late first and early second postnatal week) may produce greater (or mitigated) effects in animals subsequently exposed to AIE and/or repeated social defeat. Together, these findings suggest that early life events may be protective against acute stress-induced anhedonia but may also increase vulnerability to chronic/repeated stress-induced anhedonia in adulthood. Thus, heavy alcohol use during adolescence in humans may increase the likelihood that chronic/repeated social stress will lead to anhedonia, a core feature of depression.
The PFC, which has been implicated in depression-like behavior (Klein et al. 2010), contains reciprocal projections with key reward and stress structures such as the extended amygdala, ventral tegmental area, NAc, and HPA axis (George et al. 2012a; Herman 2012). The role of CRF neurons in the PFC is not well understood. In the present study, AIE exposure increased levels of CRFR1 mRNA in the PFC. CRFR1 gene upregulation in the PFC may be a persistent result of elevated CRF levels experienced during repeated exposure to ethanol intoxications and withdrawals throughout adolescence. Consistent with this notion, CRF neurons in the PFC were activated by ethanol withdrawal (George et al. 2012b), and CRFR1 gene expression in the PFC was increased after in vitro incubation in CRF (Meng et al. 2011). Further, AIE exposure decreased levels of CRF mRNA in the NAc and had no long-term effects either on CRF mRNA or CRFR1 mRNA levels in the CeA. The latter finding is in contrast to published work showing increased CRF immunoreactivity in the CeA after AIE exposure (Allen et al. 2011; Gilpin et al. 2012). The discrepancies between our and previously published findings may be attributed to different AIE exposure regimens (ethanol vapor in the previously published work vs intragastric gavage in the present study), as well as to the fact that the present study examined CRF mRNA, whereas other reports included CRF peptide immunoreactivity, which could include fibers containing CRF that were synthesized elsewhere and which may reflect differences in processes other than synthesis (e.g., storage, release, degradation).
Decreased dopamine turnover has been implicated in alcohol-associated depression. That is, people with depression and a history of alcoholism showed lower levels of HVA in cerebrospinal fluid (indicating lower dopamine turnover) than either depressed patients without a history of alcoholism (Sher et al. 2003) or alcoholics with no diagnosis of depression (Roy et al. 1991). In the present study, we demonstrated that AIE exposure decreased the DOPAC/DA ratio and the DOPAC/HVA ratio in the CPu, without altering levels of dopamine or its metabolites. These findings support previous suggestions that decreased dopamine turnover may contribute to the expression of depression-like behavior (Kesby et al. 2009; Westerink 1985). Consistent with our findings, exposure to ethanol during adolescence decreased dopamine D1 receptor expression in the PFC (Pascual et al. 2009) and dopamine D2 receptor expression in the CPu, NAc and hippocampus (Pascual et al. 2009), and decreased tyrosine-hydroxylase immunoreactivity in the PFC (Boutros et al. 2014b). Furthermore, exposure to chronic social defeat stress decreased dopamine transmission in the NAc (Miczek et al. 2011). Future work is warranted to determine whether AIE exposure exacerbates the effects of chronic/repeated social defeat on dopamine levels in the NAc.
CRF acts within key reward- and stress-related brain structures to increase dopamine levels (George et al. 2012a). In the NAc, CRF and dopamine are co-localized on cell bodies and terminals, and acute administration of CRF within the NAc increased dopamine release (Lemos et al. 2012). Exposure to 2 days of swim stress abolished the dopamine-increasing effects of NAc CRF (Lemos et al. 2012). In the present study, AIE-exposed rats showed decreased CRF mRNA levels in the NAc along with decreased dopamine turnover in the CPu. These results suggest that the fundamental relationships between NAc CRF, dopamine, and reward may have been compromised by AIE exposure in ways that may have contributed to the increased likelihood of stress-induced anhedonia observed in AIE-exposed rats.
Effects of social defeat that were independent of AIE-exposure were also observed. CRF expression in the CeA is required for activation of the HPA axis in response to stress (Callahan et al. 2013) and subsequent stress-induced release of glucocorticoids. In the present study, stress exposure increased both CeA CRF mRNA levels and serum corticosterone levels. However, the limitation of the present study is that it does not allow to dissect the effects of repeated defeat from the effects of the most recent acute defeat or the effects of adaptation to the most recent acute defeat.
Neuronal circuits for chronic social defeat stress involve glutamatergic projections from the PFC and limbic forebrain that modulate ascending serotonin projections (Miczek et al. 2008). Stress increases glutamatergic outflow in several depression-related brain areas, including the PFC and hippocampus, an effect that is mediated by glucocorticoids (Moghaddam et al. 1994; Popoli et al. 2012). Similarly, in our study, social defeat increased glutamate levels in the CPu of water-exposed rats. However, in AIE-exposed rats, the stress-induced increase in glutamate levels was partially blunted, suggesting disruption of the glutamatergic adaption to repeated stress in the CPu. Interestingly, chronic social defeat decreased glutamate levels in the ventral striatum, particularly in mice showing susceptibility to the stressor as determined by increased social avoidance (Bagot et al. 2015). Moreover, optogenetic stimulation of PFC glutamatergic neurons, which acts to increase glutamate release in the striatum, prevented the consequences of social defeat in susceptible mice (Covington et al. 2010), suggesting that decreased striatal glutamate is critically involved in mediating the behavioral consequences of social defeat. While the importance of glutamatergic regulation of the CPu during stress is unclear, it is possible that decreased glutamatergic outflow in this area disrupts serotonergic processing. However, additional studies are necessary to explore the functional implications of such glutamatergic and serotonergic interactions in the CPu. Social defeat is known to increase levels of serotonin, its metabolites, and serotonin turnover in a number of brain regions (Blanchard et al. 2001). In the present study, there was a tendency for exposure to social defeat to increase levels of 5HIAA, the primary metabolite of serotonin, as well as serotonin turnover in the CPu in both AIE- and water-exposed rats.
Conclusions
In summary, our results indicate that AIE exposure may increase the likelihood that exposure to chronic/repeated stress in adulthood will result in anhedonia. Moreover, this increased vulnerability to the anhedonia-producing effects of chronic/repeated stress may be mediated by alterations to parts of the brain involved in reward and stress reactivity, of which glutamate, dopamine, and CRF are crucial components. The response of serotonergic systems to social defeat was not altered by AIE exposure. These results suggest that AIE exposure may have effects on brain and behavior that endure long after termination of ethanol exposure. Thus, while individuals with a history of binge alcohol exposure during adolescence may appear to have minimal deficits in adulthood, problems may arise upon exposure to sustained stressful situations that may more likely to produce anhedonia. This increased risk of stress-induced anhedonia may contribute to the high rates of depression reported in adults with a history of heavy alcohol use during adolescence (Briere et al. 2014; Edwards et al. 2014).
References
Allen CD, Rivier CL, Lee SY (2011) Adolescent alcohol exposure alters the central brain circuits known to regulate the stress response. Neuroscience 182:162–168. https://doi.org/10.1016/j.neuroscience.2011.03.003
American Psychiatric Association (2013) Diagnostic and statistical manual of mental disorders (5th ed.). Arlington, VA.
Badanich KA, Maldonado AM, Kirstein CL (2007) Chronic ethanol exposure during adolescence increases basal dopamine in the nucleus accumbens septi during adulthood. Alcohol Clin Exp Res 31(5):895–900. https://doi.org/10.1111/j.1530-0277.2007.00370.x
Bagot RC, Parise EM, Peña CJ, Zhang HX, Maze I, Chaudhury D, Persaud B, Cachope R, Bolaños-Guzmán CA, Cheer J, Deisseroth K, Han MH, Nestler EJ (2015) Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nat Commun 6:7062. https://doi.org/10.1038/ncomms8062
Bava S, Tapert SF (2010) Adolescent brain development and the risk for alcohol and other drug problems. Neuropsychol Rev 20(4):398–413. https://doi.org/10.1007/s11065-010-9146-6
Binder EB, Nemeroff CB (2010) The CRF system, stress, depression and anxiety-insights from human genetic studies. Mol Psychiatry 15(6):574–588. https://doi.org/10.1038/mp.2009.141
Blanchard RJ, McKittrick CR, Blanchard DC (2001) Animal models of social stress: effects on behavior and brain neurochemical systems. Physiol Behav 73(3):261–271. https://doi.org/10.1016/S0031-9384(01)00449-8
Blomeyer D, Treutlein J, Esser G, Schmidt MH, Schumann G, Laucht M (2008) Interaction between CRHR1 gene and stressful life events predicts adolescent heavy alcohol use. Biol Psychiatry 63(2):146–151. https://doi.org/10.1016/j.biopsych.2007.04.026
Boden JM, Fergusson DM (2011) Alcohol and depression. Addiction 106(5):906–914. https://doi.org/10.1111/j.1360-0443.2010.03351.x
Boutros N, Semenova S, Markou A (2014a) Adolescent intermittent ethanol exposure diminishes anhedonia during ethanol withdrawal in adulthood. Eur Neuropsychopharmacol 24(6):856–864. https://doi.org/10.1016/j.euroneuro.2014.01.022
Boutros N, Semenova S, Liu W, Crews FT, Markou A (2014b) Adolescent intermittent ethanol exposure is associated with increased risky choice and decreased dopaminergic and cholinergic neuron markers in adult rats. Int J Neuropsychopharmacol 18(2). https://doi.org/10.1093/ijnp/pyu003
Briere FN, Rohde P, Seeley JR, Klein D, Lewinsohn PM (2014) Comorbidity between major depression and alcohol use disorder from adolescence to adulthood. Compr Psychiatry 55(3):526–533. https://doi.org/10.1016/j.comppsych.2013.10.007
Callahan LB, Tschetter KE, Ronan PJ (2013) Inhibition of corticotropin releasing factor expression in the central nucleus of the amygdala attenuates stress-induced behavioral and endocrine responses. Front Neurosci 7:195. https://doi.org/10.3389/fnins.2013.00195
Covington HE 3rd, Lobo MK, Maze I, Vialou V, Hyman JM, Zaman S, LaPlant Q, Mouzon E, Ghose S, Tamminga CA, Neve RL, Deisseroth K, Nestler EJ (2010) Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J Neurosci 30(48):16082–16090. https://doi.org/10.1523/JNEUROSCI.1731-10.2010
Der-Avakian A, Markou A (2010) Neonatal maternal separation exacerbates the reward-enhancing effect of acute amphetamine administration and the anhedonic effect of repeated social defeat in adult rats. Neuroscience 170(4):1189–1198. https://doi.org/10.1016/j.neuroscience.2010.08.002
Der-Avakian A, Mazei-Robison MS, Kesby JP, Nestler EJ, Markou A (2014) Enduring deficits in brain reward function after chronic social defeat in rats: susceptibility, resilience, and antidepressant response. Biol Psychiatry 76(7):542–549. https://doi.org/10.1016/j.biopsych.2014.01.013
Donahue RJ, Muschamp JW, Russo SJ, Nestler EJ, Carlezon WA Jr (2014) Effects of striatal DeltaFosB overexpression and ketamine on social defeat stress-induced anhedonia in mice. Biol Psychiatry 76(7):550–558. https://doi.org/10.1016/j.biopsych.2013.12.014
Edwards AC, Heron J, Dick DM, Hickman M, Lewis G, Macleod J, Kendler KS (2014) Adolescent alcohol use is positively associated with later depression in a population-based U.K. cohort. J Stud Alcohol Drugs 75(5):758–765. 10.15288/jsad.2014.75.758
Evrard SG, Duhalde-Vega M, Tagliaferro P, Mirochnic S, Caltana LR, Brusco A (2006) A low chronic ethanol exposure induces morphological changes in the adolescent rat brain that are not fully recovered even after a long abstinence: an immunohistochemical study. Exp Neurol 200(2):438–459. https://doi.org/10.1016/j.expneurol.2006.03.001
Fergusson DM, Boden JM, Horwood LJ (2009) Tests of causal links between alcohol abuse or dependence and major depression. Arch Gen Psychiatry 66(3):260–266. https://doi.org/10.1001/archgenpsychiatry.2008.543
Fleming RL, Acheson SK, Moore SD, Wilson WA, Swartzwelder HS (2012) In the rat, chronic intermittent ethanol exposure during adolescence alters the ethanol sensitivity of tonic inhibition in adulthood. Alcohol Clin Exp Res 36(2):279–285. https://doi.org/10.1111/j.1530-0277.2011.01615.x
Fleming RL, Li Q, Risher ML, Sexton HG, Moore SD, Wilson WA, Acheson SK, Swartzwelder HS (2013) Binge-pattern ethanol exposure during adolescence, but not adulthood, causes persistent changes in GABAA receptor-mediated tonic inhibition in dentate granule cells. Alcohol Clin Exp Res 37(7):1154–1160. https://doi.org/10.1111/acer.12087
Funk D, Li Z, Le AD (2006) Effects of environmental and pharmacological stressors on c-fos and corticotropin-releasing factor mRNA in rat brain: relationship to the reinstatement of alcohol seeking. Neuroscience 138(1):235–243. https://doi.org/10.1016/j.neuroscience.2005.10.062
George O, Le Moal M, Koob GF (2012a) Allostasis and addiction: role of the dopamine and corticotropin-releasing factor systems. Physiol Behav 106(1):58–64. https://doi.org/10.1016/j.physbeh.2011.11.004
George O, Sanders C, Freiling J, Grigoryan E, Vu S, Allen CD, Crawford E, Mandyam CD, Koob GF (2012b) Recruitment of medial prefrontal cortex neurons during alcohol withdrawal predicts cognitive impairment and excessive alcohol drinking. Proc Natl Acad Sci U S A 109(44):18156–18161. https://doi.org/10.1073/pnas.1116523109
Gilpin NW, Karanikas CA, Richardson HN (2012) Adolescent binge drinking leads to changes in alcohol drinking, anxiety, and amygdalar corticotropin releasing factor cells in adulthood in male rats. PLoS One 7(2):e31466. https://doi.org/10.1371/journal.pone.0031466
Gomez R, Behar KL, Watzl J, Weinzimer SA, Gulanski B, Sanacora G, Koretski J, Guidone E, Jiang L, Petrakis IL, Pittman B, Krystal JH, Mason GF (2012) Intravenous ethanol infusion decreases human cortical gamma-aminobutyric acid and N-acetylaspartate as measured with proton magnetic resonance spectroscopy at 4 tesla. Biol Psychiatry 71(3):239–246. https://doi.org/10.1016/j.biopsych.2011.06.026
Groves NJ, Kesby JP, Eyles DW, McGrath JJ, Mackay-Sim A, Burne TH (2013) Adult vitamin D deficiency leads to behavioural and brain neurochemical alterations in C57BL/6J and BALB/c mice. Behav Brain Res 241:120–131. https://doi.org/10.1016/j.bbr.2012.12.001
Herman JP (2012) Neural pathways of stress integration: relevance to alcohol abuse. Alcohol Res 34(4):441–447
Jacobus J, Tapert SF (2013) Neurotoxic effects of alcohol in adolescence. Annual Review Clinical Psychology 9 9:703–721. https://doi.org/10.1146/annurev-clinpsy-050212-185610
Johnston LD, O’Malley PM, Miech RA, Bachman JG, Schulenberg JE (2014) Monitoring the future national survey results on drug use: 1975–2013: overview, key findings on adolecent drug use. Institute for Social Research, The Unversity of Michigan, Ann Arbor, 88 pp
Kesby JP, Markou A, Semenova S (2016a) The effects of HIV-1 regulatory TAT protein expression on brain reward function, response to psychostimulants and delay-dependent memory in mice. Neuropharmacology 109:205–215. https://doi.org/10.1016/j.neuropharm.2016.06.011
Kesby JP, Markou A, Semenova S, Group T (2016b) Effects of HIV/TAT protein expression and chronic selegiline treatment on spatial memory, reversal learning and neurotransmitter levels in mice. Behav Brain Res 311:131–140. https://doi.org/10.1016/j.bbr.2016.05.034
Kesby JP, Cui X, Ko P, McGrath JJ, Burne TH, Eyles DW (2009) Developmental vitamin D deficiency alters dopamine turnover in neonatal rat forebrain. Neurosci Lett 461(2):155–158. https://doi.org/10.1016/j.neulet.2009.05.070
Kessler RC (1997) The effects of stressful life events on depression. Annu Rev Psychol 48(1):191–214. https://doi.org/10.1146/annurev.psych.48.1.191
Klein J, Winter C, Coquery N, Heinz A, Morgenstern R, Kupsch A, Juckel G (2010) Lesion of the medial prefrontal cortex and the subthalamic nucleus selectively affect depression-like behavior in rats. Behav Brain Res 213(1):73–81. https://doi.org/10.1016/j.bbr.2010.04.036
Koob GF, Volkow ND (2010) Neurocircuitry of addiction. Neuropsychopharmacology 35(1):217–238. https://doi.org/10.1038/npp.2009.110
Lemos JC, Wanat MJ, Smith JS, Reyes BA, Hollon NG, Van Bockstaele EJ, Chavkin C, Phillips PE (2012) Severe stress switches CRF action in the nucleus accumbens from appetitive to aversive. Nature 490(7420):402–406. https://doi.org/10.1038/nature11436
Logrip ML, Rivier C, Lau C et al (2013) Adolescent alcohol exposure alters the rat adult hypothalamic-pituitary-adrenal axis responsiveness in a sex-specific manner. Neuroscience 235:174-186. https://doi.org/10.1016/j.neuroscience.2012.12.069
Majchrowicz E (1975) Induction of physical dependence upon ethanol and the associated behavioral changes in rats. Psychopharmacologia 43(3):245–254. https://doi.org/10.1007/BF00429258
Mejia-Toiber J, Boutros N, Markou A, Semenova S (2014) Impulsive choice and anxiety-like behavior in adult rats exposed to chronic intermittent ethanol during adolescence and adulthood. Behav Brain Res 266:19–28. https://doi.org/10.1016/j.bbr.2014.02.019
Meng QY, Chen XN, Tong DL, Zhou JN (2011) Stress and glucocorticoids regulated corticotropin releasing factor in rat prefrontal cortex. Mol Cell Endocrinol 342(1-2):54–63. https://doi.org/10.1016/j.mce.2011.05.035
Miczek KA, Yap JJ, Covington HE 3rd (2008) Social stress, therapeutics and drug abuse: preclinical models of escalated and depressed intake. Pharmacol Ther 120(2):102–128. https://doi.org/10.1016/j.pharmthera.2008.07.006
Miczek KA, Nikulina EM, Shimamoto A, Covington HE 3rd (2011) Escalated or suppressed cocaine reward, tegmental BDNF, and accumbal dopamine caused by episodic versus continuous social stress in rats. J Neurosci 31(27):9848–9857. https://doi.org/10.1523/JNEUROSCI.0637-11.2011
Migliorini R, Stewart JL, May AC, Tapert SF, Paulus MP (2013) What do you feel? Adolescent drug and alcohol users show altered brain response to pleasant interoceptive stimuli. Drug Alcohol Dependence 133:661–668. https://doi.org/10.1016/j.drugalcdep.2013.08.015
Moghaddam B, Bolinao ML, Stein-Behrens B, Sapolsky R (1994) Glucocorticoids mediate the stress-induced extracellular accumulation of glutamate. Brain Res 655(1-2):251–254. https://doi.org/10.1016/0006-8993(94)91622-5
Paez-Pereda M, Hausch F, Holsboer F (2011) Corticotropin releasing factor receptor antagonists for major depressive disorder. Expert Opin Investig Drugs 20(4):519–535. https://doi.org/10.1517/13543784.2011.565330
Panksepp J, Burgdorf J, Beinfeld MC, Kroes RA, Moskal JR (2007) Brain regional neuropeptide changes resulting from social defeat. Behav Neurosci 121(6):1364–1371. https://doi.org/10.1037/0735-7044.121.6.1364
Paschall MJ, Freisthler B, Lipton RI (2005) Moderate alcohol use and depression in young adults: findings from a national longitudinal study. Am J Public Health 95(3):453–457. https://doi.org/10.2105/AJPH.2003.030700
Pascual M, Boix J, Felipo V, Guerri C (2009) Repeated alcohol administration during adolescence causes changes in the mesolimbic dopaminergic and glutamatergic systems and promotes alcohol intake in the adult rat. J Neurochem 108(4):920–931. https://doi.org/10.1111/j.1471-4159.2008.05835.x
Patrick ME, Schulenberg JE, Martz ME, Maggs JL, O’Malley PM, Johnston LD (2013) Extreme binge drinking among 12th-grade students in the United States: prevalence and predictors. JAMA Pediatr 167(11):1019–1025. https://doi.org/10.1001/jamapediatrics.2013.2392
Philpot RM, Wecker L, Kirstein CL (2009) Repeated ethanol exposure during adolescence alters the developmental trajectory of dopaminergic output from the nucleus accumbens septi. Int J Dev Neurosci 27(8):805–815. https://doi.org/10.1016/j.ijdevneu.2009.08.009
Polanczyk G, Caspi A, Williams B, Price TS, Danese A, Sugden K, Uher R, Poulton R, Moffitt TE (2009) Protective effect of CRHR1 gene variants on the development of adult depression following childhood maltreatment: replication and extension. Arch Gen Psychiatry 66(9):978–985. https://doi.org/10.1001/archgenpsychiatry.2009.114
Popoli M, Yan Z, McEwen BS, Sanacora G (2012) The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci 13(1):22–37. https://doi.org/10.1038/nrn3138
Roy A, DeJong J, Lamparski D, George T, Linnoila M (1991) Depression among alcoholics: relationship to clinical and cerebrospinal fluid variables. Arch Gen Psychiatry 48(5):428–432. https://doi.org/10.1001/archpsyc.1991.01810290040007
Russo SJ, Nestler EJ (2013) The brain reward circuitry in mood disorders. Nat Rev Neurosci 14(9):609–625. https://doi.org/10.1038/nrn3381
Schmid B, Blomeyer D, Treutlein J, Zimmermann US, Buchmann AF, Schmidt MH, Esser G, Rietschel M, Banaschewski T, Schumann G, Laucht M (2010) Interacting effects of CRHR1 gene and stressful life events on drinking initiation and progression among 19-year-olds. Int J Neuropsychopharmacol 13(06):703–714. https://doi.org/10.1017/S1461145709990290
Schuckit MA, Smith TL, Danko GP, Bucholz KK, Agrawal A, Dick DM, Nurnberger JI Jr, Kramer J, Hesselbrock M, Saunders G, Hesselbrock V (2014) Predictors of subgroups based on maximum drinks per occasion over six years for 833 adolescents and young adults in COGA. J Stud Alcohol Drugs 75(1):24–34. 10.15288/jsad.2014.75.24
Sher L, Oquendo MA, Li S, Huang YY, Grunebaum MF, Burke AK, Malone KM, Mann JJ (2003) Lower CSF homovanillic acid levels in depressed patients with a history of alcoholism. Neuropsychopharmacology 28(9):1712–1719. https://doi.org/10.1038/sj.npp.1300231
Silveri MM, Cohen-Gilbert J, Crowley DJ, Rosso IM, Jensen JE, Sneider JT (2014) Altered anterior cingulate neurochemistry in emerging adult binge drinkers with a history of alcohol-induced blackouts. Alcohol Clin Exp Res 38(4):969–979. https://doi.org/10.1111/acer.12346
Spear LP (2015) Adolescent alcohol exposure: are there separable vulnerable periods within adolescence? Physiol Behav 148:122–130. https://doi.org/10.1016/j.physbeh.2015.01.027
Squeglia LM, Rinker DA, Bartsch H, Castro N, Chung Y, Dale AM, Jernigan TL, Tapert SF (2014) Brain volume reductions in adolescent heavy drinkers. Dev Cogn Neurosci 9:117–125. https://doi.org/10.1016/j.dcn.2014.02.005
Wasserman D, Sokolowski M, Rozanov V, Wasserman J (2008) The CRHR1 gene: a marker for suicidality in depressed males exposed to low stress. Genes Brain Behav 7(0):14–19. https://doi.org/10.1111/j.1601-183X.2007.00310.x
Wasserman D, Wasserman J, Rozanov V, Sokolowski M (2009) Depression in suicidal males: genetic risk variants in the CRHR1 gene. Genes Brain Behav 8(1):72–79. https://doi.org/10.1111/j.1601-183X.2008.00446.x
Westerink BH (1985) Sequence and significance of dopamine metabolism in the rat brain. Neurochem Int 7(2):221–227. https://doi.org/10.1016/0197-0186(85)90108-1
Windle M, Spear LP, Fuligni AJ, Angold A, Brown JD, Pine D, Smith GT, Giedd J, Dahl RE (2008) Transitions into underage and problem drinking: developmental processes and mechanisms between 10 and 15 years of age. Pediatrics 121(Suppl 4):S273–S289. https://doi.org/10.1542/peds.2007-2243C
Wise RA, Morales M (2010) A ventral tegmental CRF-glutamate-dopamine interaction in addiction. Brain Res 1314:38–43. https://doi.org/10.1016/j.brainres.2009.09.101
Zorrilla EP, Logrip ML, Koob GF (2014) Corticotropin releasing factor: a key role in the neurobiology of addiction. Front Neuroendocrinol 35(2):234–244. https://doi.org/10.1016/j.yfrne.2014.01.001
Acknowledgments
This work was supported by NIH grant: U01-AA019970-NADIA (AM) and U01-AA019973-NADIA (SL). The NIH had no role in the study design, data collection, data analysis, data interpretation, writing of the report, or decision to submit the article for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Nathalie Boutros and Andre Der-Avakian are first co-authors
Athina Markou and Svetlana Semenova are senior co-authors
Electronic supplementary material
ESM 1
(PDF 77 kb)
Rights and permissions
About this article

Cite this article
Boutros, N., Der-Avakian, A., Kesby, J.P. et al. Effects of adolescent alcohol exposure on stress-induced reward deficits, brain CRF, monoamines and glutamate in adult rats. Psychopharmacology 235, 737–747 (2018). https://doi.org/10.1007/s00213-017-4789-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-017-4789-0