
Abstract
Objective
This study aims to conduct an evidence review of the effectiveness, harms, and cost-effectiveness of pharmacogenomics-guided antidepressant treatment for major depressive disorder.
Methods
We searched MEDLINE®, the Cochrane Central Registry of Controlled Trials, and PsycINFO through February 2017. We used prespecified criteria to select studies, abstract data, and rate internal validity and strength of the evidence (PROSPERO number CRD42016036358).
Results
We included two randomized trials (RCT), five controlled cohort studies, and six modeling studies of mostly women in their mid-40s with few comorbidities. CNSDose (ABCB1, ABCC1, CYP2C19, CYP2D6, UGT1A1) is the only pharmacogenomics test that significantly improved remission (one additional remitting patient in 12 weeks per three genotyped, 95% CI 1.7 to 3.5) and reduced intolerability in an RCT. ABCB1 genotyping leads to one additional remitting patient in 5 weeks per three genotyped (95% CI 3 to 20), but tolerability was not reported. In an RCT, GeneSight (CYP2D6, CYPC19, CYP1A2, SLC6A4, HTR2A) did not statistically significantly improve remission, and evidence is inconclusive about its tolerability. Evidence is generally low strength because RCTs were few and underpowered. Cost-effectiveness is unclear due to lack of directly observed cost-effectiveness outcomes. We found no studies that evaluated whether pharmacogenomics shortens time to optimal treatment, whether improvements were due to switches to genetically congruent medication, or whether effectiveness varies based on test and patient characteristics.
Conclusions
Certain pharmacogenomics tools show promise of improving short-term remission rates in women in their mid-40s with few comorbidities. But, important evidence limitations preclude recommending their widespread use and indicate a need for further research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Clinicians have an intense need to identify additional factors to help them optimize the effectiveness of available antidepressants, a mainstay of treatment for major depressive disorder (MDD). To guide the choice of antidepressants, clinicians have typically taken a “trial and error” approach, informed by a number of clinical factors thought to be associated with treatment response. However, rates of remission are low and variable, with approximately 11–30% of patients remitting, even after 1 year of antidepressant treatment (Trivedi et al. 2006; Rost et al. 2002; Rush et al. 2004; Tansey et al. 2013). This is concerning, as treatment that falls short of remission is associated with continued disabling symptoms, higher rates of depression relapse and recurrence, poorer work productivity, more impaired psychosocial functioning, higher levels of health care use, and potentially higher risk for suicide (Trivedi et al. 2006).
Genetic variation has long been explored as a potential contributor to individual differences in antidepressant treatment outcome. Whether using genetic information can help predict how an individual might respond to a particular antidepressant—referred to as “pharmacogenomics”—is of great interest for further advancing precision medicine efforts. The clinical rationale behind using pharmacogenomics data to inform antidepressant therapy is that it may shorten the time to identifying optimal treatment by using a patient’s unique genetic profile to help predict level of tolerability or response to a drug, or help tailor the dose that may have the best potential effectiveness and tolerability.
Many pharmacogenomics tests are now available for clinical use (Drozda et al. 2014; Bousman and Hopwood 2016). These tests vary widely in the number and type of genes included, which medications they target, how they are regulated, their cost, and their results’ delivery methods (e.g., whether they are drug-focused or gene-focused; how much detail is provided about therapeutic implications, categorization of interaction, and clinical impact; and whether consultation with a professional genetic counselor and/or pharmacist is available to help the treating clinician interpret the results) (Bousman and Hopwood 2016). Although depression treatment guidelines either do not reference pharmacogenomics testing at all (Bauer et al. 2015; National Institute for Health and Care Excellence 2009) or mention it only briefly as an area for future research (American Psychiatric Association 2010; Department of Veterans Affairs: The Management of MDD Working Group 2016; Bauer et al. 2013), these tests are being marketed directly to patients and clinicians (de Leon 2016; Howland 2014b). This is of concern, because as noted by Bousman and Hopwood in their recent Personal View article in Lancet Psychiatry, “the majority of psychiatrists are unfamiliar with these tools or have limited time to critically assess each of them for usefulness in their psychiatry practice” (Bousman and Hopwood 2016).
The purpose of our review is to critically appraise and synthesize the literature on the clinical utility of pharmacogenomics testing-guided antidepressant treatment of MDD. While some recent review articles partially overlap with ours (ECRI Institute 2015b; ECRI Institute 2015a; Singh et al. 2014; Howland 2014a; Bousman and Hopwood 2016; Rosenblat et al. 2017; Berm et al. 2016), none have evaluated the complete range of pharmacogenomics tools for MDD or formally graded the strength of available evidence.
Methods
We conducted this review according to a prospectively registered protocol that we developed based on established methodological standards (Agency for Healthcare Research and Quality 2014) and input from clinical content experts (PROSPERO database, CRD42016036358). The Department of Veterans Affairs (VA) Evidence-based Synthesis Program (ESP) originally conducted this review for the VA Office of Research and Development (ORD). The full evidence report (Peterson et al. 2016), published on our website, provides complete details of our methods and more comprehensive data abstraction and risk of bias and strength of evidence ratings.
Topic development
Areas of particular relevance for evaluating evidence for use of genetic tests include Analytic validity, Clinical validity, Clinical utility, and Ethical, legal, and social implications (ACCE Model, National Office of Public Health Genomics) (Centers for Disease Control and Prevention and National Office of Public Health Genomics 2007; Jonas et al. 2012). From the ACCE evaluation framework, this evidence brief focused on clinical utility and postpharmacogenomics testing analytic factors. The ESP Coordinating Center investigators worked with the VA ORD to clarify the key questions and identify the population, comparator, outcome, timing, setting, and study design characteristics of interest (Table 1). We evaluated the clinical utility of pharmacogenomics testing for predicting effectiveness and harms of antidepressant treatment for certain adults with depressive disorders, such as prior to initiation of antidepressants or after failure of one or more courses (Jonas et al. 2012).
Search strategy
To identify relevant literature, we searched MEDLINE®, Cochrane Central Registry of Controlled Trials, and PsycINFO on February 1, 2017, using terms for pharmacogenomics, pharmacogenetics, and depression from 1996 forward. We limited the search to articles involving human subjects available in the English language. Additional citations were identified from hand-searching, reference lists, and consultation with content experts. To identify additional unpublished or ongoing studies as well as guidelines on pharmacogenomics for MDD, we searched the following nonbibliographic database sources: government websites, conference proceedings, relevant genetic and psychiatric professional organizations, clinicaltrials.gov, test manufacturer websites, the VA Health Services Research & Development (HSR&D) Research Studies and Implementation Projects database, and Google.
Study selection
Study selection was based on the prespecified eligibility criteria described in Table 1. Titles and abstracts were reviewed by one investigator. Full-text articles were reviewed by one investigator and checked by another. All disagreements were resolved by consensus.
Data abstraction and quality assessment
We abstracted data from all included studies on setting, pharmacogenomics tests, patient demographics, depression characteristics, medical and psychiatric comorbidities, antidepressant treatments, and all eligible outcome data. We used predefined criteria to rate the internal validity of all randomized controlled trials (RCTs) and controlled cohort studies. We assigned ratings of good, fair, or poor quality to reflect the extent to which the methods protected against bias. For RCTs, ratings were based on assessing the adequacy of methods for randomization, allocation concealment, blinding, and outcome measurement and analysis and acceptability of levels of adherence and attrition using criteria established by the Drug Effectiveness Review Project (McDonagh et al. 2012). For controlled cohort studies, ratings were based on assessing selection, performance, detection, confounding, attrition, and reporting biases using methods established by the AHRQ Methods Guide for Comparative Effectiveness Reviews (Viswanathan et al. 2012). All data abstraction and internal validity ratings were first completed by one reviewer and then checked by another. All disagreements were resolved by consensus.
Data synthesis
Using a best-evidence approach (McDonagh et al. 2014), we prioritized RCTs when available and used controlled cohort and modeling studies to address gaps in RCT evidence (Norris et al. 2010). We used the AHRQ Methods Guide for Comparative Effectiveness Reviews to grade the strength of the body of evidence (SOE) as high, moderate, low, or insufficient. These ratings reflect our confidence that the evidence reflects the true effect (Berkman et al. 2013) based on the number and level of deficiencies (few or no, some, major or numerous, unacceptable) across five key domains: risk of bias (includes study design and aggregate quality), consistency, directness, precision, and reporting bias. Strength of evidence ratings were first completed by one reviewer and then checked by another, and we resolved disagreements using consensus.
We used StatsDirect software (StatsDirect Ltd., Version 2.8.0. (2013), England) to pool data from clinically and methodologically similar studies using random-effects models (Fu et al. 2010), to explore their statistical heterogeneity using Cochran’s Q test and to generate the forest plot to visualize the relative risks of remission rates across multiple pharmacogenomics testing tools. We used Microsoft Excel (Microsoft Corp, Redmond, WA) to calculate descriptive statistics. To summarize outcome data, we primarily used ranges but also used relative risks and their 95% confidence intervals when possible. We also synthesized the evidence qualitatively by grouping studies by pharmacogenomics test.
Results
Literature flow
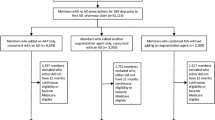
The literature flow diagram (Fig. 1) summarizes the results of the search and study selection processes. The searches resulted in 433 potentially relevant articles. Of these, we included two RCTs (Singh 2015; Winner et al. 2013), five controlled cohort studies (Breitenstein et al. 2014; Hall-Flavin et al. 2013; Hall-Flavin et al. 2012; Fagerness et al. 2014; Winner et al. 2015), and six modeling studies (Serretti et al. 2011; Perlis et al. 2009; Matchar et al. 2007; Pyne 2009; Hornberger et al. 2015; Olgiati et al. 2012). The majority of studies excluded at the full-text level were noncomparative.

Results from literature searching and screening
Overview of study characteristics
Table 2 displays the characteristics of the RCTs and controlled cohort studies. For effects on remission, response, and harms, one fair-quality cohort study evaluated ABCB1 genotype testing (Breitenstein et al. 2014), one good-quality RCT evaluated CNSDose (Singh 2015), and one fair-quality RCT and two fair-quality cohort studies evaluated GeneSight (Hall-Flavin et al. 2013; Hall-Flavin et al. 2012; Winner et al. 2013; Winner et al. 2015). For cost, one fair-quality cohort study each evaluated Genecept (Fagerness et al. 2014) and GeneSight (Winner et al. 2015). The majority of the studies were short in duration, 4 to 16 weeks, and had an average sample size of 154 participants (range 44 to 333). The exception was a single prospective controlled cohort study that assessed total medication costs over 1 year in a GeneSight-tested group (N = 2168) compared to a 5-to-1 propensity-matched large control group (N = 10,880) (Winner et al. 2015). The majority of patients were women in their mid-40s. When reported, the mean Hamilton Depression Rating Scale (HAM-D) score at baseline ranged from 20 to 26.5 points. The mean number of previous antidepressant trials was only reported in the GeneSight studies and ranged from 3.4 to 4.4. The main weakness of the fair-quality RCT was the lack of sufficient information to determine adequacy of randomization and allocation concealment or whether groups were clinically similar at baseline. Main limitations of the fair-quality cohort studies included insufficient information to determine presence and balance of comorbidities, or critical co-interventions (such as psychotherapy) that may have influenced outcomes.
Effects on remission, response, and tolerability
ABCB1 genotyping versus usual care
Compared to usual treatment, 5 weeks of ABCB1 genotyping-guided antidepressant treatment improved remission (HAM-D < 10; Fig. 2) (Breitenstein et al. 2014). Response (50% reduction in HAM-D), quality of life, functional status, and side effects and tolerability were not reported. Supporting evidence comes from one controlled cohort study of 116 adults with MDD and bipolar disorder conducted in Germany (Breitenstein et al. 2014). The mean age in the sample was 47.6 years. The study population was predominantly female, with an average number of depressive episodes of 4.24, duration of current episode of 25 weeks, and 1.3 antidepressant trials during recent admission in the experimental group, compared to 2.43 depressive episodes, 39.2 weeks for the current episode of depression, and 0.98 antidepressant trials during the recent admission for the comparison group. These differences were not statistically significant. The study has several weaknesses. First, the HAM-D remission cutoff (<10) is not considered complete remission according to typical HAM-D scoring methods (Hamilton 1960) and may have led to an overestimation of remission in this study. Also, all treatment occurred while patients were hospitalized, and clinicians were able to review weekly antidepressant plasma levels in addition to the ABCB1 genotyping. As plasma antidepressant concentration has been found to significantly interact with genotype (Breitenstein et al. 2016), the plasma monitoring may have enhanced the benefit of genotype-guided therapy. Finally, some patients had bipolar disorder, which is a relative contraindication to treatment with antidepressants.

Forest plot of remission findings from included studies. Abbreviations: RCT randomized controlled trial, RB relative benefit, NNG number needed to genotype, PGx care guided by pharmacogenomics test. a Three stars indicate moderate strength of evidence (SOE) and two stars indicate low strength of evidence. b GeneSight and CNSDose measured remission rates using the HAM-D ≤ 7 scale; ABCB1 measured remission using HAM-D < 10
CNSDose versus usual care
Compared to usual care, 12 weeks of CNSDose-guided antidepressant treatment significantly improved remission (HAM-D ≤ 7; Fig. 2). CNSDose-guided care also reduced the proportion of patients taking sick leave (usual care = 15% vs guided = 4%; P = 0.0272; low SOE) and intolerability (having an event where patients needed to reduce the dose or stop their antidepressant: usual care = 15% vs guided = 4%; RR 1.13, 95% CI 1.01 to 1.25; low SOE) (Singh 2015). Supporting evidence comes from one randomized trial conducted in Australia in 148 adults with a baseline HAM-D score of 25 taking various second-generation antidepressants (Singh 2015). The main strength of this study is its high internal validity due to its use of robust methodology. However, a potential weakness is that the applicability of its data to more general populations is likely poor, because it had a narrowly selected population of mostly employed females in their early 40s who lacked comorbid psychiatric disorders. The average number of MDD episodes was 2, with an average duration of 8.55 months; however, the number of previously failed antidepressant trials was not reported, nor was the current number of antidepressant medications or other types of concomitant treatments.
GeneSight versus unguided care
Compared to usual care, GeneSight-guided care did not significantly improve remission (HAM-D ≤ 7; Fig. 2) or response (≥50% HAM-D improvement 36 vs 21%; RR 2.14, 95% CI 0.56 to 7.69) in an RCT (Winner et al. 2013; ECRI Institute 2015b; Altar et al. 2015). The only completed, double-blind RCT was conducted in the outpatient clinics of Pine Rest Christian Mental Health Services in Grand Rapids, MI, and involved 51 patients with major depressive disorder, with a mean baseline HAM-D of 21, who had failed a mean of 4.4 previous psychiatric medication trials (Winner et al. 2013). Follow-up was 10 weeks. Types of antidepressant medications used and adverse effects were not reported.
Results from two open-label nonrandomized studies (Hall-Flavin et al. 2013; Hall-Flavin et al. 2012) were less informative than the findings of the RCT (Winner et al. 2013). Ideally, nonrandomized trials can address gaps in RCTs such as evaluating a broader spectrum of patients, providing longer-term follow-up, and contributing data on missing outcomes (Norris et al. 2010). However, these studies were short-term (8 weeks), did not evaluate adverse effects, and included mostly females in their mid-40s with unknown comorbidities. These open-label nonrandomized studies found that GeneSight-guided care significantly improved response (ESP-pooled, 40 vs 23%; RR 1.73, 95% CI 1.09 to 2.73; Cochran Q = 1.04, P = 0.31) but not remission (ESP-pooled, 28 vs 19%; RR 1.47, 95% CI 0.89 to 2.41, Cochran Q = 0.16, P = 0.69). However, because the patients knew whether or not their medication selection was being guided by GeneSight, this raises the likelihood that the increased response could have been biased by their expectations. Also, in the case of the Hall-Flavin 2013 study, the patients in the GeneSight group may have had a more positive prognosis at baseline due to fewer previously failed psychiatric medication trials (4.7 vs 3.6; P = 0.021) (Hall-Flavin et al. 2013). Also, because groups were not matched on psychiatric and medical comorbidities, concomitant medications, medication adherence, and health and lifestyle characteristics, significant differences in these characteristics could have confounded the effects of the GeneSight guiding. When data from the double-blind RCT (Winner et al. 2013) and these two open-label nonrandomized studies (Hall-Flavin et al. 2013; Hall-Flavin et al. 2012) were combined in a meta-analysis (Altar et al. 2015), the improved response with GeneSight-guided care reached statistical significance (RR 1.71, 95% CI 1.17 to 2.49). However, the limitations of the open-label, nonrandomized studies weaken the validity of this meta-analysis.
We identified three ongoing clinical trials assessing the efficacy of GeneSight-guided management of depressive disorders (NCT02189057, NCT02466477, NCT02109939). All studies are double-blind RCTs that are expected to address some gaps in the existing evidence by increasing precision with larger sample sizes and providing longer follow-up (ECRI Institute 2015a). The studies are expected to be completed between 2015 and 2018.
Improving time to antidepressant effectiveness
We found no studies that evaluated the impact of pharmacogenomics-guided treatment on time to antidepressant effectiveness in patients with MDD or number of failed antidepressant trials.
Association of improvements in remission and response with switches to genetically congruent medication
In establishing the clinical utility of pharmacogenomics-guided treatment, a first step is to demonstrate an overall improvement in the key outcomes of remission, response, and tolerability for guided versus unguided care. An essential second step is to demonstrate that the improvement on those key outcomes is due to a greater incidence in the guided group of actually implementing recommended medication changes to more genetically suitable regimens. At the time of this report, no pharmacogenomics-guided treatment strategy has met both of these criteria.
Guided care with GeneSight is the only strategy with any evidence for the second step of showing that symptom reduction was associated with switches to more genetically suitable regimens. In the randomized trial (Winner et al. 2013), compared to usual care, twice as many patients in the GeneSight group were switched to genetically congruent medication (100 vs 50%; P = 0.02) and, among those patients, there was a greater mean HAM-D score improvement (33.1 vs 0.8%; P = 0.06). However, the clinical meaningfulness of the evidence is unclear because it was measured based on mean change in depression symptoms, rather than remission and/or response (Winner et al. 2013).
Optimal clinical scenarios for using pharmacogenomics-guided treatment
We found no studies that evaluated whether the impact of using pharmacogenomics-guided treatment on the effectiveness and harms of antidepressants differs according to the following key patient characteristics: demographics, psychiatric and medical comorbidities, depression symptomatology, depression severity and duration, history of antidepressant treatment resistance, concomitant medication, polypharmacy, medication side effects, nonadherence, or other health or lifestyle behaviors.
Cost-effectiveness
We found little evidence of the cost-effectiveness of pharmacogenomics-guided care for MDD. No study has prospectively or retrospectively compared directly observed cost-effectiveness outcomes of pharmacogenomics-guided care versus usual care specifically in patients with depressive disorders. For evaluation of directly observed cost-effectiveness, we identified an RCT of YouScript® that evaluated cost-effectiveness in polypharmacy home health patients, but it did not provide information specifically about antidepressant use in patients with depressive disorders (Elliott et al. 2017). Available controlled cohort studies of GeneSight (Winner et al. 2015) and Genecept (Fagerness et al. 2014) also did not provide information about cost-effectiveness of pharmacogenomics-guided care for MDD because they only measured cost savings, were comprised of populations using antidepressant medication primarily for diagnoses other than depressive disorders (i.e., anxiety, ADHD, other mood disorder, dementia, personality disorder, “all other psych”), and did not evaluate the subgroups of patients with depressive disorders (14–39%).
We also identified modeling studies that evaluated potential cost-effectiveness outcomes of GeneSight (Hornberger et al. 2015), 5-HTTLPR (Olgiati et al. 2012; Serretti et al. 2011), HTR2A (Perlis et al. 2009), and CYP450 polymorphisms (Matchar et al. 2007; Pyne 2009) for guiding antidepressant treatment (Table 3). Although a recent systematic review by Berm et al. evaluated the majority of these studies, its conclusions are not generalizable to depression because they were based on the combined findings from these plus 74 additional studies, the majority of which were about other pharmacogenomics tests used in a variety of clinical areas (e.g., oncology, cardiology, neurology, etc.) (Berm et al. 2016). Among modeling studies focused on antidepressant treatment, GeneSight was found to have the strongest evidence of estimated cost-effectiveness. Compared to treatment as usual, Genesight-guided care was more effective and more cost-saving than treatment as usual, with a 94.5% probability of being cost-effective at the willingness-to-pay (WTP) threshold of $50,000/quality-adjusted life-years (QALYs), a finding that persisted in 75.7% of 10,000 simulations that varied input parameters (Hornberger et al. 2015). These findings should be considered preliminary, however, as they rely on inferences of potential outcomes rather than provide precise estimates of directly observed outcomes and do not include assessments of commonly used higher WTP thresholds of $100,000 to $150,000/QALY (Alagoz et al. 2016; Ubel et al. 2003; Marseille et al. 2015).
Discussion
This evidence review formally critically appraised and rated the strength of the complete body of available evidence that compares use of single- and multi-gene testing-guided antidepressant treatment selection for depressive disorders to treatment as usual. While there is a plausible clinical rationale for expecting benefits from pharmacogenomics-guided treatment, the actual impact has not been well-established. We identified three pharmacogenomics-guided treatment strategies that have been evaluated in published studies that compare pharmacogenomics-guided care to usual care. Of the three pharmacogenomics-guided treatment strategies, CNSDose has the most favorable preliminary findings because it is the only one with evidence of both a significant improvement in remission (one additional patient had a remission by 12 weeks for every three genotyped; 95% CI 1.7 to 3.5) and improved antidepressant tolerability. ABCB1 genotyping also improved the chance of remission, with one additional remission at 5 weeks for every 3 to 20 patients genotyped, but data on tolerability was lacking. In the best study available for GeneSight, an RCT, its effects on remission and response were not statistically significant and left unclear whether the chance of remission was substantially better or worse than usual care. There is some doubt about the stability of all these findings, however, because there is only a single, small, short-term study of each strategy, and the majority has numerous minor methodological limitations. Cost-effectiveness of pharmacogenomics is unclear because of the uncertain effectiveness and the lack of studies evaluating directly observed cost-effectiveness outcomes. We found no studies that evaluated whether pharmacogenomics shortens time to optimal treatment, whether improvements were due to switches to genetically congruent medication, or to what extent variation in test and results’ delivery methods, patient comorbidities and/or health or lifestyle behaviors, may modify effectiveness of pharmacogenomics.
Additional single-arm studies of the clinical utility of pharmacogenomics tools are available but were excluded from this review due to their inability to distinguish the specific effects of the pharmacogenomics guiding as distinct from what may have naturally occurred over time regardless of the intervention (Effective Practice and Organization of Care 2013). For example, the clinical utility of CYP2D6- and CYP2C19-guided (Müller et al. 2013) and Genecept-guided antidepressant treatment (Brennan et al. 2015) has been evaluated in single-group before-after studies. Additionally, one ongoing double-blind randomized controlled trial of 8 weeks comparing Genecept-guided versus usual care in adults with MDD is expected to assess response, remission, and safety outcomes (NCT02634177). This study was expected to be completed in October of 2016 and will hopefully provide more relevant and higher-quality evidence with which to evaluate the clinical utility of Genecept.
Our findings are consistent with previous reviews in suggesting that evidence on clinical utility is still in its early developmental stages and is currently inadequate to precisely determine the overall balance of comparative benefits and harms across the full range of key outcomes (ECRI Institute 2015a, b; Singh et al. 2014; Howland 2014a; Bousman and Hopwood 2016; Rosenblat et al. 2017). However, our review adds significant depth by updating searches, evaluating single- and multi-gene testing panels, evaluating adverse consequences, adding formal critical appraisal of internal validity and strength of evidence of the complete body of available evidence, and providing specific suggestions for future research.
There are numerous gaps in the evidence. New research would be more meaningful if it (1) included a broader population; (2) recorded what medication changes were recommended and how often following the recommendations resulted in improvement on key outcomes; (3) evaluated multiple key outcomes of remission, response, quality of life, functional capacity, and tolerability and by how much the pharmacogenomics testing-guided care reduced time to these outcomes; (4) obtained longer-term follow-up of at least 6 months to a year; and (5) evaluated to what extent the complexity of interacting factors may impact the utility of pharmacogenetics in MDD treatment, including patients’ prior experience with antidepressants, plasma level, demographics, psychiatric and medical comorbidities, depression characteristics, concomitant medication, or other health or lifestyle behaviors.
We suggest future studies also consider level of clinician education about pharmacogenomics testing. Studies have shown that despite patients’ expectations of clinicians’ competency in explaining, interpreting, and applying pharmacogenomics test results in clinical decision-making (Squassina et al. 2010), a majority of previously surveyed clinicians acknowledged that they may be inadequately informed to do so (Prainsack and Wolinsky 2010). Therefore, we recommend future studies explore whether competency and clinical expertise (e.g., primary care, psychiatry) may affect skill in utilizing pharmacogenomics data and potentially antidepressant treatment outcomes. Also, there may be a need to identify available, and ideally validated, educational materials on the utilization and potential harms of pharmacogenomics data in clinical decision-making and compare the effects of different educational approaches on patient outcomes.
Another consideration for facilitating accurate translation of pharmacogenomics into clinical practice is the format and complexity of results delivery (Drozda et al. 2014). The complexity in interpreting results of gene-panel tests may increase as the numbers of genes and gene variants increase, and there may be challenges in finding the appropriate balance between the level of detail in results delivery and information overload for busy practitioners and patients (Hornberger et al. 2015). We noted that available pharmacogenomics testing results varied in (1) how much detail was provided about the gene result, categorization of gene-drug interaction, therapeutic implications, and clinical impact; (2) the format of the interpretive information (e.g., length of report, computer-based or paper-based components); (3) turnaround time (e.g., at point of care, days, weeks); and (4) whether or not a consultation with a professional genetic counselor and/or a pharmacist was available. To assist with interpretation and replication, more details are needed about methods used to predict phenotype (e.g., poor metabolizer) from genotype, algorithms used to combine phenotype information across multiple variants to make drug selection and dosing recommendations, and which guidelines are used to inform dosing recommendations. However, this may not be realistic given the commercial context of these tests. To assess if and how such differences in format of pharmacogenomics testing results delivery may affect the accuracy of their interpretation and use, we suggest direct comparison of a few different approaches.
Potential limitations of our review methods include language bias and use of sequential rather than independent dual review of investigator judgments. Although we would expect the potential impact of excluding non-English studies to be minimal, there is a chance this exclusion may have biased our findings (Higgins and Green 2011). Although compared to dual independent review sequential dual review may conceivably increase risk of reviewer bias and error, this has not been empirically evaluated. Considering that the body of available evidence is sparse in general, there is limited potential for sequential dual review to have dramatically altered our overall low confidence in the stability of the evidence.
Conclusions
In conclusion, limited evidence suggests that certain pharmacogenomics tools show promise for improving short-term remission rates in women in their mid-40s with few comorbidities, but provided little to no information about if and how they impact quality of life, functional capacity, and tolerability, or whether they reduced time to these outcomes. New research would be more meaningful if it included a broader population; recorded what medication changes were recommended and how often following the recommendations resulted in remission, and by how much the time to remission was reduced; identified optimal clinical scenarios for use; and obtained longer follow-up.
References
Agency for Healthcare Research and Quality (2014) Methods guide for effectiveness and comparative effectiveness reviews. AHRQ Publication No. 10(14)-EHC063-EF, Rockville
Alagoz O, Durham D, Kasirajan K (2016) Cost-effectiveness of one-time genetic testing to minimize lifetime adverse drug reactions. Pharmacogenomics J 16:129–136
Altar CA, Carhart J, Allen JD, Hall-Flavin D, Winner J, Dechairo B (2015) Clinical utility of combinatorial pharmacogenomics-guided antidepressant therapy: evidence from three clinical studies. Mol Neuropsychiatry 1:145–155
American Psychiatric Association (2010) Practice guideline for the treatment of patients with major depressive disorder. Am Psychiatric Assoc
Bauer M, Pfennig A, Severus E, Whybrow PC, Angst J, Möller H-J (2013) World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of unipolar depressive disorders, part 1: update 2013 on the acute and continuation treatment of unipolar depressive disorders. World J Biol Psychiatry 14:334–385
Bauer M, Whybrow PC, Angst J, Versiani M, Möller H-J (2015) World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of unipolar depressive disorders, part 2: maintenance treatment of major depressive disorder - update 2015. World J Biol Psychiatry 16:76–95
Berkman ND, Lohr KN, Ansari M et al. (2013) Grading the strength of a body of evidence when assessing health care interventions for the effective health care program of the agency for healthcare research and quality: an update methods guide for effectiveness and comparative effectiveness reviews
Berm EJ, Looff M, Wilffert B, Boersma C, Annemans L, Vegter S, Boven JF, Postma MJ (2016) Economic evaluations of pharmacogenetic and pharmacogenomic screening tests: a systematic review. Second update of the literature. PLoS One 11:e0146262
Bousman CA, Hopwood M (2016) Commercial pharmacogenetic-based decision-support tools in psychiatry. The Lancet Psychiatry
Breitenstein B, Scheuer S, Pfister H, Uhr M, Lucae S, Holsboer F, Ising M, Bruckl TM (2014) The clinical application of ABCB1 genotyping in antidepressant treatment: a pilot study. Cns Spectrums 19:165–175
Breitenstein B, Scheuer S, Bruckl TM, Meyer J, Ising M, Uhr M, Holsboer F (2016) Association of ABCB1 gene variants, plasma antidepressant concentration, and treatment response: results from a randomized clinical study. J Psychiatr Res 73:86–95
Brennan FX, Gardner KR, Lombard J et al. (2015) A naturalistic study of the effectiveness of pharmacogenetic testing to guide treatment in psychiatric patients with mood and anxiety disorders. The primary care companion for CNS disorders 17
Centers for Disease Control and Prevention, National Office of Public Health Genomics. 2007. ACCE model process for evaluating genetic tests [online]. Available: http://www.cdc.gov/genomics/gtesting/ACCE/index.htm [Accessed March 2016]
de Leon J (2016) Pharmacogenetic tests in psychiatry: from fear to failure to hype. J Clin Psychopharmacol 36:299–304
Department of Veterans Affairs: The Management of MDD Working Group (2016) VA/DoD clinical practice guideline for the management of major depressive disorder
Drozda K, Müller DJ, Bishop JR (2014) Pharmacogenomic testing for neuropsychiatric drugs: current status of drug labeling, guidelines for using genetic information, and test options. Pharmacother: J Hum Pharmacol Drug Ther 34:166–184
ECRI Institute (2015a) GeneSight psychotropic pharmacogenomic testing (Assurex Health, Inc.) for guiding medication selection for patients with neuropsychiatric disorders. Genetic Test Product Brief
ECRI Institute (2015b) Pharmacogenetic testing to guide treatment of behavioral and mental health disorders. Genetic Test Hotline Response
Effective Practice and Organization of Care (2013) What study designs should be included in an EPOC review? EPOC Resources for review authors
Elliott LS, Henderson JC, Neradilek MB, Moyer NA, Ashcraft KC, Thirumaran RK (2017) Clinical impact of pharmacogenetic profiling with a clinical decision support tool in polypharmacy home health patients: a prospective pilot randomized controlled trial. PLoS One 12:e0170905
Fagerness J, Fonseca E, Hess GP, Scott R, Gardner KR, Koffler M, Fava M, Perlis R, Brennan FX, Lombard J (2014) Pharmacogenetic-guided psychiatric intervention associated with increased adherence and cost savings. Am J Manag Care 20:e146–e156
Fu R, Gartlehner G, Grant M et al. (2010) Chapter 12. Conducting quantitative synthesis when comparing medical interventions. Methods guide for effectiveness and comparative effectiveness reviews. Agency for Healthcare Research and Quality
Hall-Flavin DK, Winner JG, Allen JD, Jordan JJ, Nesheim RS, Snyder KA, Drews MS, Eisterhold LL, Biernacka JM, Mrazek DA (2012) Using a pharmacogenomic algorithm to guide the treatment of depression. Transl Psychiatry 2:e172
Hall-Flavin DK, Winner JG, Allen JD, Carhart JM, Proctor B, Snyder KA, Drews MS, Eisterhold LL, Geske J, Mrazek DA (2013) Utility of integrated pharmacogenomic testing to support the treatment of major depressive disorder in a psychiatric outpatient setting. Pharmacogenet Genomics 23:535–548
Hamilton SP (1960) A rating scale for depression. J Neurol Neurosurg Psychiatry 23:56–62
Higgins J, Green S (2011) Cochrane handbook for systematic reviews of interventions. Version 5.1.0.
Hornberger J, Li Q, Quinn B (2015) Cost-effectiveness of combinatorial pharmacogenomic testing for treatment-resistant major depressive disorder patients. Am J Manag Care 21:e357–e365
Howland RH (2014a) Pharmacogenetic testing in psychiatry: not (quite) ready for primetime. J Psychosoc Nurs Ment Health Serv 52:13–16
Howland RH (2014b) Pharmacogenetic testing in psychiatry: not (quite) ready for primetime. J Psychosoc Nurs Ment Health Serv 52:13–16
Jonas DE, Wilt TJ, Taylor BC, Wilkins TM, Matchar DB (2012) Challenges in and principles for conducting systematic reviews of genetic tests used as predictive indicators. J Gen Intern Med 27:83–93
Marseille E, Larson B, Kazi DS, Kahn JG, Rosen S (2015) Thresholds for the cost–effectiveness of interventions: alternative approaches. Bull World Health Organ 93:118–124
Matchar DB, Thakur ME, Grossman I et al. (2007) Testing for cytochrome P450 polymorphisms in adults with non-psychotic depression treated with selective serotonin reuptake inhibitors (SSRIs)
McDonagh M, Jonas D, Gartlehner G, Little A, Peterson K, Carson S, Gibson M, Helfand M (2012) Methods for the drug effectiveness review project. BMC Med Res Methodol 12:1
McDonagh M, Peterson K, Raina P et al. (2014) Methods guide for effectiveness and comparative effectiveness reviews: chapter 7: avoiding bias in selecting studies. Agency for Healthcare Research and Quality
Müller DJ, Kekin I, Kao AC, Brandl EJ (2013) Towards the implementation of CYP2D6 and CYP2C19 genotypes in clinical practice: update and report from a pharmacogenetic service clinic. Int Rev Psychiatry 25:554–571
National Institute for Health and Care Excellence (2009) Depression in adults: recognition and management
Norris S, Atkins D, Bruening W et al. (2010) Chapter 8. Selecting observational studies for comparing medical interventions. Methods guide for effectiveness and comparative effectiveness reviews. Agency for Healthcare Research and Quality
Olgiati P, Bajo E, Bigelli M, De Ronchi D, Serretti A (2012) Should pharmacogenetics be incorporated in major depression treatment? Economic evaluation in high- and middle-income European countries. Prog Neuro-Psychopharmacol Biol Psychiatry 36:147–154
Perlis RH, Patrick A, Smoller JW, Wang PS (2009) When is pharmacogenetic testing for antidepressant response ready for the clinic? A cost-effectiveness analysis based on data from the STAR D study. Neuropsychopharmacology 34:2227–2236
Peterson K, Dieperink E, Ferguson L et al. (2016) Evidence brief: the comparative effectiveness, harms, and cost-effectiveness of pharmacogenomic testing guided versus unguided antidepressant treatment for major depressive disorder
Prainsack B, Wolinsky H (2010) Direct-to-consumer genome testing: opportunities for pharmacogenomics research? Pharmacogenomics 11:651–655
Pyne, J (2009) Genetic testing decision analysis model for antidepressant treatment
Rosenblat, JD, Lee, Y, McIntyre, RS (2017) Does pharmacogenomic testing improve clinical outcomes for major depressive disorder? A systematic review of clinical trials and cost-effectiveness studies. J Clin Psychiatry
Rost K, Nutting P, Smith JL, Elliott CE, Dickinson M (2002) Managing depression as a chronic disease: a randomised trial of ongoing treatment in primary care. BMJ 325:934
Rush AJ, Trivedi M, Carmody TJ, Biggs MM, Shores-Wilson K, Ibrahim H, Crismon ML (2004) One-year clinical outcomes of depressed public sector outpatients: a benchmark for subsequent studies. Biol Psychiatry 56:46–53
Serretti A, Olgiati P, Bajo E, Bigelli M, De Ronchi D (2011) A model to incorporate genetic testing (5-HTTLPR) in pharmacological treatment of major depressive disorders. World J Biol Psychiatry 12:501–515
Singh AB (2015) Improved antidepressant remission in major depression via a pharmacokinetic pathway polygene pharmacogenetic report. Clin Psychopharmacol Neurosci 13:150–156
Singh AB, Bousman CA, Ng C, Berk M (2014) Antidepressant pharmacogenetics. Curr Opin Psychiatry 27:43–51
Squassina A, Manchia M, Manolopoulos VG, Artac M, Lappa-Manakou C, Karkabouna S, Mitropoulos K, Zompo MD, Patrinos GP (2010) Realities and expectations of pharmacogenomics and personalized medicine: impact of translating genetic knowledge into clinical practice. Pharmacogenomics 11:1149–1167
Tansey KE, Guipponi M, Hu X, Domenici E, Lewis G, Malafosse A, Wendland JR, Lewis CM, McGuffin P, Uher R (2013) Contribution of common genetic variants to antidepressant response. Biol Psychiatry 73:679–682
Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, Norquist G, Howland RH, Lebowitz B, McGrath PJ, Shores-Wilson K, Biggs MM, Balasubramani GK, Fava M (2006) Evaluation of outcomes with citalopram for depression using measurement-based care in STAR* D: implications for clinical practice. Am J Psychiatr 163:28–40
Ubel PA, Hirth RA, Chernew ME, Fendrick AM (2003) What is the price of life and why doesn’t it increase at the rate of inflation? Arch Intern Med 163:1637–1641
Viswanathan, M, Ansari, M, Berkman, ND et al. (2012) Assessing the risk of bias of individual studies in systematic reviews of health care interventions:methods guide for comparative effectiveness reviews, Rockville, MD
Winner JG, Carhart JM, Altar CA, Allen JD, Dechairo BM (2013) A prospective, randomized, double-blind study assessing the clinical impact of integrated pharmacogenomic testing for major depressive disorder. Discov Med 16:219–227
Winner, JG, Carhart, JM, Altar, CA et al. (2015) Combinatorial pharmacogenomic guidance for psychiatric medications reduces overall pharmacy costs in a 1 year prospective evaluation. Current medical research and opinion
Acknowledgements
We would like to thank Julia Haskin, MA, for editorial support. This material is based upon work supported by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Quality Enhancement Research Initiative (QUERI), Evidence-Based Synthesis Program (ESP).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Peterson, K., Dieperink, E., Anderson, J. et al. Rapid evidence review of the comparative effectiveness, harms, and cost-effectiveness of pharmacogenomics-guided antidepressant treatment versus usual care for major depressive disorder. Psychopharmacology 234, 1649–1661 (2017). https://doi.org/10.1007/s00213-017-4622-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-017-4622-9