
Abstract
Rationale
Sex-related differences in the clinical profiles of some insomnia medications have been previously reported.
Objective
To evaluate the clinical profile of suvorexant, a novel orexin receptor antagonist approved for treating insomnia at doses up to 20 mg, by sex subgroups.
Methods
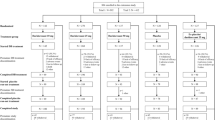
Efficacy analyses by sex were based on pooled data from two similar phase 3, randomized, double-blind, placebo-controlled, 3-month trials in elderly (≥65 years) and non-elderly (18–64 years) insomnia patients. Two age-adjusted (non-elderly/elderly) dose regimes of 40/30 and 20/15 mg were evaluated, with fewer patients assigned to 20/15 mg. Efficacy was assessed by patient-reported outcomes (N = 1264 women, 707 men) and by polysomnography endpoints in ~75% of patients. Safety analyses by sex (N = 1744 women, 1065 men) included pooled data from the two 3-month trials plus 3-month data from a safety trial of 40/30 mg.
Results
The sex subgroup efficacy analyses mirrored the improvements seen for suvorexant 40/30 and 20/15 mg over placebo on patient-reported outcomes and polysomnography sleep maintenance and onset endpoints in the primary analyses; 95% CIs excluded zero in favor of suvorexant for most endpoints in both sexes, and similar efficacy was observed between sexes (95% CIs overlapped). Suvorexant was well-tolerated in women and men, although women in all treatment groups (including placebo) reported more adverse events than men. The most frequent adverse event was somnolence (women: 11.1% for 40/30 mg, 8.5% for 20/15 mg, 2.3% for placebo; men: 10.1% for 40/30 mg, 3.4% for 20/15 mg, 4.2% for placebo).
Conclusion
Suvorexant was generally effective and well-tolerated in both women and men with insomnia.
ClinicalTrials.gov trial registration numbers: NCT01097616, NCT01097629, NCT01021813.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Insomnia is common in both women and men, but the incidence in women is higher by approximately 50% (Zhang and Wing 2006). Furthermore, differences between the sexes in sleep patterns and EEG profiles during sleep have been reported (Mallampalli and Carter 2014; Svetnik et al. 2017). Women’s sleep may be influenced by menstrual cycle, menopausal status, and other hormonal, biological, and psychosocial factors (Mong et al. 2011; Nowakowski et al. 2013; Boivin et al. 2016).
Trials of medications for insomnia have typically looked at treatment effects in a combined sex population and often have not evaluated potential differences between women and men. Sex-related differences in neurobiology, hormones, body composition, and/or metabolism may make women more (or less) sensitive to the effects of some psychotropic drugs compared to men (Gandhi et al. 2004; Cosgrove et al. 2007; Franconi et al. 2007; Jacobson 2014). A notable example in the sleep field is slower metabolism in women of the benzodiazepine receptor agonist hypnotic zolpidem, which led to sex-specific dosing recommendations (halving of the dose in women) due to a possible increased risk of next-morning impairment and motor vehicle accidents (Farkas et al. 2013). Furthermore, sex-related differences in the efficacy and EEG power spectral profile of the investigational insomnia treatment gaboxadol were reported in the absence of pharmacokinetic differences; the differences were hypothesized to be due to differential affinity for GABA(A) receptor subtypes and/or their modulation by neurosteroids (Dijk et al. 2010; Roth et al. 2010).
Suvorexant is a first-in-class orexin receptor antagonist (ORA) approved for treating insomnia at doses of 10–20 mg (Cox et al. 2010; Winrow et al. 2011; Herring et al. 2012; Sun et al. 2013). Suvorexant provides a novel approach to treating insomnia by blocking orexin-mediated wake signaling (Mieda and Sakurai 2013; Winrow and Renger 2014). This approach is distinct from commonly used benzodiazepine receptor agonist sleep medications (e.g., zolpidem, zaleplon, eszopiclone, temazepam) which act by enhancing the broad central nervous system depressant effects of GABA inhibition, through effects on the benzodiazepine receptor at the GABA-A complex (Roehrs and Roth 2012).
The suvorexant phase 3 development program included both women and men with insomnia. The program consisted of two 3-month pivotal trials, each of which evaluated two age-adjusted (non-elderly/elderly) suvorexant dose regimes of 40/30 and 20/15 mg (Herring et al. 2016) and a 1-year trial of 40/30 mg (Michelson et al. 2014). A subgroup analysis by sex of pooled data from the suvorexant phase 3 trials was pre-specified and is reported here.
Phase 1 pharmacokinetic data showed that suvorexant exposure is slightly higher in women than in men. In women, the area-under-the-curve and maximum plasma concentrations are increased by 17 and 9%, respectively, for a 40-mg dose, and the average concentration of suvorexant 9 h after dosing is ~5% higher for women over the 10–40-mg dose range (these data were not adjusted for weight since this was found not to be a modifying factor in a population pharmacokinetic model). Based on these pharmacokinetic data, no differences were expected in the clinical profile of suvorexant in women and men with insomnia in the phase 3 trials. However, as noted above, sex-related differences in clinical profiles have been noted for another insomnia treatment in the absence of pharmacokinetic differences.
Materials and methods
Overview
Full details of trial methods and results in the overall population (women and men combined) are in the primary publications (Michelson et al. 2014; Herring et al. 2016). The following sections summarize key information relevant to understanding the present sex subgroup analyses.
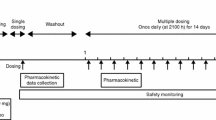
The efficacy subgroup analyses by sex included pooled data from two phase 3 randomized, double-blind, placebo-controlled, parallel-group, 3-month efficacy, and safety trials in non-elderly (18–64 years) and elderly (≥65 years) patients with primary insomnia (P028 and P029) (Herring et al. 2016). Suvorexant doses of 40/30 mg (non-elderly/elderly) and 20/15 mg (non-elderly/elderly) were evaluated, with fewer patients randomized to 20/15 mg than 40/30 mg or placebo. Doses differed by age to adjust for previously observed plasma exposure differences (<65: 40 mg or 20 mg; ≥65: 30 mg or 15 mg).
The safety subgroup analyses by sex included pooled data from the two 3-month phase 3 trials (P028 and P029) (Herring et al. 2016) and also 3-month data on 40/30 mg and placebo from a 1-year safety trial (P009; 20/15 mg was not included in the trial) (Michelson et al. 2014). It should be noted that each trial incorporated a 1-week, randomized, double-blind run-out after double-blind treatment (3 months in P029, 3 or 6 months in P028, 12 months in P009) to assess withdrawal and rebound insomnia; data from the run-out periods were not included in the present analysis.
Patients
Non-elderly (18–64) and elderly (≥65) women and men who met DSM-IV-TR criteria for primary insomnia (APA 2000) and were otherwise in good physical and mental health were enrolled. Women of reproductive potential had to have a negative serum pregnancy test at screening and agree to use adequate contraception, which could include hormonal contraception, during the trial. All patients in the two pivotal efficacy studies provided subjective sleep estimates using an electronic sleep diary/questionnaire. Approximately 75% of those patients also underwent polysomnography (PSG) over 8 h (subset determined by whether the site had the capacity to perform PSG or not). Patients who completed only self-report assessments are referred to as the questionnaire (Q)-cohort; those who completed self-report and PSG assessments are referred to as the PSG + questionnaire (PQ)-cohort. To enter the Q-cohort, patients had to report a total sleep time (sTST) <6.5 h and time to sleep onset (sTSO) ≥30 min, both on ≥4 of 7 nights during the last week of a 2-week placebo run-in before randomization. For the PQ-cohort, patients had to meet the following PSG criteria for screening and baseline PSG nights: latency to onset of persistent sleep (LPS) >20 min and mean (across screening and baseline) wakefulness after persistent sleep onset (WASO) ≥60 min with neither night ≤45 min. PQ-cohort patients were not required to also meet the Q-cohort diary entry criteria.
Design and procedure
Patients were discontinued from hypnotic medications prior to entering the trials. During the trials, patients were asked to limit alcohol consumption (≤2 drinks per day and ≥3 h before bedtime or ≥24 h before on PSG nights), caffeine consumption (≤5 cups per day and none after 4 pm, or after 1 pm on PSG nights), and smoking (≤15 cigarettes per day and none during the night).
Patients were randomized to treatment with suvorexant 40/30 mg, suvorexant 20/15 mg, or placebo in a 3:2:3 ratio in P028, and a 1:1:1 ratio (Q-cohort) or a 2:1:2 ratio (PQ-cohort) in P029. In P009, patients were randomized to treatment with suvorexant 40/30 mg or placebo in a 2:1 ratio. Randomization was stratified by age-category (non-elderly vs elderly) in all trials and also by cohort (Q vs PQ) in P028 and P029 and by region in P009. Patients were assigned to treatment groups using an allocation-schedule system that provided a computer-generated randomization schedule based upon input from a statistician from Merck & Co., Inc., from whom treatment allocation was masked. Treatment assignment was implemented through an interactive voice response system. Study investigators, site staff, patients, PSG scorers, and monitoring staff from Merck & Co., Inc. remained blinded to treatment allocation throughout the study.
The trials were conducted in accordance with principles of Good Clinical Practice and were approved by the appropriate institutional review boards and regulatory agencies for each site. Informed consent was obtained from all patients. The trials were registered at ClinicalTrials.gov (NCT01097616, NCT01097629, NCT01021813).
Assessments
Patients used an electronic diary each morning to report measures of the previous night’s sleep including sTST (min) and sTSO (min). PSG measures included WASO (min) and LPS (min) assessed at night 1, month 1, and month 3. Safety assessments included open-ended questioning for adverse events.
Efficacy endpoints
The efficacy endpoints selected for the subgroup analysis were those pre-specified as primary in the original trials (Herring et al. 2016): change from baseline in sleep diary and PSG measures of sleep maintenance (sTST, WASO) and sleep onset (sTSO, LPS). Monthly values for the self-report endpoints were the mean of the daily values for the last week (week 1, month 1) or 2 weeks (month 3) of the month. In the original trials, the primary time points were month 1 and month 3, while week 1 (self-report measures) and night 1 (PSG measures) were secondary time points.
Statistical analysis
The pooled efficacy analyses by sex were pre-specified to allow a more robust evaluation than in each individual trial and included all patients in P028 + P029 who took ≥1 dose of treatment, had ≥1 post-treatment efficacy measure, and had baseline data. Efficacy endpoints (i.e., change from baseline in sTST, sTSO, WASO, LPS) were assessed using a longitudinal data analysis model with terms for study, baseline value (of the response variable), age category (<65, ≥65), region, sex, treatment, time point, treatment-by-time point, and sex-by-treatment-by-time point interaction as covariates; cohort was also included in the models for sTST and sTSO. Estimates and comparisons of the treatment differences versus placebo in women and men were obtained using appropriate contrasts of the sex-by-treatment-by-time point interaction term.
No formal multiplicity strategy was employed for these pooled subgroup analyses since the primary purpose was to provide improved precision in the estimates of the treatment group differences from placebo; nominal p values for these treatment differences were computed as a measure of strength of evidence for the effect rather than a formal test of hypothesis.
The safety analysis included all patients in P028 + P029 + P009 who took ≥1 dose of treatment. The percentages of women and men with adverse events were calculated.
Power
No power calculations were made for this pooled subgroup analysis. In each of the primary studies, there was greater power to declare all primary sleep maintenance measures significant as compared to sleep onset measures, and greater power for the higher 40/30 mg dose comparisons to placebo versus the 20/15 mg dose comparisons to placebo due to the smaller sample size allocated to the 20/15 mg group.
Results
Patients
Patient baseline characteristics and baseline symptom severity were generally similar among treatment groups and are summarized in Table 1 (pooled P028 + P029 efficacy population). Approximately 65% of the patients were women, and the percentages were similar across treatment groups. There were no striking differences in baseline sleep measures between women and men or between treatment groups. Patient baseline characteristics were generally similar among treatment groups in the pooled P028 + P029 + P009 safety population (Supplementary Material Table S1).
Efficacy
A total of 1264 women and 707 men had efficacy data (at the earliest time point) for the analysis of patient-reported subjective (sleep diary) endpoints, and 963 women and 528 men had efficacy data (at the earliest time point) for the analysis of objective (PSG) endpoints. Mean changes from baseline in each treatment group for diary and PSG sleep onset and maintenance measures by sex are shown in Supplementary Material Table S2. Suvorexant differences from placebo in change-from-baseline are summarized in Table 2 and Fig. 1. Mean differences favored suvorexant and 95% CIs generally excluded zero except for sleep onset measures at some time points with suvorexant 20/15 mg in both sexes, and LPS in men at month 3 with suvorexant 40/30 mg. Efficacy was generally seen at the first assessment time point (night 1 for PSG measures, week 1 for subjective measures) and was usually maintained over 3 months except for LPS at month 3; examination of the change-from-baseline LPS values in Table S2 suggests that this reflected an increasing response in the placebo group over time rather than a reduced response in the suvorexant group. The point estimates and 95% CIs for all measures were similar (overlapping) for women and men (Fig. 1), with more precise CIs for women due to the larger sample size.

Effect of suvorexant 20/15 mg (a) and 40/30 mg (b) on sleep maintenance and onset measures in women and men in P028 + P029: estimate (95% CI) of difference in least squares mean change from baseline for suvorexant versus placebo
Safety
A total of 1744 women and 1065 men were included in the safety analyses. Adverse events over 3 months are summarized in Table 3. The percentages of patients with any adverse event were higher with suvorexant 40/30 mg than with placebo for both women (52.4 vs 48.8%) and men (48.7 vs 43.0%), but comparable for suvorexant 20/15 mg and placebo (women = 49.5 vs 48.8%; men = 40.8 vs 43.0%). The percentages of women with any adverse event were higher across all treatment groups, including placebo, compared with men.
The most frequent adverse event in both sexes was somnolence which was generally transient and mild to moderate in intensity. In women, the incidence of somnolence was higher for suvorexant 20/15 mg (8.5%) and suvorexant 40/30 mg (11.1%) versus placebo (2.3%). In men, the incidence of somnolence was higher for suvorexant 40/30 mg versus placebo (10.1 vs 4.2%) but similar for suvorexant 20/15 mg and placebo (3.4 vs 4.2%). Women had a higher incidence of somnolence than men for suvorexant 20/15 mg (8.5 vs 3.4%) and, to a lesser extent, for suvorexant 40/30 mg (11.1 vs 10.1%). Because the 20/15-mg dose was not included in one of the trials used for the pooled analysis (P009), we repeated the analysis of somnolence for 20/15 mg versus placebo using P028 + P029 data only, and the results were similar (women: 27/319 [8.5%] versus 15/492 [3.0%]; men: 6/174 [3.4%] versus 10/275 [3.6%]).
Of the other common adverse events shown in Table 3, the following occurred in women at an incidence greater than placebo and at least twice than that in men for the approved 20/15 mg dose: headache, abnormal dreams, dry mouth, cough, and upper respiratory tract infection. However, the differences were small (e.g., 2.2 vs 1.1% for abnormal dreams, dry mouth, and cough).
Discussion
The efficacy analyses by sex subgroup mirrored the improvements previously reported for suvorexant 20/15 and 40/30 mg over placebo on subjective and PSG sleep maintenance and onset endpoints in the combined sex primary analyses (Herring et al. 2016). Efficacy was generally seen at the first assessment time point (night 1 for PSG measures, week 1 for subjective measures) and was usually maintained over 3 months except for LPS at month 3. The non-approved 40/30 mg dose appeared somewhat more effective than the approved 20/15 mg dose in both sexes, although no formal testing of dose response was performed. The 95% confidence intervals for differences from placebo overlapped for women and men, indicating no important sex differences in the efficacy of suvorexant. Thus, our analysis shows that suvorexant is similarly effective in women and men.
With regard to safety, women reported more adverse events than men across all treatment groups, including placebo, consistent with previous observations of increased adverse event reporting by women for other conditions (Zopf et al. 2008). The adverse event profile appeared generally similar in women and men. The most frequent adverse event in both sexes was somnolence which was typically transient and mild-to-moderate in intensity. Of note, women taking suvorexant 20/15 mg showed an increase in somnolence versus placebo (8.5 vs 3.4%) whereas men did not (3.4 vs 4.2%). This could be a chance finding given that both men and women showed an increase in somnolence for suvorexant 40/30 mg versus placebo, albeit the difference was somewhat larger for women (11.1 vs 2.3%) than men (10.1 vs 4.2%). The data could also be interpreted as suggesting that women taking suvorexant may be more prone to next-day somnolence than men. Whether this increase could be related to slightly higher suvorexant exposure in women than men (~5% higher 9 h after dosing across doses of 10–40 mg) is unknown.
The following limitations to our analysis should be noted. First, this was a generally healthy population without significant comorbidities that could have contributed to insomnia. Results may differ in real-world use in a more general insomnia population with comorbidities that might differentially affect the sexes such as depression, or differential use of concomitant medications, alcohol, or stimulants. Second, our analysis did not account for body mass which has an inversely related effect on the apparent clearance of suvorexant in both sexes; the effect is most notable in obese women where suvorexant area-under-the-curve and maximum plasma concentrations are increased by 46 and 25% compared to non-obese women. Third, we did not account for other factors which could have potentially modified the findings in women such as stage of menstrual cycle, menopausal status, etc.
In conclusion, suvorexant 20/15 and 40/30 mg were generally effective and well-tolerated by women and men with insomnia, with no apparent major differences between the sexes. Consequently, the prescribing instructions for suvorexant do not recommend any dose adjustment based on the patient’s sex. Given that the approved doses are 10–20 mg, the 20/15 mg data are the most clinically relevant.
References
American Psychiatric Association (2000) Diagnostic and statistical manual of mental disorders, 4th edn. American Psychiatric Association, Washington
Boivin DB, Shechter A, Boudreau P, Begum EA, Ng Ying-Kin NM (2016) Diurnal and circadian variation of sleep and alertness in men vs. naturally cycling women. Proc Natl Acad Sci U S A 113:10980–10985
Cosgrove KP, Mazure CM, Staley JK (2007) Evolving knowledge of sex differences in brain structure, function, and chemistry. Biol Psychiatry 62:847–855
Cox CD, Breslin MJ, Whitman DB, Schreier JD, McGaughey GB, Bogusky MJ, Roecker AJ, Mercer SP, Bednar RA, Lemaire W, Bruno JG, Reiss DR, Harrell CM, Murphy KL, Garson SL, Doran SM, Prueksaritanont T, Anderson WB, Tang C, Roller S, Cabalu TD, Cui D, Hartman GD, Young SD, Koblan KS, Winrow CJ, Renger JJ, Coleman PJ (2010) Discovery of the dual orexin receptor antagonist [(7R)-4-(5-chloro-1,3-benzoxazol-2-yl)-7-methyl-1,4-diazepan-1-yl][5-methyl-2-(2H −1,2,3-triazol-2-yl)phenyl]methanone (MK-4305) for the treatment of insomnia. J Med Chem 53:5320–5332
Dijk DJ, James LM, Peters S, Walsh JK, Deacon S (2010) Sex differences and the effect of gaboxadol and zolpidem on EEG power spectra in NREM and REM sleep. J Psychopharmacol 24:1613–1618
Farkas RH, Unger EF, Temple R (2013) Zolpidem and driving impairment—identifying persons at risk. N Engl J Med 369:689–691
Franconi F, Brunelleschi S, Steardo L, Cuomo V (2007) Gender differences in drug responses. Pharmacol Res 55:81–95
Gandhi M, Aweeka F, Greenblatt RM, Blaschke TF (2004) Sex differences in pharmacokinetics and pharmacodynamics. Annu Rev Pharmacol Toxicol 44:499–523
Herring WJ, Snyder E, Budd K, Hutzelmann J, Snavely D, Liu K, Lines C, Roth T, Michelson D (2012) Orexin receptor antagonism for treatment of insomnia: a randomized clinical trial of suvorexant. Neurology 79:2265–2274
Herring WJ, Connor KM, Ivgy-May N, Snyder E, Liu K, Snavely DB, Krystal AD, Walsh JK, Benca RM, Rosenberg R, Sangal RB, Budd K, Hutzelmann J, Leibensperger H, Froman S, Lines C, Roth T, Michelson D (2016) Suvorexant in patients with insomnia: results from two 3-month randomized controlled clinical trials. Biol Psychiatry 79:136–148
Jacobson R (2014) Psychotropic drugs affect men and women differently. Scientific American Mind July 1 2014. http://www.scientificamerican.com/article/psychotropic-drugs-affect-men-and-women-differently/ Accessed November 18, 2016.
Mallampalli MP, Carter CL (2014) Exploring sex and gender differences in sleep health: a Society for Women's Health Research report. J Women's Health (Larchmt) 23:553–562
Michelson D, Snyder E, Paradis E, Chengan-Liu M, Snavely DB, Hutzelmann J, Walsh JK, Krystal AD, Benca RM, Cohn M, Lines C, Roth T, Herring WJ (2014) Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol 13:461–471
Mieda M, Sakurai T (2013) Orexin (hypocretin) receptor agonists and antagonists for treatment of sleep disorders. Rationale for development and current status. CNS Drugs 27:83–90
Mong JA, Baker FC, Mahoney MM, Paul KN, Schwartz MD, Semba K, Silver R (2011) Sleep, rhythms, and the endocrine brain: influence of sex and gonadal hormones. J Neurosci 31:16107–16116
Nowakowski S, Meers J, Heimbach E (2013) Sleep and women's health. Sleep Med Res 4:1–22
Roth T, Lines C, Vandormael K, Ceesay P, Anderson D, Snavely D (2010) Effect of gaboxadol on patient-reported measures of sleep and waking function in patients with primary insomnia: results from two randomized, controlled, 3-month studies. J Clin Sleep Med 6:30–39
Roehrs T, Roth T (2012) Insomnia pharmacotherapy. Neurotherapeutics 9:728–738
Sun H, Kennedy WP, Wilbraham D, Lewis N, Calder N, Li X, Ma J, Yee KL, Ermlich S, Mangin E, Lines C, Rosen L, Chodakewitz J, Murphy GM (2013) Effects of suvorexant, an orexin receptor antagonist, on sleep parameters as measured by polysomnography in healthy men. Sleep 36:259–267
Svetnik V, Snyder ES, Ma J, Tao P, Lines C, Herring WJ (2017) EEG spectral analysis of NREM sleep in a large sample of patients with insomnia and good sleepers: effects of age, sex and part of the night. J Sleep Res 26:92–104
Winrow CJ, Gotter AL, Cox CD, Doran SM, Tannenbaum PL, Breslin MJ, Garson SL, Fox SV, Harrell CM, Stevens J, Reiss DR, Cui D, Coleman PJ, Renger JJ (2011) Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet 25:52–61
Winrow CJ, Renger JJ (2014) Discovery and development of orexin receptor antagonists as therapeutics for insomnia. Br J Pharmacol 171:283–293
Zhang B, Wing YK (2006) Sex differences in insomnia: a meta-analysis. Sleep 29:85–93
Zopf Y, Rabe C, Neubert A, Gassmann KG, Rascher W, Hahn EG, Brune K, Dormann H (2008) Women encounter ADRs more often than do men. Eur J Clin Pharmacol 64:999–1004
Acknowledgements
These studies were funded by Merck & Co., Inc., Kenilworth, NJ, USA. Mingqui Wu, formerly from Merck & Co., Inc., contributed to the statistical analysis. Sheila Erespe from Merck & Co., Inc. assisted with the submission.
Contributors
WJH was involved in study concept and design, interpretation of data, and drafting of manuscript. KMC, DBS, YZ, RMB, ADK, JKW, TR, and DM were involved in study concept and design and interpretation of data. JH was involved in study concept and design and acquisition of data. ES was involved in study concept and design, analysis and interpretation of data, and drafting of manuscript. DM-W was involved in interpretation of data. CL was involved in interpretation of data and drafting of manuscript. All authors reviewed and/or revised the manuscript for intellectual content and approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The trials were conducted in accordance with principles of Good Clinical Practice and were approved by the appropriate institutional review boards and regulatory agencies for each site. Informed consent was obtained from all patients. The trials were registered at ClinicalTrials.gov (NCT01097616, NCT01097629, NCT01021813).
Conflict of interests
WJH, KMC, ES, DBS, YZ, JH, DM-W, CL, and DM are current or former employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ USA and own or owned stock/stock options in the Company.
RMB has served as a consultant to and receives research support from Merck & Co., Inc., and has served as a consultant to Janssen and Jazz over the past 2 years.
ADK has received grants/research support from NIH, Teva, Sunovion, Astellas, Abbott, Neosync, Brainsway, Janssen, ANS St. Jude, Novartis. He has served as a consultant to Abbott, Astellas, AstraZeneca, Attentiv, BMS, Teva, Eisai, Eli Lilly, GlaxoSmithKline, Jazz, Janssen, Merck, Neurocrine, Novartis, Otsuka, Lundbeck, Roche, Sanofi-Aventis, Somnus, Sunovion, Somaxon, Takeda, Transcept, Vantia.
JKW has received research support from the following in the past 2 years: Apnex, Merck & Co., Inc., Novo Nordisk, Respironics, Vanda, and has provided consulting services to Merck & Co., Inc., Somnus, Transcept, Vanda, Ventus, and Vivus.
TR has received grants/research support from Aventis, Cephalon, GlaxoSmithKline, Neurocrine, Pfizer, Sanofi, Schering-Plough, Sepracor, Somaxon, Syrex, Takeda, TransOral, Wyeth and Xenoport; has acted as a consultant for Abbott, Acadia, Acoglix, Actelion, Alchemers, Alza, Ancil, Arena, AstraZeneca, Aventis, AVER, BMS, BTG, Cephalon, Cypress, Dove, Elan, Eli Lilly, Evotec, Forest, GlaxoSmithKline, Hypnion, Impax, Intec, Intra-Cellular, Jazz, Johnson–Johnson, King, Lundbeck, McNeil, MediciNova, Merck & Co., Inc., Neurim, Neurocrine, Neurogen, Novartis, Orexo, Organon, Prestwick, Procter–Gamble, Pfizer, Purdue, Resteva, Roche, Sanofi, Schering-Plough, Sepracor, Servier, Shire, Somaxon, Syrex, Takeda, TransOral, Vanda, Vivometrics, Wyeth, Yamanuchi, and Xenoport; and has participated in speaking engagements supported by Cephalon, Sanofi, and Takeda.
Funding
The studies were funded by Merck & Co., Inc., Kenilworth, NJ, USA. A Scientific Advisory Committee comprised Merck and non-Merck investigators contributed to the development of the protocols, statistical analysis plans, analysis and interpretation of data, served as authors of the manuscript and was responsible for the decision to submit the manuscript for publication. The funding organization was involved in the design and conduct of the studies, the collection, management, analysis, interpretation of data, and preparation, review, and approval of the manuscript.
Rights and permissions
About this article

Cite this article
Herring, W., Connor, K.M., Snyder, E. et al. Clinical profile of suvorexant for the treatment of insomnia over 3 months in women and men: subgroup analysis of pooled phase-3 data. Psychopharmacology 234, 1703–1711 (2017). https://doi.org/10.1007/s00213-017-4573-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-017-4573-1