
Abstract
Rationale
Evidence suggests that neuronal nicotinic acetylcholine receptor (nAChR) ligand lobeline has antidepressant-like properties.
Objectives
The present study investigated the effects of lobeline on nicotine withdrawal-induced depression-like behavior.
Methods
Adult C57BL/6J mice were exposed to nicotine (200 μg/ml) in drinking solution for 3 weeks. During withdrawal, depression-like behavior was measured by the forced swim test (FST). We also determined norepinephrine (NE) levels in the prefrontal cortex (PFC) and hippocampus during nicotine withdrawal. Furthermore, we determined the effects of repeated treatment with lobeline or a selective α4β2 nAChR ligand 3-(pyridine-3́-yl)-cytisine on brain-derived neurotrophic factor (BDNF) and phosphorylated cAMP-responsive element binding (p-CREB) protein expression in the hippocampus.
Results
Withdrawal from chronic nicotine increased immobility time in the FST, a measure for depression-like behavior. Pretreatment with lobeline significantly decreased immobility time during nicotine withdrawal. In addition, pretreatment with lobeline attenuated nicotine withdrawal-induced increased NE levels in the PFC and hippocampus. Further, repeated treatment with lobeline or 3-(pyridine-3́-yl)-cytisine decreased immobility time in the FST and reduced withdrawal-induced increased BDNF and p-CREB expression in the hippocampus.
Conclusions
Taken together, our results indicate that lobeline attenuated nicotine withdrawal-induced depression-like behavior likely by targeting brain nAChRs, noradrenergic neurotransmission, and/or hippocampal BDNF. Thus, lobeline may have some potential to prevent smoking relapse by counteracting nicotine withdrawal-induced depression in humans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tobacco smoking behavior is a leading preventable cause of premature death in the USA and other countries (Benowitz 2010). Smoking cessation is believed to be linked to the heightened depressive states (Zaniewska et al. 2010). Relapse is thought to arise, in part, from the need to counteract the negative affective symptoms such as depression (Hughes et al. 2007). Evidence suggests that the negative affective symptoms of nicotine withdrawal are mediated by neuronal nicotinic acetylcholine receptors (nAChRs) (Watkins et al. 2000; Jackson et al. 2008, 2009). One of the most extensively studied effects of chronic nicotine treatment in humans and rodents is the upregulation of nAChR expression and function (Nguyen et al. 2003). Chronic oral nicotine administration increases density of neuronal nAChRs in mice (Sparks and Pauly 1999). Chronic nicotine causes the largest increase in [3H]-cytisine binding (marker for α4β2 nAChRs) in the hippocampus and cortex. The nAChR upregulation can contribute to the mechanisms that maintain nicotine consumption, as well as withdrawal symptoms including depression by influencing release of major neurotransmitters and diverse signaling pathways. For example, neuronal nAChRs are thought to regulate release of monoamine neurotransmitters such as norepinephrine (NE) and serotonin (Rahman et al. 2008; Sajja et al. 2010; Tani et al. 1997). Monoamine neurotransmitters play important roles in depression or nicotine withdrawal (Gäddnäs et al. 2000; Ressler and Nemeroff 2000). For instance, Gäddnäs et al. (2000) reported that withdrawal from chronic nicotine increases NE levels in the hypothalamus. Altered noradrenergic neurotransmission could be related, in part, to the neuroadaptations observed during nicotine withdrawal. A previous report indicates that NE induces cell signaling pathways of neuroplasticity including cAMP-responsive element binding protein (CREB) in the hippocampus (Chen et al. 2007). The activation of CREB signaling pathways leads to the gene expression of brain-derived neurotrophic factor (BDNF), promoting neuroplastic changes. In addition to a downstream target of CREB signaling, BDNF is an upstream activator of CREB (Kivinummi et al. 2011). Moreover, BDNF is involved in the neuroadaptations underlying both depression and addictive behaviors (see Duman and Aghajanian 2012; McClung and Nestler 2007 for reviews). The above findings suggest that neuronal nAChRs are potential targets for the treatment of nicotine withdrawal due to critical involvements in the neurochemical and neuroadaptive functions.
Evidence also suggests that blockade of neuronal nAChR function may produce antidepressant-like effects (Sanberg et al. 2012). For example, nAChR antagonists such as mecamylamine and dihydro-β-erythroidine (DHβE) produce antidepressant-like effects in mice (Rabenstein et al. 2006). Recently, we found that nAChR ligand lobeline has antidepressant-like and anxiolytic-like properties (Roni and Rahman 2011, 2013) in mice. Lobeline is considered as a partial agonist or an antagonist at nAChRs. The observations from receptor binding studies suggest that lobeline is a nonselective nAChR antagonist with high affinity for α4β2 and α3β2 nAChRs (Dwoskin and Crooks 2002; Parker et al. 1998). Lobeline inhibits the effects of nicotine in a concentration-dependent manner in voltage-clamped Xenopus oocytes expressing α4β2 nAChRs (Damaj et al. 1997). Lobeline blocks nicotine-evoked [3H]NE release from rat locus coeruleus cells and nicotine-evoked [3H]dopamine overflow from rat striatal slices (Gallardo and Leslie 1998; Miller et al. 2000). Behavioral effects of lobeline were neither enhanced nor blocked by β2-selective nAChR antagonist DHβE (Damaj et al. 1997). Similarly, behavioral effects of lobeline remained unchanged or not inhibited in the presence of nonselective antagonist mecamylamine (Damaj et al. 1997; Stolerman et al. 1995). Additionally, lobeline reduces forced swim stress (FSS)-induced increased NE levels in the prefrontal cortex (PFC) (Roni and Rahman 2013). Besides, our laboratory has recently studied lobeline for the treatment of alcohol dependence (Sajja et al. 2010; Sajja and Rahman 2011, 2012). We found that lobeline decreased voluntary ethanol intake in rats (Bell et al. 2009) and mice (Sajja and Rahman 2011, 2012). In addition, lobeline decreased ethanol-induced midbrain dopamine function and metabolism in mice (Sajja et al. 2010). Similarly, lobeline was shown to decrease behavioral and neurochemical effects of psychostimulants (Harrod et al. 2001; Miller et al. 2001). While previous studies indicate the potential efficacy of lobeline in the treatment of depression or smoking cessation (Glover et al. 1998), no study examined the effects of lobeline on nicotine withdrawal-induced depression.
The goal of the present study was to investigate the effects of lobeline in mice withdrawn from chronic oral nicotine. We selected oral nicotine administration because it produces numerous small peaks in plasma nicotine levels, an effect that closely resembles the condition found in human users (Sparks and Pauly 1999). Moreover, oral nicotine administration effectively produces dependence, upregulates neuronal nAChRs (Sparks and Pauly 1999), increases striatal dopamine levels (Pietilä et al. 1995), and produces less stress than invasive nicotine administration methods. We hypothesized that lobeline will decrease depression-like behaviors during nicotine withdrawal. In addition, we anticipated that the changes in depression-like behaviors would be correlated with changes in NE neurotransmitter and relevant neuroadaptive markers such as CREB and BDNF in relevant brain regions. Therefore, we studied the effects of lobeline on FSS-induced NE levels in the PFC and hippocampus. Finally, in order to understand the effects of lobeline on neuroadaptive changes related to nicotine withdrawal, we measured phosphorylated-CREB (p-CREB) and BDNF expressions in the PFC and hippocampus.
Materials and methods
Animals
Male C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). These mice were selected because they consume more nicotine than any other inbred strains (Robinson et al. 1996). They were single-housed in standard shoebox cages (29 × 18 × 12 cm), under standard laboratory conditions (22 ± 2 °C, relative humidity 50–60 %) and maintained on a 12-h light/dark cycle (lights on at 0600 hours) with free access to food and water. Mice were 10–12 weeks of age at the start of the experiment. The behavioral experiments were conducted between 0900 and 1600 hours, and mice were allowed to habituate to the testing room for at least 30 min. Counterbalanced design was used to control for any order effects. All procedures were in compliance with the National Institutes of Health guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at South Dakota State University.
Drugs and nicotine solution
Nicotine hydrogen tartrate, lobeline hydrochloride, and bupropion hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO, USA). The 3-(pyridine-3́-yl)-cytisine (3-pyr-cyt) was purchased from Tocris Bioscience (Ellisville, MO, USA). Nicotine hydrogen tartrate was dissolved in 2 % saccharine solution. The nicotine solution was replenished every other day with fresh solution. Lobeline (1 or 4 mg/kg), bupropion (8 mg/kg), and 3-pyr-cyt (0.5 mg/kg) were dissolved in saline before subcutaneous injection in a volume of 0.01 ml/g body weight of animal. Doses, expressed as salt form of the drugs, were selected based on our previous studies (Roni and Rahman 2013).
Establishment of nicotine dependence
Mice were allowed to drink from 200 μg/ml nicotine (as free base) solution for 21 days (Grabus et al. 2005). The sweetened nicotine solution was the only fluid available for drinking during that period. Control mice received only 2 % saccharine solution. Nicotine solution was given in 15-ml plastic centrifuge tubes fitted with stainless steel straight sipper tubes with balls. Daily consumption of nicotine solution (ml) was measured from graduated centrifuge tubes. Four tubes were kept in empty cages to measure loss of solution by leakage or evaporation. The average loss of solution was subtracted from the test values.
Measurement of cotinine levels
Serum cotinine levels were measured after 2 weeks of nicotine exposure. About 40 μl blood samples were collected from the tail vein of mice in a microvette tube (Microvette 500 Z-Gel, Sarstedt, Germany) during the first hour of dark cycle. Serums were separated by centrifugation at 10,000×g for 5 min at 20 °C. Serum cotinine levels were measured by enzyme immunosorbent assay (OraSure Technologies, Bethlehem, PA, USA) according to manufacturer’s instructions (Klein et al. 2004).
Experimental procedure
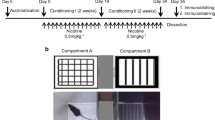
In experiment 1, we have determined the acute effects of lobeline during nicotine withdrawal (Fig. 1a). After 21 days of drinking, nicotine solutions were replaced with tap water to initiate withdrawal. Saccharine solutions were replaced with tap water in the control group. Body weights of mice were measured every week throughout the experiment. Lobeline (1 or 4 mg/kg) or saline was administered 24 h following withdrawal. Immediately following lobeline or saline treatment, somatic signs and locomotor activities were measured consecutively. Seventy-two hours following above tests, mice were administered with lobeline, saline, or bupropion, and the forced swim test (FST) was performed 20 min later. Bupropion was used as a positive control since it is prescribed as an antidepressant as well as smoking cessation agent. Previous report suggests that only high dose of bupropion (30 mg/kg) increases locomotor activity in nicotine-exposed rats (Wilkinson et al. 2006). We selected a low dose of bupropion (8 mg/kg) in the present study to avoid potential generalized motor activity from influencing the FST results.

Experimental timeline. a Mice were exposed to oral nicotine solution for 3 weeks. Somatic sign measurement (SS), locomotor activity (LMA), and forced swim test (FST) were carried out during withdrawal. Black arrow indicates acute injection of drug or saline. b Following withdrawal from chronic oral nicotine, mice were administered drug or saline for 1 week. The FST was performed 24 h after last drug injections. See section “Experimental procedure” for further details
Nicotine withdrawal-induced increased depression-like behavior in the FST persists for about 2–9 weeks (Mannucci et al. 2006; Zaniewska et al. 2010). To determine the relationships of increased depression-like behavior with NE and serotonin levels, we sacrificed the mice after 2 weeks of withdrawal. Mice received lobeline or saline treatment before a FST session, and brain tissue was immediately harvested for NE and serotonin assay.
In experiment 2, we have determined the effects of repeated lobeline treatment during nicotine withdrawal (Fig. 1b). After 21 days of drinking, nicotine solutions were replaced with tap water to initiate withdrawal. Saccharine solutions were replaced with tap water in the control group. Following 24 h of withdrawal, mice received once daily treatment with lobeline, 3-pyr-cyt, or saline for seven consecutive days (Harrod and Van Horn 2009). The FST was performed 24 h after the last drug injection. Mice were sacrificed on the next day to collect brain samples for Western blot analysis. Due to lack of a specific antagonist, we used a selective α4β2 nAChR partial agonist (3-pyr-cyt) for comparing with the lobeline treatment. A single dose of 3-pyr-cyt was used based on our previous studies (Roni and Rahman 2013).
Somatic signs
Twenty-four hours following withdrawal, mice were observed for 20 min in home cages to detect somatic signs. Frequency of somatic signs such as paw tremor (forelimb shakes), head shakes, and body tremor was counted (Grabus et al. 2005; Mannucci et al. 2006).
Locomotor activity
Each mouse was placed in the periphery of a square chamber (40 × 40 × 35 cm) and allowed to explore freely for 10 min (after a 5-min chamber acclimation period) (Grabus et al. 2005). All sessions were recorded and analyzed by video tracking system (ANY-maze-Stoelting, Inc., Wood Dale, IL, USA) to measure the total distance traveled (m) (Roni and Rahman 2011).
Forced swimming test
The FST, a widely used behavioral test to assess the pharmacological efficacy of antidepressants in rodents, was performed with minor modifications (Porsolt et al. 1977). Each mouse was placed in a cylindrical Plexiglas tank (45-cm height × 20-cm diameter), filled with 25 cm of water (20–22 °C), for 15 min (Roni and Rahman 2013). The experiments were video recorded and immobility time was measured by two skilled observers. Immobility was counted when no additional activities were observed other than that required to keep the head above water.
Monoamine assay
Monoamine assay was performed as described previously (Roni and Rahman 2013). Twenty minutes after saline or drug treatment, mice were forced to swim for 10 min in the FST apparatus. Mice were sacrificed by rapid decapitation immediately after the FSS. The PFC and hippocampus were dissected from 1-mm coronal sections using Allen Brain Atlas and mouse brain stereotaxic coordinates (Franklin and Paxinos 2007). Samples were stored at −80 °C until analysis. Upon assay, tissues were diluted with 0.1 N perchloric acid (1:10 as g/ml); samples were homogenized and centrifuged (14,000×g for 30 min at 4 °C). Resulting supernatants (20 μl) were injected onto a high-performance liquid chromatography system coupled with electrochemical detection unit (ESA Inc., Chelmsford, MA, USA). The guard cell potential was set at +350 mV. The gain of the detector was set at 100 nA for both electrodes (electrode 1, −150 mV; electrode 2, +220 mV). The mobile phase (pH = 3.0) consisted of 75 mM NaH2PO4, 1.7 mM 1-octane sulfonic acid, 25 μM EDTA, 100 μl/l triethylamine, and 100 ml/l of acetonitrile. The C-18 analytical column (BetaBasic-18 column, 150 × 3 mm, Thermo Hypersil-Keystone, PA, USA) was used as stationary phase, and the flow rate was 0.5 ml/min. Data were collected in ESA chromatography data system (EZChrom Elite, Chelmsford, MA, USA). Peak heights and calibration factors based on the standard solutions of NE and serotonin (1–100 ng/ml) were used to calculate the amount of NE and serotonin, and the values were expressed as nanograms per milligram weight of tissue.
Western blot analysis
The PFC and hippocampus were dissected, frozen in liquid nitrogen, and stored at −80 °C until analysis. Western blot analysis was performed as described previously with some modifications (Xu et al. 2006). Tissue samples were homogenized in modified RIPA buffer containing Dulbecco’s phosphate-buffered saline (pH 7.4), 1 % Igepal CA-630, 0.1 % sodium dodecyl sulfate (SDS), and protease inhibitor mix (cOmplete, Mini, Roche, Indianapolis, IN, USA). The samples were centrifuged (16,000×g, 20 min at 4 °C) and supernatants were collected. Protein concentration was determined by bicinchoninic acid assay (Pierce, Rockford, IL, USA) using albumin as standard. Equal amounts of protein (60 μg) were loaded onto 10 % gels for SDS polyacrylamide gel electrophoresis. Separated proteins were transferred onto nitrocellulose membranes at 80 V for 90 min. Membranes were blocked on a gyro-rocker with 5 % nonfat dry milk in Tris-buffered saline/0.1 % Tween-20 (TBST) for 1 h, rinsed with TBST, and subsequently incubated overnight at 4 °C with primary antibodies for Ser-133 p-CREB (1:500, rabbit polyclonal, Santa Cruz Biotech, USA), BDNF (H-117, 1:300, rabbit polyclonal, Santa Cruz Biotech, USA), or β-tubulin (E7, 1:5,000, mouse monoclonal, University of Iowa, USA). After incubation, membranes were washed in TBST, followed by incubation with appropriate horseradish peroxide-conjugated secondary antibodies, diluted in blocking buffer at a concentration of 1:5,000. Bound antibodies were detected with ECL Prime reagent (Amersham, Buckinghamshire, UK), and protein quantification was performed using densitometric analysis.
Statistical analyses
Data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for multiple comparisons using GraphPad Prism (GraphPad Inc., San Diego, CA, USA). Data from Western blot studies were expressed as p-CREB or BDNF/β-tubulin expression (% control). The difference between treatments was considered significant at p <0.05. Results were expressed as mean ± SEM.
Results
Nicotine consumption and cotinine levels
Daily average consumption of oral nicotine solution (200 μg/ml, 21 days) was 6.18 ± 0.11 ml which corresponds to about 49 mg/kg nicotine consumption by oral route. Serum cotinine level, measured during the first hour of dark phase, was 142.86 ± 3.7 ng/ml for mice receiving oral nicotine for 2 weeks. Cotinine levels were below detection level (<8 ng/ml) during nicotine withdrawal. The body weight of nicotine-treated mice did not differ significantly from that of control during nicotine withdrawal.
Effects of lobeline on the somatic signs
As shown in Fig. 2 and Table 1, mice withdrawn from 200 μg/ml oral nicotine exhibited significantly higher numbers of somatic signs such as paw tremors, body tremors, and head shakes compared to saccharine-exposed controls (Fig. 2; F 3,21 = 25.56; p < 0.0001). Multiple comparisons of means revealed that both 1- and 4-mg/kg lobeline treatments significantly decreased withdrawal-induced somatic signs (p < 0.05 or p < 0.0001, respectively) compared to saline-treated group.

Effects of lobeline (Lob) on somatic signs of withdrawal. Somatic signs were counted about 24 h after cessation of chronic nicotine administration (200 μg/ml, 3 weeks). Mice (n = 6–7) received Lob (1 or 4 mg/kg, s.c.) or saline (Sal, s.c.) injections immediately before somatic sign measurements. Control group was exposed to 2 % saccharine solution. Data are presented as mean ± SEM. *p < 0.05; **p < 0.0001
Effects of lobeline on the locomotor activity
As shown in Fig. 3, withdrawal from chronic oral nicotine did not produce significant change in the locomotor activity compared to saccharine-exposed controls. In addition, lobeline treatment had no effect on locomotor activity during withdrawal.

Lobeline (Lob) had no effect on locomotor activity during nicotine withdrawal in mice. Locomotor activities were measured about 24 h after cessation of chronic nicotine administration (200 μg/ml, 3 weeks). Control group was exposed to 2 % saccharine solution. Data are presented as mean ± SEM
Effects of lobeline in the FST
The effects of acute lobeline treatment in the FST are shown in Fig. 4. There was a main effect of treatment on immobility time in the FST (F 4,27 = 12; p < 0.0001). Multiple comparisons of means revealed that immobility time was significantly higher in mice withdrawn from chronic oral nicotine compared to saccharine-exposed control (p < 0.05). Conversely, pretreatment with lobeline (1 or 4 mg/kg) significantly decreased immobility time during nicotine withdrawal (p < 0.0001 or p < 0.05, respectively). As a positive control, bupropion also significantly decreased immobility time during nicotine withdrawal (p < 0.0001).

Effects of acute lobeline (Lob) treatment on immobility time in the forced swim test (FST) during nicotine withdrawal in mice. The FST was performed about 96 h after cessation of chronic nicotine administration (200 μg/ml, 3 weeks). Mice (n = 6–7) received Lob (1 or 4 mg/kg, s.c.), bupropion (Bup, 8 mg/kg, s.c.), or saline (Sal, s.c.) injections 20 min before the FST. Control group was exposed to 2 % saccharine solution. Data are presented as mean ± SEM. *p < 0.05; **p < 0.0001
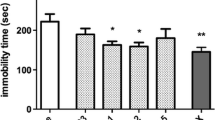
The effects of repeated lobeline treatment for 7 days in the FST are shown in Fig. 5. There was a significant overall effect of repeated treatment on immobility time (F 3,20 = 20.8; p < 0.0001). Multiple comparisons of means showed that immobility time remained significantly high in mice withdrawn from chronic nicotine compared to saccharine-exposed control (p < 0.01). Repeated lobeline treatment significantly decreased immobility time during withdrawal (p < 0.0001). Similarly, immobility time was reduced by 3-pyr-cyt, a selective α4β2 nAChR ligand and partial agonist (p < 0.0001).

Effects of repeated lobeline (Lob) treatment on immobility time in the forced swim test (FST) during nicotine withdrawal in mice. Mice (n = 6) received Lob (1 or 4 mg/kg, s.c.), 3-pyr-cyt (0.5 mg/kg, s.c.), or saline (Sal, s.c.) injections for 7 days following cessation of nicotine administration (200 μg/ml, 3 weeks). The FST was performed 24 h after last injections. Control group was exposed to 2 % saccharine solution. Data are presented as mean ± SEM. *p < 0.01; **p < 0.0001
Effects of lobeline on brain monoamines
Effects of lobeline treatment on NE levels in the hippocampus are shown in Fig. 6a. One-way ANOVA indicated that there was a treatment effect on NE levels (F 3,14 = 6.6; p < 0.01). Multiple comparisons revealed that NE levels were significantly higher in mice withdrawn from chronic oral nicotine compared to saccharine-exposed controls (p < 0.05). Conversely, pretreatment with 1 mg/kg lobeline significantly reduced nicotine withdrawal-induced increased NE levels in the hippocampus (p < 0.01). However, 4 mg/kg lobeline did not significantly change NE levels.

a Effects of lobeline (Lob) on hippocampal norepinephrine (NE) levels after the forced swim test (FST) during nicotine withdrawal. b Effects of Lob on NE levels in the prefrontal cortex after the FST during nicotine withdrawal. Mice (n = 4–5) received Lob (1 or 4 mg/kg, s.c.) or saline (Sal, s.c.) injections 20 min before the FST. Control group was exposed to 2 % saccharine solution. Data are presented as mean ± SEM. *p < 0.05; **p < 0.01
Effects of lobeline treatment on NE levels in the PFC are shown in Fig. 6b. There was a significant main effect of treatment on NE levels in the PFC (F 3,13 = 4; p < 0.05). Similar to the hippocampus, increased NE levels were observed in the PFC of mice withdrawn from chronic oral nicotine compared to saccharine-exposed controls (p < 0.05). Pretreatment with 1 mg/kg lobeline decreased nicotine withdrawal-induced increased NE levels in the PFC (p < 0.05). Pretreatment with 4 mg/kg lobeline did not have a significant effect on NE levels in the PFC.
Nicotine withdrawal did not significantly change the serotonin levels in the hippocampus (control 0.35 ± 0.1 ng/mg, nicotine withdrawn 0.41 ± 0.05 ng/mg) or the PFC (control 0.45 ± 0.1 ng/mg, nicotine withdrawn 0.43 ± 0.1 ng/mg) compared to control. In addition, pretreatment with lobeline 1 or 4 mg/kg did not change serotonin levels in both brain regions during nicotine withdrawal (data not shown).
Effects of lobeline on BDNF and p-CREB
As shown in Fig. 7, one-way ANOVA indicated a significant overall treatment effect on BDNF expression in the hippocampus (F 3,17 = 7.4; p < 0.01) following repeated administration of saline or drugs during withdrawal. Further post hoc analysis indicated that withdrawal from chronic oral nicotine significantly increased BDNF expression compared to control (p < 0.05). Conversely, lobeline (1 mg/kg) pretreatment for 7 days significantly suppressed nicotine withdrawal-induced upregulation of BDNF in the hippocampus (p < 0.01). Likewise, pretreatment with 3-pyr-cyt significantly decreased BDNF expression in the same region (p < 0.01). However, no significant treatment effect was observed on BDNF expression in the PFC (data not shown).

Effects of repeated treatment with lobeline (Lob) on expression of BDNF in the hippocampus. Mice (n = 5–6) received Lob (1 mg/kg, s.c.), 3-pyr-cyt (0.5 mg/kg, s.c.), or saline (Sal, s.c.) injections for 7 days following cessation of nicotine administration (200 μg/ml, 3 weeks). Hippocampi were collected 48 h after last injections. Control group was exposed to 2 % saccharine solution. Data are presented as mean ± SEM (n = 5–6). *p < 0.05; **p < 0.01
One-way ANOVA revealed a significant main effect of treatment on p-CREB expression in the hippocampus (F 3,18 = 5; p < 0.01) following repeated administration of saline or drugs during withdrawal. As shown in Fig. 8, although there was a trend of p-CREB upregulation in nicotine-withdrawn mice compared to control, multiple comparisons of means did not reveal significant difference. Lobeline (1 mg/kg) pretreatment for 7 days did not significantly change p-CREB expression in the hippocampus. However, pretreatment with α4β2 nAChR-selective ligand 3-pyr-cyt decreased p-CREB expression in the hippocampus compared to nicotine-withdrawn mice receiving saline treatment (p < 0.01). Conversely, there was no significant effect of treatment on p-CREB expression in the PFC (data not shown).

Repeated treatment with lobeline (Lob) had modest effect on p-CREB expression in the hippocampus. Mice (n = 5–6) received Lob (1 mg/kg, s.c.), 3-pyr-cyt (0.5 mg/kg, s.c.), or saline (Sal, s.c.) injections for 7 days following cessation of nicotine administration (200 μg/ml, 3 weeks). Hippocampi were collected 48 h after last injections. Control group was exposed to 2 % saccharine solution. Data are presented as mean ± SEM
Discussion
The important findings of the present study are that lobeline at lower dose significantly reduced nicotine withdrawal-induced depression-like behavior in mice. In addition, pretreatment with lobeline significantly decreased swim stress-induced increased NE levels in the PFC and hippocampus during nicotine withdrawal. Further, repeated lobeline treatment reduced nicotine withdrawal-induced increased BDNF and p-CREB expression in the hippocampus. Overall, the results indicate that lobeline suppressed nicotine withdrawal-induced depression-like behavior likely by targeting brain nAChRs, noradrenergic neurotransmission, and/or hippocampal BDNF.
In the present study, nicotine withdrawal increased immobility time in the FST, indicating depression-like behavior. To the best of our knowledge, this is the first report of depression-like behavior induced by withdrawal from chronic oral nicotine. Interestingly, Andreasen et al. (2009) did not observe depression-like behavior in mice during oral nicotine withdrawal. The apparent discrepancy could be due to methodological differences such as mice strain (NMRI vs. C57BL/6J), length of nicotine access (21 vs. 3 weeks), or nicotine concentration (400 vs. 200 μg/ml).
Repeated lobeline treatment produced antidepressant-like effects without development of tolerance. Similarly, repeated treatment with 3-pyr-cyt, a α4β2-selective nAChR partial agonist, produced antidepressant-like effects during withdrawal likely by inhibiting α4β2 nAChR. The 3-pyr-cyt is likely to compete with endogenous ACh in vivo which is similar to competitive antagonism (Mineur et al. 2009). Previous studies indicate that β2- or α6-containing nAChRs are associated with nicotine withdrawal-induced negative mood (Jackson et al. 2008, 2009). Given that lobeline has high affinity for β2-containing nAChR subtypes (such as α4β2, α3β2, or α6β2) (Dwoskin and Crooks 2002; Parker et al. 1998), the antidepressant-like effects of lobeline are likely mediated by inhibiting those particular nAChR subtypes.
The antidepressant-like effects of lobeline during nicotine withdrawal were similar (1 mg/kg) or less (4 mg/kg) than our previous studies with nicotine-naïve mice (Roni and Rahman 2013). However, nAChR expression and function are believed to be different between nicotine and non-nicotine groups. Ribeiro-Carvalho et al. (2009) showed that nicotine-induced upregulation of α4β2 nAChRs was maintained even after 5 days of withdrawal. Therefore, nicotine-withdrawn mice were expected to respond more to lobeline treatment than nicotine-naïve mice. In contrast, nicotine-withdrawn mice did not show increased response to lobeline treatment in the FST. The possible explanation could be associated with hypothalamic-pituitary-adrenal (HPA) axis hypoactivity during nicotine withdrawal (Semba et al. 2004). The hypoactivity of the HPA axis to stress during nicotine withdrawal could make lobeline less effective as the effects of lobeline partly depend on the normal stress response of the HPA axis. Previously, we reported that lobeline decreased swim stress-induced increased corticosterone levels, a measure of HPA axis activity, in mice (Roni and Rahman 2013). Additionally, we found that nicotine-dependent mice had increased somatic signs, 24 h after withdrawal from nicotine, which are in agreement with previous studies (Grabus et al. 2005; Mannucci et al. 2006). Evidence suggests that somatic signs of nicotine withdrawal are mediated by central and partly by peripheral nAChRs (Watkins et al. 2000). It has been reported that non-β2-containing nAChRs are associated with nicotine withdrawal signs (Jackson et al. 2008; Salas et al. 2004). Being a nonselective antagonist, the effects of lobeline on somatic signs could be mediated by interaction with non-β2-containing nAChRs.
Interestingly, the lower dose of lobeline was more effective in reducing depression-like behavior but less effective in reducing somatic signs of withdrawal. The apparent discrepancy could be explained by the diverse nature of nAChRs. As discussed earlier, β2-containing nAChRs are believed to be associated with depression-like behaviors and non-β2-containing nAChRs are associated with somatic signs. Therefore, lower dose of lobeline might target β2-containing nAChRs to produce antidepressant-like effects, but higher dose of lobeline might interact with non-β2-containing nAChR subtypes to alleviate somatic signs. Although the same group of mice was used for the somatic sign measurement and the FST with a 72-h interval, a residual effect is less likely due to the short half-life of lobeline (50 min) in mice (Miller et al. 2003).
Further, we found that lobeline (1 mg/kg) significantly reduced nicotine withdrawal-induced increased NE levels in the PFC and hippocampus, the brain regions which are thought to be affected by stress and depression (Campbell and MacQueen 2004; George et al. 1994). The higher dose (4 mg/kg) of lobeline failed to reduce NE levels during nicotine withdrawal likely due to nonspecific effects, consistent with our previous studies on nicotine-naïve mice (Roni and Rahman 2013). Conversely, there was no effect of nicotine withdrawal on serotonin levels in the PFC and hippocampus. Taken together, our results suggest that antidepressant-like effects of lobeline are partly mediated by nAChR interaction with noradrenergic, but not serotonergic, function. The results support clinical findings that showed antidepressants other than selective serotonin reuptake inhibitors were effective in smoking cessation (Hughes et al. 2007). However, a recent study indicates the involvement of serotonin receptors (5-HT2C or 5-HT2A) in nicotine withdrawal-induced depression-like behaviors in rats (Zaniewska et al., 2010). Therefore, further studies are needed to confirm the role of serotonergic function in nicotine dependence.
Another important finding of the present study was that withdrawal from chronic oral nicotine increased BDNF expression in the hippocampus. Our results are in agreement with a previous study where BDNF upregulation was observed in mice withdrawn from chronic oral nicotine (Kivinummi et al. 2011). Moreover, withdrawal-induced increased plasma BDNF levels were reported in human smokers (Kim et al. 2007; Bhang et al. 2010). Withdrawal from chronic cocaine also increases BDNF levels in certain brain areas which were correlated with cocaine craving and depression-like behavior (Grimm et al. 2003; Filip et al. 2006). We found that repeated lobeline treatment decreased BDNF expression in the hippocampus, correlating with noradrenergic function and behavior during nicotine withdrawal. Therefore, antidepressant-like effects of lobeline could be mediated by regulating hippocampal BDNF levels in nicotine-withdrawn mice. Likewise, repeated 3-pyr-cyt treatment decreased BDNF levels in nicotine-withdrawn mice, indicating possible involvement of α4β2-containing nAChRs in BDNF expression.
The effects of lobeline on p-CREB levels mirrored BDNF expression but did not reach statistical significance. In contrast to the hippocampus, p-CREB or BDNF expression in the PFC remained unchanged during nicotine withdrawal. Our results are consistent with a previous study where p-CREB levels in the PFC remained unchanged after withdrawal from chronic oral nicotine (Brunzell et al. 2003).
Although we have found that increased BDNF in the hippocampus is somewhat pro-depressant during nicotine withdrawal, previous evidence indicates that chronic antidepressant treatment acts by elevating BDNF levels and promoting neurogenesis in the hippocampus (Duman and Aghajanian 2012; Krishnan and Nestler 2008). Therefore, further studies are warranted for these paradoxical findings using direct lobeline injection into the hippocampus to determine the role of hippocampal BDNF in depression during nicotine withdrawal. In addition, future studies are necessary during nicotine withdrawal to correlate directly with changes in BDNF expression in other brain regions.
In summary, the present study shows that lobeline decreases nicotine withdrawal-induced depression-like behaviors in mice. The antidepressant-like effects of lobeline are likely mediated by neuronal nAChRs and/or modulation of noradrenergic systems. The effects of lobeline treatment could also be mediated by modulation of BDNF expression in the hippocampus. Thus, lobeline may have some potential to prevent smoking relapse by counteracting nicotine withdrawal-induced depression in humans.
References
Andreasen JT, Nielsen EØ, Redrobe JP (2009) Chronic oral nicotine increases brain [3H] epibatidine binding and responsiveness to antidepressant drugs, but not nicotine, in the mouse forced swim test. Psychopharmacology (Berl) 205:517–528
Bell RL, Eilier BA, Cook JB, Rahman S (2009) Nicotinic receptor ligands reduce ethanol intake by high alcohol-drinking HAD-2 rats. Alcohol 43:581–592
Benowitz NL (2010) Nicotine addiction. N Engl J Med 362:2295–2303
Bhang SY, Choi SW, Ahn JH (2010) Changes in plasma brain-derived neurotrophic factor levels in smokers after smoking cessation. Neurosci Lett 468:7–11
Brunzell DH, Russell DS, Picciotto MR (2003) In vivo nicotine treatment regulates mesocorticolimbic CREB and ERK signaling in C57Bl/6J mice. J Neurochem 84:1431–1441
Campbell S, MacQueen G (2004) The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci 29:417–426
Chen MJ, Nguyen TV, Pike CJ, Russo-Neustadt AA (2007) Norepinephrine induces BDNF and activates the PI-3K and MAPK cascades in embryonic hippocampal neurons. Cell Signal 19:114–128
Damaj M, Patrick G, Creasy K, Martin B (1997) Pharmacology of lobeline, a nicotinic receptor ligand. J Pharmacol Exp Ther 282:410–419
Duman RS, Aghajanian GK (2012) Synaptic dysfunction in depression: potential therapeutic targets. Science 338:68–72
Dwoskin LP, Crooks PA (2002) A novel mechanism of action and potential use for lobeline as a treatment for psychostimulant abuse. Biochem Pharmacol 63:89–98
Filip M, Faron-Górecka A, Kuśmider M, Gołda A, Frankowska M, Dziedzicka-Wasylewska M (2006) Alterations in BDNF and trkB mRNAs following acute or sensitizing cocaine treatments and withdrawal. Brain Res 1071:218–225
Franklin KBG, Paxinos G (2007) The mouse brain in stereotaxic coordinates, 3rd edn. Academic Press, Inc., New York
Gäddnäs H, Pietilä K, Ahtee L (2000) Effects of chronic oral nicotine treatment and its withdrawal on locomotor activity and brain monoamines in mice. Behav Brain Res 113:65–72
Gallardo KA, Leslie FM (1998) Nicotine-stimulated release of [3H]norepinephrine from fetal rat locus coeruleus cells in culture. J Neurochem 70:663–670
George MS, Ketter TA, Post RM (1994) Prefrontal cortex dysfunction in clinical depression. Depression 2:59–72
Glover ED, Leischow SJ, Rennard SI, Glover PN, Daughton D, Quiring JN, Schneider FH, Mione PJ (1998) A smoking cessation trial with lobeline sulfate: a pilot study. Am J Health Behav 22:62–74
Grabus S, Martin B, Imad Damaj M (2005) Nicotine physical dependence in the mouse: involvement of the α7 nicotinic receptor subtype. Eur J Pharmacol 515:90–93
Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, Shaham Y (2003) Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J Neurosci 23:742–747
Harrod SB, Van Horn ML (2009) Sex differences in tolerance to the locomotor depressant effects of lobeline in periadolescent rats. Pharmacol Biochem Behav 94:296–304
Harrod SB, Dwoskin LP, Crooks PA, Klebaur JE, Bardo MT (2001) Lobeline attenuates d-methamphetamine self-administration in rats. J Pharmacol Exp Ther 298:172–179
Hughes, J.R., Stead, L.F., Lancaster, T. (2007) Antidepressants for smoking cessation. Cochrane Database Syst Rev 1:CD000031.
Jackson KJ, Martin BR, Changeux JP, Damaj MI (2008) Differential role of nicotinic acetylcholine receptor subunits in physical and affective nicotine withdrawal signs. J Pharmacol Exp Ther 325:302–312
Jackson KJ, McIntosh JM, Brunzell DH, Sanjakdar SS, Damaj MI (2009) The role of α6-containing nicotinic acetylcholine receptors in nicotine reward and withdrawal. J Pharmacol Exp Ther 331:547–554
Kim TS, Kim DJ, Lee H, Kim YK (2007) Increased plasma brain-derived neurotrophic factor levels in chronic smokers following unaided smoking cessation. Neurosci Lett 423:53–57
Kivinummi T, Kaste K, Rantamäki T, Castrén E, Ahtee L (2011) Alterations in BDNF and phospho-CREB levels following chronic oral nicotine treatment and its withdrawal in dopaminergic brain areas of mice. Neurosci Lett 491:108–112
Klein LC, Stine MM, Vandenbergh DJ, Whetzel CA, Kamens HM (2004) Sex differences in voluntary oral nicotine consumption by adolescent mice: a dose–response experiment. Pharmacol Biochem Behav 78:13–25
Krishnan V, Nestler EJ (2008) The molecular neurobiology of depression. Nature 455:894–902
Mannucci C, Tedesco M, Bellomo M, Caputi A, Calapai G (2006) Long-term effects of nicotine on the forced swimming test in mice: an experimental model for the study of depression caused by smoke. Neurochem Int 49:481–486
McClung CA, Nestler EJ (2007) Neuroplasticity mediated by altered gene expression. Neuropsychopharmacology 33:3–17
Miller DK, Crooks P, Dwoskin LP (2000) Lobeline inhibits nicotine-evoked [3H]dopamine overflow from rat striatal slices and nicotine-evoked 86Rb+ efflux from thalamic synaptosomes. Neuropharmacology 39:2654–2662
Miller DK, Crooks PA, Teng L, Witkin JM, Munzar P, Goldberg SR, Acri JB, Dwoskin LP (2001) Lobeline inhibits the neurochemical and behavioral effects of amphetamine. J Pharmacol Exp Ther 296:1023–1034
Miller DK, Harrod SB, Green TA, Wong MY, Bardo MT, Dwoskin LP (2003) Lobeline attenuates locomotor stimulation induced by repeated nicotine administration in rats. Pharmacol Biochem Behav 74:279–286
Mineur YS, Eibl C, Young G, Kochevar C, Papke RL, Gündisch D, Picciotto MR (2009) Cytisine-based nicotinic partial agonists as novel antidepressant compounds. J Pharmacol Exp Ther 329:377–386
Nguyen HN, Rasmussen BA, Perry DC (2003) Subtype-selective up-regulation by chronic nicotine of high-affinity nicotinic receptors in rat brain demonstrated by receptor autoradiography. J Pharmacol Exp Ther 307:1090–1097
Parker MJ, Beck A, Luetje CW (1998) Neuronal nicotinic receptor β2 and β4 subunits confer large differences in agonist binding affinity. Mol Pharmacol 54:1132–1139
Pietilä K, Laakso I, Ahtee L (1995) Chronic oral nicotine administration affects the circadian rhythm of dopamine and 5-hydroxytryptamine metabolism in the striata of mice. Naunyn-Schmiedeberg Arch Pharmacol 353:110–115
Porsolt RD, Le Pichon M, Jalfre M (1977) Depression: a new animal model sensitive to antidepressant treatments. Nature 266:730–732
Rabenstein RL, Caldarone BJ, Picciotto MR (2006) The nicotinic antagonist mecamylamine has antidepressant-like effects in wild-type but not beta2- or alpha7-nicotinic acetylcholine receptor subunit knockout mice. Psychopharmacology (Berl) 189:395–401
Rahman S, Neugebauer NM, Zhang Z, Crooks PA, Dwoskin LP, Bardo MT (2008) The novel nicotinic receptor antagonist N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide decreases nicotine-induced dopamine metabolism in rat nucleus accumbens. Eur J Pharmacol 601:103–105
Ressler KJ, Nemeroff CB (2000) Role of serotonergic and noradrenergic systems in the pathophysiology of depression and anxiety disorders. Depress Anxiety 12:2–19
Ribeiro-Carvalho A, Lima CS, Medeiros AH, Siqueira NR, Filgueiras CC, Manhaes AC, Abreu-Villaça Y (2009) Combined exposure to nicotine and ethanol in adolescent mice: effects on the central cholinergic systems during short and long term withdrawal. Neurosci 162:1174–1186
Robinson SF, Marks MJ, Collins AC (1996) Inbred mouse strains vary in oral self-selection of nicotine. Psychopharmacology (Berl) 124:332–339
Roni MA, Rahman S (2011) Neuronal nicotinic receptor antagonist reduces anxiety-like behavior in mice. Neurosci Lett 504:237–241
Roni MA, Rahman S (2013) Antidepressant-like effects of lobeline in mice: behavioral, neurochemical, and neuroendocrine evidence. Prog Neuropsychopharmacol Biol Psychiatry 41:44–51
Sajja RK, Rahman S (2011) Lobeline and cytisine reduce voluntary ethanol drinking behavior in male C57BL/6J mice. Prog Neuro-Psychopharmacol Biol Psychiatry 35:257–264
Sajja RK, Rahman S (2012) Neuronal nicotinic receptor ligands modulate chronic nicotine-induced ethanol consumption in C57BL/6J mice. Pharmacol Biochem Behav 102:36–43
Sajja RK, Dwivedi C, Rahman S (2010) Nicotinic ligands modulate ethanol-induced dopamine function in mice. Pharmacology 86:168–173
Salas R, Pieri F, De Biasi M (2004) Decreased signs of nicotine withdrawal in mice null for the β4 nicotinic acetylcholine receptor subunit. J Neurosci 24:10035–10039
Sanberg PR, Vindrola-Padros C, Shytle RD (2012) Translating laboratory discovery to the clinic: from nicotine and mecamylamine to tourettes, depression, and beyond. Physiology Behav 107:801–808
Semba J, Wakuta M, Maeda J, Suhara T (2004) Nicotine withdrawal induces subsensitivity of hypothalamic–pituitary–adrenal axis to stress in rats: implications for precipitation of depression during smoking cessation. Psychoneuroendocrinology 29:215–226
Sparks JA, Pauly JR (1999) Effects of continuous oral nicotine administration on brain nicotinic receptors and responsiveness to nicotine in C57Bl/6 mice. Psychopharmacology (Berl) 14:145–153
Stolerman IP, Garcha HS, Mirza NR (1995) Dissociations between the locomotor stimulant and depressant effects of nicotinic agonists in rats. Psychopharmacology (Berl) 117:430–437
Tani Y, Saito K, Tsuneyoshi A, Imoto M, Ohno T (1997) Nicotinic acetylcholine receptor (nACh-R) agonist-induced changes in brain monoamine turnover in mice. Psychopharmacology (Berl) 129:225–232
Watkins SS, Stinus L, Koob GF, Markou A (2000) Reward and somatic changes during precipitated nicotine withdrawal in rats: centrally and peripherally mediated effects. J Pharmacol Exp Ther 292:1053–1064
Wilkinson JL, Palmatier MI, Bevins RA (2006) Preexposure to nicotine alters the subsequent locomotor stimulant effects of bupropion in rats. Nicotine Tob Res 8:141–146
Xu Y, Ku B, Tie L, Yao H, Jiang W, Ma X, Li X (2006) Curcumin reverses the effects of chronic stress on behavior, the HPA axis, BDNF expression and phosphorylation of CREB. Brain Res 1122:56–64
Zaniewska M, McCreary AC, Wydra K, Filip M (2010) Effects of serotonin 5-HT 2 receptor ligands on depression-like behavior during nicotine withdrawal. Neuropharmacology 58:1140–1146
Acknowledgments
The authors wish to thank Dr. Chandradhar Dwivedi and Dr. Ruth Guillermo for their help in Western blot studies. The anti-β-tubulin antibody developed by Dr. Michael Klymkowsky was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biology, Iowa City, IA 52242. This study was supported in part by the funds from the College of Pharmacy and grants from SDSU Research Foundation and RSSF (SR). All experiments were in compliance with the National Institutes of Health guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at South Dakota State University in the USA.
Conflict of interest
The authors declare that there is no conflict of interest related to this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Roni, M.A., Rahman, S. The effects of lobeline on nicotine withdrawal-induced depression-like behavior in mice. Psychopharmacology 231, 2989–2998 (2014). https://doi.org/10.1007/s00213-014-3472-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-014-3472-y