
Abstract
Many patients with depression fail to derive sufficient benefit from available treatment options, with up to a third never reaching remission despite multiple trials of appropriate treatment. Novel antidepressant agents are needed, and drugs targeting nicotinic acetylcholine receptors (nAChRs) appear to hold promise in this regard. nAChRs are involved in a variety of neurobiological systems implicated in the pathophysiology of depression. In addition to their role in cholinergic neurotransmission, they modulate dopamine function and influence inflammation and hypothalamic–pituitary–adrenal axis activity. Preclinical studies have suggested antidepressant-like effects of drugs targeting nAChRs, with the most consistent results observed with α4β2 nAChR modulators such as varenicline and nonspecific nAChR antagonists such as mecamylamine. These agents appear to offer the most potential antidepressant-like efficacy when used in conjunction with other established antidepressant treatments. nAChR modulators also influence neural processes that appear to mediate the behavioral effects of antidepressants, such as hippocampal cell proliferation. Clinical evidence, while limited, shows preliminary efficacy for mecamylamine and varenicline. Taken together, the preclinical and clinical evidence suggests that drugs targeting nAChRs may represent an important new approach to the treatment of depression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Major depression is one of the most common psychiatric illnesses worldwide, with significant public health impact. The World Health Organization designated major depression as the most common cause of disease burden in North America (Mathers and Loncar 2006). Despite numerous treatment options, many patients do not achieve relief with currently available medications. The Sequenced Treatment of Alternatives to Relieve Depression (STAR*D) trial showed that only about half of patients receiving initial treatment with antidepressants respond to treatment, and only a third reach remission. Even after multiple levels of treatment, patients in the STAR*D study reached a cumulative remission rate of only 67% (Trivedi et al. 2006). Clearly, new pharmacologic treatments are needed.
Targeting the cholinergic system, and particularly the nicotinic cholinergic receptor (nAChR), holds promise as a novel therapeutic approach to depression. Speculation that acetylcholine might be involved in depression is not new, as early work suggested that a cholinergic/adrenergic imbalance might lead to depressive symptoms (Janowsky et al. 1972, 1974). However, these early studies were performed before the availability of drugs targeting specific receptors within the cholinergic system, making the results difficult to interpret.
This paper provides an overview of nAChRs and their relationships to other neurobiological systems relevant to depression and examines preclinical and clinical data on the antidepressant effects of drugs acting on nAChRs. The clinical efficacy of such drugs in the treatment of smoking cessation (Gonzales et al. 2006; Jorenby et al. 2006) and their potential role in the treatment of cognitive disorders, autism, attention deficit hyperactivity disorder, and schizophrenia have been addressed elsewhere (Bacher et al. 2009; Cincotta et al. 2008; Ochoa and Lasalde-Dominicci 2007; Sabri et al. 2008). Since there are multiple effects of different compounds acting on the nAChR, such as antagonism, partial agonism, and desensitization, we will refer to drugs that target nAChRs system in general as nAChR modulators after specifically discussing each drug.
Overview of the nAChR system
nAChRs are part of the larger system of cholinergic receptors (AChRs). AChRs are divided into two receptor systems, muscarinic (mAChRs) and nicotinic (nAChRs). Muscarinic receptors are G-protein-coupled acetylcholine receptors, distributed in the central and parasympathetic nervous systems (Eglen 2006). The nAChR is a ligand-gated, i.e., ionotropic, receptor for acetylcholine and can also bind to exogenous ligands such as nicotine. When activated, nAChRs open nonselective cation channels that can affect membrane polarity as well as influence intracellular messenger cascades under the control of calcium concentration. nAChRs share structural similarity with γ-amino butyric acid (GABA), glycine, and serotonin type 3 (5-HT3) receptors. They are composed of five pentameric units, formed from up to 17 different subunits coded by an extensive family of coding DNA (Albuquerque et al. 2009). There are two classes of nAChRs in the central nervous system, high- and low-affinity. High-affinity nAChRs are composed of heteromers of α and β subunits, which are antagonized by dihydro-β-erythriodine and mecamylamine and stimulated in low doses by the α4β2 partial agonist varenicline. Low-affinity nAChRs are α homopentamers, which are blocked by the snake venom α-bungarotoxin and by methyllycacotinine. nAChRs are highly conserved over the course of evolution, although studies of nAChR knockout mice suggest that brain nAChRs are not required for survival or for execution of basic behaviors but rather are important for control of complex behaviors (Gotti and Clementi 2004; Ross et al. 2000).
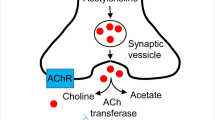
With respect to depression, the α4β2 nAChR subtype has received the most attention as a potential target. α4β2 nAChRs are widely distributed in neuroanatomic regions implicated in depression, including basal ganglia, striatum, thalamus, hypothalamus, amygdala, ventral tegmental area, locus coeruleus, and dorsal raphe nucleus. Through their actions in these areas, they are thought to regulate the release of other monoamine neurotransmitters (Albuquerque et al. 2009; Gotti et al. 2006), particularly dopamine, which is strongly implicated in affect regulation and reward processing and reinforcement. nAChRs modulate dopamine neurotransmission by direct action from cholinergic projections, as well as by indirect excitatory (glutamatergic) and inhibitory (GABAergic) influences on the dopamine neuron (Fig. 1) (Albuquerque et al. 2009; Dunlop and Nemeroff 2007; Li et al. 1998; Mihailescu et al. 2002; Mihailescu et al. 1998). There is neuroimaging evidence of decreased α4β2 nAChR binding in Parkinson’s patients with depression, and this decreased binding is independent of cognitive symptoms (Meyer et al. 2009).

Modulation of dopamine with nAChRs. A rendition of nAChR modulation of dopamine neurotransmission with a focus on α4β2 and α7 nAChRs. Adapted from Albuquerque et al. (2009). ACh acetylcholine, DA dopamine, Glu glutamate, GABA γ-aminobutryic acid
nAChRs are also involved in neuroendocrine systems implicated in depression, particularly the hypothalamic–pituitary–adrenal (HPA) axis. Altered HPA axis function is one of the most consistent biological findings in depression, with demonstrated effects including changes in corticotropin releasing factor (CRF) secretion, glucocorticoid receptor sensitivity, and pituitary and adrenocortical structure and function (Pariante and Lightman 2008; Tsigos and Chrousos 2002). nAChRs are located on the presynaptic terminals of CRF-releasing neurons (Okuda et al. 1993), and drugs that act on nAChRs have been shown to affect key components of the HPA axis. Mecamylamine, a nonspecific nAChR antagonist approved for use as an antihypertensive, can prevent CRF release from the hypothalamus (Raber et al. 1995). Mecamylamine can also block pharmacologically induced secretion of corticosterone (Rhodes et al. 2001). Nicotine, the classic nAChR ligand, has similarly been shown to alter HPA activity. Nicotine can stimulate CRF release (Fuxe et al. 1989) and can influence the secretion of adrenocorticotropin (ACTH) independent of its effects on CRF, possibly by modulating catecholamine activity in the tractus solitarius ascending from the locus coeruleus (Matta et al. 1998). In humans, nicotine delivered by cigarette smoke results in acute increases of salivary cortisol (Kudielka et al. 2009). Chronic administration of nicotine through cigarette smoke may result in down-regulation of the HPA axis, which may explain why habitual smokers manifest a blunted salivary cortisol response to the Trier Social Stress Test (Rohleder and Kirschbaum 2006), a paradigm often used to measure HPA axis responsivity (Kirschbaum et al. 1993).
Recent work has suggested that agents affecting the HPA axis might also affect nAChRs. For example, in mice, CRF-1 antagonists improve deficits in brain reward function during nicotine withdrawal (Bruijnzeel et al. 2009). This deficit is thought to be a proxy for the negative affective state associated with nicotine withdrawal, in which nAChRs are presumably unbound. While this evidence is indirect, it supports the possibility of a reciprocal relationship between nAChRs and a neuroendocrine system consistently associated with depression.
nAChRs play a role in inflammation, which has also been implicated in the pathophysiology of depression (Miller et al. 2009). α7 nAChRs mediate the effects of the vagus cholinergic anti-inflammatory pathway. When the vagus nerve is stimulated, it releases peripheral acetylcholine, which binds to α7 nAChRs on macrophages, resulting in decreased macrophage activity (Gallowitsch-Puerta and Pavlov 2007; Shytle et al. 2004). This was demonstrated by assessing levels of inflammatory cytokines (such as tumor necrosis factor (TNF), interleukin-1 (IL-1), and interleukin-6 (IL-6)) in α7 nAChR wild-type and knockout mice during stimulation of the vagus nerve. Stimulation decreased cytokine secretion in wild-type mice, whereas in knockout mice cytokine levels were unchanged (Wang et al. 2003). The same authors showed that vagotomized wild-type mice had greater levels of TNF during challenge with endotoxin compared to wild-type mice with an intact vagus nerve (Wang et al. 2003).
α7 nAChRs have been shown to mediate inflammatory regulation in a variety of inflammatory states, such as sepsis, endotoxic shock, and colitis (Pavlov 2008), and α7 agonists are under development as treatments for inflammation. GTS-21, an α7 agonist, decreases TNF release from endotoxin-stimulated macrophages and suppresses activation of NF-κB, a transcription factor regulating TNF synthesis (Pavlov et al. 2007)
Given the mounting evidence for a relationship between inflammation and depression, the role of nAChRs in modulating inflammatory responses is of particular interest.
nAChRs and depression: history
In the early 1970s, Janowsky proposed a cholinergic/adrenergic theory of depression, invoking a pathogenic imbalance between these two systems that was driven by cholinergic over-activity (Janowsky et al. 1972). Evidence for this theory was supported by experiments with physostigmine, a nonspecific acetylcholinesterase inhibitor. When given to healthy controls, physostigmine induced feelings of depression, anxiety, and irritability. Similarly, administration of physostigmine to depressed patients exacerbated depressive symptoms (Janowsky et al. 1974; Janowsky and Risch 1983). The theory was given further support by animal studies showing that mice bred specifically for sensitivity to cholinergic agents demonstrated depression-like behaviors (Overstreet 1993). Other studies found that learned helplessness and swim stress, widely used preclinical models of depression, could induce sensitivity to cholinergic agents (Dilsaver and Alessi 1987; Dilsaver et al. 1986).
There are some significant limitations to the theory as it was initially formulated. This theory was advanced before the modern understanding of the role of serotonin in depression, and it relied heavily on a single experimental paradigm. However, the cholinergic/adrenergic theory suggested that antagonizing acetylcholine receptors could have antidepressant effects. This hypothesis is supported by recent clinical investigations of intravenous scopolamine, an anti-emetic and anesthetic that is a nonspecific muscarinic acetylcholine receptor antagonist, in adults with major depressive episodes.
In an initial placebo-controlled dose-finding study, Furey and Drevets (2006) administered intravenous infusions of scopolamine 2.0, 3.0, and 4.0 mcg/kg to eight depressed patients; depressive symptoms robustly improved only at the highest dose over the 3–5 days after infusion, suggesting a degree of dose-dependent effect. The authors subsequently gave intravenous scopolamine 4.0 mcg/kg to 18 depressed patients (nine unipolar, nine bipolar) in a double-blind, placebo-controlled, crossover design following a single-blind placebo lead-in. Again, depressive and anxiety symptoms, as assessed by the Montgomery–Asberg Depression Rating Scale (MADRS) and the Hamilton Anxiety Rating Scale (HARS), respectively, improved significantly more with scopolamine than with placebo. Eleven of 18 patients had a full response (defined as a >50% improvement in MADRS score), and 10 of 18 experienced remission (defined as a MADRS score <10). These findings were replicated in a study of 23 unipolar depressed patients using a similar design, which showed a 32% improvement in MADRS scores in patients receiving scopolamine compared to 6.5% improvement with placebo (Drevets and Furey 2010). Of the total 40 participants in these studies with unipolar depression, 29 (72.5%) had a history of comorbid anxiety disorders, chronic medical illness, or treatment resistance, suggesting these patients would have a relatively poor prognosis with standard monoaminergic antidepressant medications and highlighting the therapeutic potential for drugs acting on cholinergic neurotransmission.
While scopolamine’s intravenous formulation and sedative and amnestic properties limit its potential for clinical use, the drug’s putative antidepressant effects constitute important support for the involvement of the cholinergic system in depression. Moreover, the clinical characteristics of scopolamine responders suggest that the cholinergic system may play an important role in treatment-resistant patients (Philip et al. 2010). In light of this evidence, the introduction of drugs that can specifically target individual receptors in the cholinergic system suggests a significant opportunity for the development of novel therapeutic approaches.
nAChRs and depression: smoking and nicotine
Another body of work that supports the relationship between nAChRs and depression involves tobacco smoking. Smoking rates in major depression are higher than in the general population, generally between 40% and 60% vs. 22%, respectively (Kalman et al. 2005). Smoking cessation has also been found to influence mood. Smokers with a history of major depression have a harder time quitting smoking, and patients with depression are at risk for developing a major depressive episode during smoking cessation (Dani and Harris 2005; Hughes 2007). Smokers also have lower levels of monoamine oxidase A (MAO-A), an enzyme involved in the degradation of norepinephrine, serotonin, and dopamine, which has been hypothesized to lead to depressed mood (Fowler et al. 2003).
The link between smoking and depression is likely to be through nicotine, the classic nAChR ligand that is the primary active ingredient in cigarette smoke. Nicotine has been shown to have antidepressant properties in a number of studies. Initial work in the late 1990s showed that giving nicotine over 1–2 weeks to rodents produces decreased immobility time in both the forced swim and tail suspension tests (Andreasen and Redrobe 2009b; Djuric et al. 1999; Semba et al. 1998; Tizabi et al. 1999), two laboratory paradigms that are frequently used to evaluate a candidate drug for potential antidepressant effects (Porsolt et al. 1977; Steru et al. 1985). In humans, a brief, open-label trial showed transdermal nicotine could decrease depressive symptoms in depressed nonsmokers (Salin-Pascual 2002). A second trial replicated this finding of mood benefits following a month of double-blind transdermal nicotine treatment in mildly depressed nonsmokers (McClernon et al. 2006).
Antidepressant medications used clinically for smoking cessation have antagonist properties at the nAChR. Bupropion, a norepinephrine and dopamine reuptake inhibitor, has nAChR antagonist properties, as does nortriptyline, a tricyclic antidepressant (TCA) that inhibits norepinephrine and serotonin reuptake. Indeed, it has been suggested that this property accounts for the efficacy of these drugs in smoking cessation and contributes to their antidepressant effects (Arias 2009; Damaj et al. 2004; Schofield et al. 1981).
Taken together, these data lead to an apparent paradox: nicotine, a potent nAChR agonist, manifests antidepressant effects, whereas antagonists of the nAChR, such as mecamylamine, also manifest antidepressant effects. This might be explained by a closer consideration of nicotine’s effects at the nAChR. Nicotine initially activates the nAChR, but this is then followed by a rapid desensitization (Gentry and Lukas 2002; Paradiso and Steinbach 2003). Sustained binding of nicotine to the nAChR leads to continued desensitization, which is hypothesized to result in chronic antagonism (Mineur and Picciotto 2009; Picciotto et al. 2008; Shytle et al. 2002a).
nAChRs and depression: pharmacologic evidence
Most antidepressant research targeting nAChRs has focused on two kinds of ligands: mecamylamine and the plant alkaloid cytisine and cytisine-based molecules, such as varenicline.
Preclinical findings
Preclinical studies using nAChR modulators have resulted in a variety of different, and at times discordant, findings. Table 1 summarizes the preclinical literature on this topic. Popik et al. (2003) demonstrated that mecamylamine could increase the antidepressant-like effects of the TCA imipramine and the selective serotonin reuptake inhibitor (SSRI) citalopram during the tail suspension test. Nicotine also enhanced the antidepressant effects of both imipramine and citalopram in this paradigm. In contrast, dihydrobetaerythiodine, a selective α4β2 nAChR antagonist, increased the antidepressant-like effects of imipramine but not those of citalopram in this study, making the overall findings more difficult to interpret. This was the first study to show that nAChR antagonists might enhance the activity of primary antidepressants.
Rabenstein et al. (2006) demonstrated that mecamylamine had antidepressant-like effects on wild-type mice during the forced swim and tail suspension tests. When the experiments were repeated with either β2 or α7 nAChR knockout mice, antidepressant-like effects were lost. Based on these findings, the authors hypothesized that these nAChR subunits are important for mood regulation.
Caldarone et al. (2004) showed that high-affinity nAChRs are required for the antidepressant-like effects of amitriptyline in mice. They examined performance on the forced swim test in mice treated with pharmacologic doses of mecamylamine alone and compared that with mice given sub-pharmacologic doses of mecamylamine plus amitriptyline. Pharmacologic doses of mecamylamine alone produced improved performance, suggesting clinical antidepressant potential. While neither sub-pharmacologic mecamylamine alone nor sub-pharmacologic amitriptyline alone improved test performance, the combination of the two decreased immobility time, signaling a possible potentiating action of nAChR modulators during treatment with a conventional antidepressant such as amitriptyline. When nAChR knockout mice were subjected to the forced swim test, the previously observed antidepressant-like effects of amitriptyline at pharmacologic doses were lost, providing additional evidence of a potential synergy between the agents.
Caldarone et al. also investigated hippocampal cell proliferation, since this phenomenon has been observed with chronic successful antidepressant treatment (Malberg and Duman 2003; Malberg et al. 2000) and has been postulated as a necessary condition for the positive behavioral effects of antidepressant pharmacotherapy (Santarelli et al. 2003). They found that nAChR knockout mice did not demonstrate hippocampal cell proliferation when treated with amitriptyline, suggesting that nAChRs may play a role in the biological changes underlying the antidepressant activity of TCAs.
Andreasen and colleagues investigated nAChR modulators in two preclinical studies. In the first, nAChR antagonists, but not agonists, exhibited antidepressant-like effects during forced swim and tail suspension tests (Andreasen et al. 2009). The authors tested several different compounds, including the α4β2 agonist RJR-2403, the α4β2 antagonist dihydrobetaerythiodine, the α7 agonist PNU-282987, and the α7 antagonist methyllycacotinine. They also included tests of hexamethonium, a nonspecific nAChR antagonist with limited penetration of the blood–brain barrier, as well as nicotine and mecamylamine, in their experimental design. Mecamylamine and antagonists of the α4β2 and α7 receptors demonstrated improved performance on the forced swim and tail suspension tests. Hexamethonium had no activity, suggesting that peripheral nAChRs are not involved in responses to these tests. However, in this study, nicotine also had no effect on test performance, a finding discordant with previous evidence showing antidepressant-like effects of nicotine in mouse models (Djuric et al. 1999; Semba et al. 1998; Suemaru et al. 2006; Tizabi et al. 1999). This may be explained by a significant increase in locomotor activity during nicotine treatment compared to saline, which could confound results from the forced swim test (Porsolt et al. 1977).
In a subsequent study, the same group examined the effects of nAChR antagonists as “augmenting” agents for enhancing the antidepressant-like effects from several commonly used drugs (Andreasen and Redrobe 2009b). They investigated whether nicotine or mecamylamine could augment the antidepressant effects of SSRIs or selective norepinephrine reuptake inhibitors (SNRI) on the mouse forced swim test and the tail suspension test. Pharmacologic and sub-pharmacologic doses of the SSRI citalopram and SNRI reboxetine, alone and in combination with nicotine or mecamylamine, were administered. Nicotine improved performance on the forced swim and tail suspension tests in conjunction with both pharmacologic and sub-pharmacologic doses of citalopram and reboxetine. Mecamylamine monotherapy improved performance in some tests, but did not augment the effects produced by sub-pharmacologic or pharmacologic doses of citalopram or reboxetine. One reason for the negative results using mecamylamine in this study may have been under-dosing; in their 2006 study, this group found improved test performance with at least 3 mg/kg of mecamylamine, and in this 2008 study, the upper limit of mecamylamine dose was 3 mg/kg. Another reason for discrepant results may have been the use of different mouse strains in different experiments, resulting in different responses to the behavioral paradigms administered (Andreasen and Redrobe 2009a).
Mineur et al. (2007) demonstrated that the alkaloid cytisine, the α4β2 partial agonist from which varenicline was derived, has antidepressant-like effects in mouse models. They administered cytisine and measured acute antidepressant-like effects via the forced swim and tail suspension tests and chronic antidepressant-like effects via the novelty-suppressed feeding test, comparing the results to similar experiments conducted with mecamylamine. They also assessed expression of c-fos, a genetic marker of neuronal activity associated with antidepressant action (Beck and Fibiger 1995). C-fos has been shown to upregulate during acute antidepressant treatment and downregulate with more chronic treatment (Slattery et al. 2005). Mice treated with cytisine had significantly improved performance on all three tests, and results with mecamylamine were comparable to those found with cytisine. Cytisine resulted in an overall reduction of c-fos activity after 21 days. The greatest reduction in c-fos was seen in the basolateral amygdala region. Mecamylamine also reduced c-fos activity, although its effects were more pronounced in the hypothalamus, nucleus accumbens, and suprachiasmatic nucleus. The authors hypothesized that these drugs might exert antidepressant effects by influencing neuronal activity in these areas.
The same group then conducted a series of experiments with other compounds derived from cytisine to assess for specific antidepressant effects (Mineur et al. 2009). They examined cytisine and two derivatives with more specific antagonism at the α4β2 receptors, 3-(pyridin-3′-yl)-cytisine (3-pyr-Cyt) and 5-bromo-cytisine (5-Br-Cyt). 3-pyr-Cyt and cytisine, but not 5-Br-Cyt, had generally dose-dependent antidepressant-like effects in both the tail suspension and forced swim tests using intraperitoneal injections. Because 5-Br-Cyt does not penetrate the blood–brain barrier, they repeated the tail suspension test using intraventricular administration of 5-Br-Cyt and found significant improvement with this compound. Based on their findings, the authors suggested that more specific α4β2 nAChR antagonists might be candidates for drug development for depression.
Lippiello et al. (2008) tested the s-enantiomer of mecamylamine in rodent models for potential antidepressant-like effects. They found that s-mecamylamine improved performance on the forced swim and behavioral despair tests. They subsequently assessed s-mecamylamine in a social paradigm model for anxiety-like behavior and also found significant improvement associated with the s-enantiomer on that behavioral assay. The drug was well tolerated without any acute or chronic toxicity.
Rollema et al. (2009) examined whether varenicline had effects on the forced swim test and whether it could augment the antidepressant-like effects of the SSRI sertraline. These investigators evaluated varenicline alone, sertraline alone, and combination treatment of varenicline plus sertraline. All three groups were compared with a control group of rats treated with amitriptyline alone. Varenicline and sertraline monotherapies significantly improved performance on the forced swim test, although these drugs’ effects were inferior relative to the effect observed in the amitriptyline control condition. The combination of sertraline plus varenicline did result in significantly improved test performance, achieving an effect comparable to that observed in rats treated with amitriptyline. Augmentation-like effects were significantly more prominent at lower doses of varenicline, suggesting an inverse dose-dependent effect of varenicline augmentation.
Clinical findings
Clinical data targeting nAChRs are limited; most research has been done with the two compounds available for clinical study, mecamylamine and varenicline. These studies are summarized in Table 2.
Mecamylamine
Shytle et al. (2002b) reported that mecamylamine reduced depression and irritability in four children and adolescents (ages 8–17) with Tourette disorder and comorbid major depression. Following this preliminary report, George et al. (2008) conducted an 8-week, double-blind, placebo-controlled trial of mecamylamine 10 mg/day augmentation in 21 depressed adults who were currently taking an SSRI for at least 3 months with either no or partial response and baseline Hamilton Depression Rating Scale (HDRS) scores of 12 or higher. The investigators hypothesized that antidepressant response to mecamylamine would be more robust in patients who smoked, given that nAChRs may be up-regulated in smokers due to chronic nicotine administration (Dani and Harris 2005). Patients taking mecamylamine had a significant improvement in HDRS score by the 8-week endpoint compared with those on placebo. Counter to their hypothesis, there was a trend-level (p = 0.06) positive association between nonsmoking status and antidepressant response. The authors discussed how larger trials would be needed to replicate this effect and suggested that smokers may have chronically up-regulated levels of nAChRs which may make them less likely to respond to nAChR antagonist treatment. Larger trials of mecamylamine and s-mecamylamine antidepressant augmentation are not yet publicly available, but commercial press releases have reported efficacy with these compounds (Targacet IW-S, NC); at present, it is not possible to objectively evaluate these claims.
Varenicline
To evaluate the effects of varenicline on mood and cognition during smoking cessation, Patterson et al. (2009) performed a double-blind placebo-controlled study in 67 subjects undergoing smoking cessation with a scheduled smoking relapse. The authors assessed two phases of treatment with varenicline. In an initial phase, they first compared changes in affect (as measured by the Positive and Negative Affect Schedule), attention, and working memory during varenicline treatment compared to placebo. On day 14 of treatment, they exposed patients to a scheduled cigarette smoking “relapse” and measured levels of satisfaction with smoking during this relapse. During the cessation phase, varenicline treatment decreased withdrawal symptoms, increased positive affect, and decreased negative affect. Compared with placebo, patients treated with varenicline also had improved performance on tests of attention and working memory during cessation. During the scheduled relapse, patients treated with varenicline reported less intense feelings of reward compared to the placebo group. The authors concluded that varenicline improved mood and cognition during smoking cessation and suggested varenicline be explored as a potential treatment for mood disorders.
Following similar clinical observations, our group conducted an open-label study of varenicline augmentation in 18 adult smokers with an Axis I depressive disorder and pharmacotherapy-resistant symptoms (Philip et al. 2009). Depressive symptoms (using the Quick Inventory of Depressive Symptoms, Self-Report (QIDS-SR16)), anhedonia, and overall illness severity were measured. Varenicline produced a robust antidepressant response that was generally early and sustained. Forty-four percent of patients met criteria for categorical response (defined as a greater than 50% improvement in the QIDS-SR), and 33% achieved remission (QIDS-SR score less than five). Improvement in depressive symptoms correlated with cessation status late in the trial. There was also significant improvement in overall depressive illness severity, but no changes in anhedonia were observed. No evidence of treatment-emergent suicidality was found.
Although there have been few clinical studies of varenicline specifically for treatment of depression, the drug has seen widespread clinical use for smoking cessation. Some case reports described worsened mood in patients with schizophrenia, bipolar disorder, and unipolar depressive disorders (Freedman 2007; Kohen and Kremen 2007; Popkin 2008). These reports led to a black-box warning from the US Food and Drug Administration advising clinicians to monitor for changes in behavior, hostility, agitation, depressed mood, suicidal thoughts and behavior, and attempted suicide in patients receiving varenicline and sustained-release bupropion (Administration 2009). In meta-analyses of phase 2 and 3 trials of varenicline (n = 5,096), the drug has not been shown to induce these effects in otherwise healthy smokers (Tonstad et al. 2010), and other studies have not confirmed a varenicline-associated increase in adverse mood and behavioral symptoms in at-risk populations. A large (n = 1,117) open-label trial used telephone reports of mood state during smoking cessation to investigate this issue in individuals with a possible history of depression and compared them with another group of individuals without such a history (McClure et al. 2009). Significant improvement in depressive symptoms at 21 days and 3 months of treatment was seen for the pooled sample, although the authors found that patients with a probable history of depression were more likely to report depressive symptoms during the trial. There was no significant difference in adverse mood or behavioral symptoms between the two groups. Another report of 208 smokers showed that varenicline did not produce worsened mood or emergence of suicidality when used for smoking cessation in smokers with comorbid psychiatric illnesses including unipolar depression, bipolar disorder, and psychosis (Stapleton et al. 2008). In the largest study to date, comprising about 80,000 primary care patients who received new prescriptions for smoking cessation products over a 2-year period (including n = 10,973 that received varenicline), there was no difference in suicidal behavior or depression between patients given nicotine replacement products, bupropion, or varenicline (Gunnell et al. 2009). There was also no difference between the three groups in the rate of new antidepressant prescriptions, another marker for emerging depressive symptoms. About 11,000 patients in this study received varenicline, 10% of whom had a previous history of self-harm or suicidal thoughts before study entry. An important consideration is the confounding effect of smoking cessation on mood, which has been shown to induce major depressive episodes and suicidality in susceptible individuals (Hughes 2007).
Depression exacerbation and suicidality have not been reported during mecamylamine treatment. This may be due to several factors. Poor tolerability has limited its use as an antihypertensive, and other side effects might be observed if the drug were more widely used for treatment of depression. Additionally, these drugs have substantially different effects on nAChRs, as mecamylamine is a nonspecific antagonist and varenicline is a partial agonist, and the consequences of such differences with respect to the reported behavioral toxicity is unknown.
Conclusions
nAChRs are located in areas of the brain implicated in depression, and are involved in regulating neurobiological systems that are similarly implicated, including monoamine neurotransmitters, the HPA axis, and certain inflammatory processes. Preclinical studies consistently demonstrate beneficial effects of nAChR modulators in animal models of depression. Mecamylamine, cytosine, and varenicline have all been shown to have antidepressant-like effects in such models, and negative results in preclinical models appear to be driven by either inadequate dosing or confounding effects of control conditions rather than lack of efficacy. The most consistent preclinical support for antidepressant effects involves α4β2 antagonism in the context of concomitant treatment with an established monoaminergic-mechanism antidepressant.
Clinical evidence, while more limited, also suggests that the nAChR may be a viable target for the development of novel antidepressant treatments. There are preliminary data supporting the use of mecamylamine as an antidepressant-augmenting agent, and positive results of larger trials with mecamylamine and s-mecamylamine are currently pending peer review and publication. It is unlikely that there will be appreciable differences in clinical efficacy between racemic mecamylamine and its individual constituents, since there are no significant preclinical differences in efficacy between S-mecamylamine, R-mecamylamine, or their racemate (Papke et al. 2001). Preliminary work in open trials and in samples of smokers with psychiatric comorbidity suggests that varenicline may have efficacy in enhancing antidepressant response. The available literature for varenicline has significant limitations, as studies were either not in populations with depression or were limited by small sample size and open-label design. However, these clinical results mirror preclinical findings and should be examined in larger, controlled trials.
The concern surrounding varenicline and suicidality may require more study, since reports from trials of mecamylamine have not generated similar reports. This may be due to different mechanisms of action or limited use of mecamylamine. The possible relationship between nAChR modulators and induction of suicidal thoughts/actions or other adverse behavioral effects merits attention as the field moves forward, although this relationship may also reveal an endophenotype of depression characterized by disturbances in nAChR function. A confounding feature in the available safety literature from clinical trials of varenicline is the effect of tobacco dependence and nicotine withdrawal, which produce independent effects on mood and suicidality (Hughes 2007; Wilhelm et al. 2006). This potential confound should be addressed explicitly in future studies by comparing behavioral health outcomes in varenicline-treated individuals with and without tobacco dependence.
An intriguing question is how targeting nAChRs mediate potential antidepressant effects. It appears that blockade of the nAChR is the necessary component for antidepressant activity, whether obtained through constant agonism (such as with a nicotine patch), initial agonism and likely subsequent desensitization (such as with varenicline), or direct antagonism (such as with mecamylamine). Another question is why nAChR modulators appear to function better as augmenting or adjuvant agents rather than as a monotherapy. An unsatisfying hypothesis is that nAChR modulators may simply be weak antidepressants, building additional antidepressant efficacy in an incremental fashion, but this would not explain why significant additional efficacy was gained when continuing patients on medications with limited antidepressant results. There are several other possibilities regarding mechanism of action of nAChRs and depression. One hypothesis is that there is a relationship between the nAChR and monoamine or other neurotransmitter systems. nAChR modulators work by increasing dopamine neurotransmission (Albuquerque et al. 2009), likely through the dopamine-reward processing pathway, which may lead to antidepressant effects in and of itself. Another hypothesis is that antidepressant medications may act nonspecifically on nAChRs (Arias et al. 2010; Santamaria and Arias 2010), and nAChR modulators trophic to mood-regulating regions in the brain may potentiate this effect, leading to efficacy with a combination of drugs. An alternative mechanism of action may involve a relationship between cholinergic and N-methyl-d-aspartate (NMDA) receptors. Both nicotinic and muscarinic receptors have been shown to interact with NMDA receptors (Figueredo et al. 2008; Livingstone et al. 2010), which may suggest a convergence of cholinergic modulation resulting in antidepressant effects mediated by the NMDA system. This may explain the antidepressant efficacy of both muscarinic and nicotinic systems, and the relationship between cholinergic and glutamatergic systems in depression has not been addressed in the literature.
While it is still unclear which exact properties of the nAChR are necessary for antidepressant activity, the concordance between preclinical and clinical evidence suggests this is an area with tremendous potential. Based on the available evidence, targeting the α4β2 nAChR for depression appears to yield the best results, although to be clinically useful, positive findings for the available medications need to be replicated and consideration should be given to the potential use of these drugs as antidepressant monotherapies in selected patients. Investigation into a potential role of the α7 nAChR and other receptor subunit conformations should also be pursued, and characterization of mutations in the nAChR and their effect on response to nAChR modulators may also be a fruitful avenue for future research. The growing body of preclinical evidence and the promising preliminary clinical findings in this area constitute a compelling argument for further evaluation of the nAChR as a target for novel antidepressant drug development.
References
Administration USFaD (2009) Varenicline (Marketed as Chantix) Available at: www.fda.gov/CDER/Drug/Infopage/varenicline/default.htm [accessed 12/2009].
Albuquerque EX, Pereira EF, Alkondon M, Rogers SW (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89:73–120
Andreasen JT, Redrobe JP (2009a) Antidepressant-like effects of nicotine and mecamylamine in the mouse forced swim and tail suspension tests: role of strain, test and sex. Behav Pharmacol 20:286–295
Andreasen JT, Redrobe JP (2009b) Nicotine, but not mecamylamine, enhances antidepressant-like effects of citalopram and reboxetine in the mouse forced swim and tail suspension tests. Behav Brain Res 197:150–156
Andreasen JT, Olsen GM, Wiborg O, Redrobe JP (2009) Antidepressant-like effects of nicotinic acetylcholine receptor antagonists, but not agonists, in the mouse forced swim and mouse tail suspension tests. J Psychopharmacol 23:797–804
Arias HR (2009) Is the inhibition of nicotinic acetylcholine receptors by bupropion involved in its clinical actions? Int J Biochem Cell Biol 41:2098–2108
Arias HR, Feuerbach D, Targowska-Duda KM, Russell M, Jozwiak K (2010) Interaction of selective serotonin reuptake inhibitors with neuronal nicotinic acetylcholine receptors. Biochemistry (in press)
Bacher I, Wu B, Shytle DR, George TP (2009) Mecamylamine—a nicotinic acetylcholine receptor antagonist with potential for the treatment of neuropsychiatric disorders. Expert Opin Pharmacother 10:2709–2721
Beck CH, Fibiger HC (1995) Chronic desipramine alters stress-induced behaviors and regional expression of the immediate early gene, c-fos. Pharmacol Biochem Behav 51:331–338
Bruijnzeel AW, Prado M, Isaac S (2009) Corticotropin-releasing factor-1 receptor activation mediates nicotine withdrawal-induced deficit in brain reward function and stress-induced relapse. Biol Psychiatry 66:110–117
Caldarone BJ, Harrist A, Cleary MA, Beech RD, King SL, Picciotto MR (2004) High-affinity nicotinic acetylcholine receptors are required for antidepressant effects of amitriptyline on behavior and hippocampal cell proliferation. Biol Psychiatry 56:657–664
Cincotta SL, Yorek MS, Moschak TM, Lewis SR, Rodefer JS (2008) Selective nicotinic acetylcholine receptor agonists: potential therapies for neuropsychiatric disorders with cognitive dysfunction. Curr Opin Investig Drugs 9:47–56
Damaj MI, Carroll FI, Eaton JB, Navarro HA, Blough BE, Mirza S, Lukas RJ, Martin BR (2004) Enantioselective effects of hydroxy metabolites of bupropion on behavior and on function of monoamine transporters and nicotinic receptors. Mol Pharmacol 66:675–682
Dani JA, Harris RA (2005) Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat Neurosci 8:1465–1470
Dilsaver SC, Alessi NE (1987) Chronic inescapable footshock produces cholinergic system supersensitivity. Biol Psychiatry 22:914–918
Dilsaver SC, Snider RM, Alessi NE (1986) Stress induces supersensitivity of a cholinergic system in rats. Biol Psychiatry 21:1093–1096
Djuric VJ, Dunn E, Overstreet DH, Dragomir A, Steiner M (1999) Antidepressant effect of ingested nicotine in female rats of Flinders resistant and sensitive lines. Physiol Behav 67:533–537
Drevets WC, Furey ML (2010) Replication of Scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry 67:432–438
Dunlop BW, Nemeroff CB (2007) The role of dopamine in the pathophysiology of depression. Arch Gen Psychiatry 64:327–337
Eglen RM (2006) Muscarinic receptor subtypes in neuronal and non-neuronal cholinergic function. Auton Autacoid Pharmacol 26:219–233
Figueredo LZ, Moreira KM, Ferreira TL, Fornari RV, Oliveira MG (2008) Interaction between glutamatergic-NMDA and cholinergic-muscarinic systems in classical fear conditioning. Brain Res Bull 77:71–76
Fowler JS, Logan J, Wang GJ, Volkow ND (2003) Monoamine oxidase and cigarette smoking. Neurotoxicology 24:75–82
Freedman R (2007) Exacerbation of schizophrenia by varenicline. Am J Psychiatry 164:1269
Furey ML, Drevets WC (2006) Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry 63:1121–1129
Fuxe K, Andersson K, Eneroth P, Harfstrand A, Agnati LF (1989) Neuroendocrine actions of nicotine and of exposure to cigarette smoke: medical implications. Psychoneuroendocrinology 14:19–41
Gallowitsch-Puerta M, Pavlov VA (2007) Neuro-immune interactions via the cholinergic anti-inflammatory pathway. Life Sci 80:2325–2329
Gentry CL, Lukas RJ (2002) Regulation of nicotinic acetylcholine receptor numbers and function by chronic nicotine exposure. Curr Drug Targets CNS Neurol Disord 1:359–385
George TP, Sacco KA, Vessicchio JC, Weinberger AH, Shytle RD (2008) Nicotinic antagonist augmentation of selective serotonin reuptake inhibitor-refractory major depressive disorder: a preliminary study. J Clin Psychopharmacol 28:340–344
Gonzales D, Rennard SI, Nides M, Oncken C, Azoulay S, Billing CB, Watsky EJ, Gong J, Williams KE, Reeves KR (2006) Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation: a randomized controlled trial. JAMA 296:47–55
Gotti C, Clementi F (2004) Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol 74:363–396
Gotti C, Zoli M, Clementi F (2006) Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci 27:482–491
Gunnell D, Irvine D, Wise L, Davies C, Martin RM (2009) Varenicline and suicidal behaviour: a cohort study based on data from the General Practice Research Database. BMJ 339:b3805
Hughes JR (2007) Depression during tobacco abstinence. Nicotine Tob Res 9:443–446
Janowsky DS, Risch CR (1983) Cholinomimetic and anticholinergic drugs used to investigate an acetylcholine hypothesis of affective disorders and stress. Drug Devel Res 4:125–142
Janowsky DS, el-Yousef MK, Davis JM, Sekerke HJ (1972) A cholinergic-adrenergic hypothesis of mania and depression. Lancet 2:632–635
Janowsky DS, el-Yousef MK, Davis JM (1974) Acetylcholine and depression. Psychosom Med 36:248–257
Jorenby DE, Hays JT, Rigotti NA, Azoulay S, Watsky EJ, Williams KE, Billing CB, Gong J, Reeves KR (2006) Efficacy of varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation: a randomized controlled trial. JAMA 296:56–63
Kalman D, Morissette SB, George TP (2005) Co-morbidity of smoking in patients with psychiatric and substance use disorders. Am J Addict 14:106–123
Kirschbaum C, Pirke KM, Hellhammer DH (1993) The ‘Trier Social Stress Test’—a tool for investigating psychobiological stress responses in a laboratory setting. Neuropsychobiology 28:76–81
Kohen I, Kremen N (2007) Varenicline-induced manic episode in a patient with bipolar disorder. Am J Psychiatry 164:1269–1270
Kudielka BM, Hellhammer DH, Wust S (2009) Why do we respond so differently? Reviewing determinants of human salivary cortisol responses to challenge. Psychoneuroendocrinology 34:2–18
Li X, Rainnie DG, McCarley RW, Greene RW (1998) Presynaptic nicotinic receptors facilitate monoaminergic transmission. J Neurosci 18:1904–1912
Lippiello PM, Beaver JS, Gatto GJ, James JW, Jordan KG, Traina VM, Xie J, Bencherif M (2008) TC-5214 (S-(+)-mecamylamine): a neuronal nicotinic receptor modulator with antidepressant activity. CNS Neurosci Ther 14:266–277
Livingstone PD, Dickinson JA, Srinivasan J, Kew JN, Wonnacott S (2010) Glutamate-dopamine crosstalk in the rat prefrontal cortex is modulated by Alpha7 nicotinic receptors and potentiated by PNU-120596. J Mol Neurosci 40:172–176
Malberg JE, Duman RS (2003) Cell proliferation in adult hippocampus is decreased by inescapable stress: reversal by fluoxetine treatment. Neuropsychopharmacology 28:1562–1571
Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000) Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20:9104–9110
Mathers CD, Loncar D (2006) Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 3:e442
Matta SG, Fu Y, Valentine JD, Sharp BM (1998) Response of the hypothalamo-pituitary-adrenal axis to nicotine. Psychoneuroendocrinology 23:103–113
McClernon FJ, Hiott FB, Westman EC, Rose JE, Levin ED (2006) Transdermal nicotine attenuates depression symptoms in nonsmokers: a double-blind, placebo-controlled trial. Psychopharmacology (Berl) 189:125–133
McClure JB, Swan GE, Jack L, Catz SL, Zbikowski SM, McAfee TA, Deprey M, Richards J, Javitz H (2009) Mood, side-effects and smoking outcomes among persons with and without probable lifetime depression taking varenicline. J Gen Intern Med 24:563–569
Meyer PM, Strecker K, Kendziorra K, Becker G, Hesse S, Woelpl D, Hensel A, Patt M, Sorger D, Wegner F, Lobsien D, Barthel H, Brust P, Gertz HJ, Sabri O, Schwarz J (2009) Reduced alpha4beta2*-nicotinic acetylcholine receptor binding and its relationship to mild cognitive and depressive symptoms in Parkinson disease. Arch Gen Psychiatry 66:866–877
Mihailescu S, Palomero-Rivero M, Meade-Huerta P, Maza-Flores A, Drucker-Colin R (1998) Effects of nicotine and mecamylamine on rat dorsal raphe neurons. Eur J Pharmacol 360:31–36
Mihailescu S, Guzman-Marin R, Dominguez Mdel C, Drucker-Colin R (2002) Mechanisms of nicotine actions on dorsal raphe serotoninergic neurons. Eur J Pharmacol 452:77–82
Miller AH, Maletic V, Raison CL (2009) Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65:732–741
Mineur YS, Picciotto MR (2009) Biological basis for the co-morbidity between smoking and mood disorders. J Dual Diagn 5:122–130
Mineur YS, Somenzi O, Picciotto MR (2007) Cytisine, a partial agonist of high-affinity nicotinic acetylcholine receptors, has antidepressant-like properties in male C57BL/6J mice. Neuropharmacology 52:1256–1262
Mineur YS, Eibl C, Young G, Kochevar C, Papke RL, Gundisch D, Picciotto MR (2009) Cytisine-based nicotinic partial agonists as novel antidepressant compounds. J Pharmacol Exp Ther 329:377–386
Ochoa EL, Lasalde-Dominicci J (2007) Cognitive deficits in schizophrenia: focus on neuronal nicotinic acetylcholine receptors and smoking. Cell Mol Neurobiol 27:609–639
Okuda H, Shioda S, Nakai Y, Nakayama H, Okamoto M, Nakashima T (1993) The presence of corticotropin-releasing factor-like immunoreactive synaptic vesicles in axon terminals with nicotinic acetylcholine receptor-like immunoreactivity in the median eminence of the rat. Neurosci Lett 161:183–186
Overstreet DH (1993) The Flinders sensitive line rats: a genetic animal model of depression. Neurosci Biobehav Rev 17:51–68
Papke RL, Sanberg PR, Shytle RD (2001) Analysis of mecamylamine stereoisomers on human nicotinic receptor subtypes. J Pharmacol Exp Ther 297:646–656
Paradiso KG, Steinbach JH (2003) Nicotine is highly effective at producing desensitization of rat alpha4beta2 neuronal nicotinic receptors. J Physiol 553:857–871
Pariante CM, Lightman SL (2008) The HPA axis in major depression: classical theories and new developments. Trends Neurosci 31:464–468
Patterson F, Jepson C, Strasser AA, Loughead J, Perkins KA, Gur RC, Frey JM, Siegel S, Lerman C (2009) Varenicline improves mood and cognition during smoking abstinence. Biol Psychiatry 65:144–149
Pavlov VA (2008) Cholinergic modulation of inflammation. Int J Clin Exp Med 1:203–212
Pavlov VA, Ochani M, Yang LH, Gallowitsch-Puerta M, Ochani K, Lin X, Levi J, Parrish WR, Rosas-Ballina M, Czura CJ, Larosa GJ, Miller EJ, Tracey KJ, Al-Abed Y (2007) Selective alpha7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit Care Med 35:1139–1144
Philip NS, Carpenter LL, Tyrka AR, Whiteley LB, Price LH (2009) Varenicline augmentation in depressed smokers: an 8-week, open-label study. J Clin Psychiatry 70:1026–1031
Philip NS, Carpenter LL, Tyrka AR, Price LH (2010) Pharmacologic approaches to treatment resistant depression: a re-examination for the modern era. Expert Opin Pharmacother 11:709–722
Picciotto MR, Addy NA, Mineur YS, Brunzell DH (2008) It is not “either/or”: activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol 84:329–342
Popik P, Kozela E, Krawczyk M (2003) Nicotine and nicotinic receptor antagonists potentiate the antidepressant-like effects of imipramine and citalopram. Br J Pharmacol 139:1196–1202
Popkin MK (2008) Exacerbation of recurrent depression as a result of treatment with varenicline. Am J Psychiatry 165:774
Porsolt RD, Le Pichon M, Jalfre M (1977) Depression: a new animal model sensitive to antidepressant treatments. Nature 266:730–732
Rabenstein RL, Caldarone BJ, Picciotto MR (2006) The nicotinic antagonist mecamylamine has antidepressant-like effects in wild-type but not beta2- or alpha7-nicotinic acetylcholine receptor subunit knockout mice. Psychopharmacology (Berl) 189:395–401
Raber J, Koob GF, Bloom FE (1995) Interleukin-2 (IL-2) induces corticotropin-releasing factor (CRF) release from the amygdala and involves a nitric oxide-mediated signaling; comparison with the hypothalamic response. J Pharmacol Exp Ther 272:815–824
Rhodes ME, O’Toole SM, Wright SL, Czambel RK, Rubin RT (2001) Sexual diergism in rat hypothalamic-pituitary-adrenal axis responses to cholinergic stimulation and antagonism. Brain Res Bull 54:101–113
Rohleder N, Kirschbaum C (2006) The hypothalamic-pituitary-adrenal (HPA) axis in habitual smokers. Int J Psychophysiol 59:236–243
Rollema H, Guanowsky V, Mineur YS, Shrikhande A, Coe JW, Seymour PA, Picciotto MR (2009) Varenicline has antidepressant-like activity in the forced swim test and augments sertraline’s effect. Eur J Pharmacol 605:114–116
Ross SA, Wong JY, Clifford JJ, Kinsella A, Massalas JS, Horne MK, Scheffer IE, Kola I, Waddington JL, Berkovic SF, Drago J (2000) Phenotypic characterization of an alpha 4 neuronal nicotinic acetylcholine receptor subunit knock-out mouse. J Neurosci 20:6431–6441
Sabri O, Kendziorra K, Wolf H, Gertz HJ, Brust P (2008) Acetylcholine receptors in dementia and mild cognitive impairment. Eur J Nucl Med Mol Imaging 35(Suppl 1):S30–S45
Salin-Pascual RJ (2002) Relationship between mood improvement and sleep changes with acute nicotine administration in non-smoking major depressed patients. Rev Invest Clin 54:36–40
Santamaria A, Arias HR (2010) Neurochemical and behavioral effects elicited by bupropion and diethylpropion in rats. Behav Brain Res 211:132–139
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301:805–809
Schofield GG, Witkop B, Warnick JE, Albuquerque EX (1981) Differentiation of the open and closed states of the ionic channels of nicotinic acetylcholine receptors by tricyclic antidepressants. Proc Natl Acad Sci USA 78:5240–5244
Semba J, Mataki C, Yamada S, Nankai M, Toru M (1998) Antidepressantlike effects of chronic nicotine on learned helplessness paradigm in rats. Biol Psychiatry 43:389–391
Shytle RD, Silver AA, Lukas RJ, Newman MB, Sheehan DV, Sanberg PR (2002a) Nicotinic acetylcholine receptors as targets for antidepressants. Mol Psychiatry 7:525–535
Shytle RD, Silver AA, Sheehan KH, Sheehan DV, Sanberg PR (2002b) Neuronal nicotinic receptor inhibition for treating mood disorders: preliminary controlled evidence with mecamylamine. Depress Anxiety 16:89–92
Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, Ehrhart J, Silver AA, Sanberg PR, Tan J (2004) Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem 89:337–343
Slattery DA, Morrow JA, Hudson AL, Hill DR, Nutt DJ, Henry B (2005) Comparison of alterations in c-fos and Egr-1 (zif268) expression throughout the rat brain following acute administration of different classes of antidepressant compounds. Neuropsychopharmacology 30:1278–1287
Stapleton JA, Watson L, Spirling LI, Smith R, Milbrandt A, Ratcliffe M, Sutherland G (2008) Varenicline in the routine treatment of tobacco dependence: a pre-post comparison with nicotine replacement therapy and an evaluation in those with mental illness. Addiction 103:146–154
Steru L, Chermat R, Thierry B, Simon P (1985) The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology (Berl) 85:367–370
Suemaru K, Yasuda K, Cui R, Li B, Umeda K, Amano M, Mitsuhashi H, Takeuchi N, Inoue T, Gomita Y, Araki H (2006) Antidepressant-like action of nicotine in forced swimming test and brain serotonin in mice. Physiol Behav 88:545–549
Targacet IW-S, NC. http://www.targacept.com/wt/page/pr_1247655940 [Last Accessed 2/2010].
Tizabi Y, Overstreet DH, Rezvani AH, Louis VA, Clark E Jr, Janowsky DS, Kling MA (1999) Antidepressant effects of nicotine in an animal model of depression. Psychopharmacology (Berl) 142:193–199
Tonstad S, Davies S, Flammer M, Russ C, Hughes J (2010) Psychiatric adverse events in randomized, double-blind, placebo-controlled clinical trials of varenicline: a pooled analysis. Drug Saf 33:289–301
Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, Norquist G, Howland RH, Lebowitz B, McGrath PJ, Shores-Wilson K, Biggs MM, Balasubramani GK, Fava M (2006) Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 163:28–40
Tsigos C, Chrousos GP (2002) Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J Psychosom Res 53:865–871
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421:384–388
Wilhelm K, Wedgwood L, Niven H, Kay-Lambkin F (2006) Smoking cessation and depression: current knowledge and future directions. Drug Alcohol Rev 25:97–107
Acknowledgements
Supported in part by NIMH T32MH067553
Conflict of interest statement
The authors attest, to the best of their knowledge, that they have no conflicts of interest in relation to this article to disclose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Philip, N.S., Carpenter, L.L., Tyrka, A.R. et al. Nicotinic acetylcholine receptors and depression: a review of the preclinical and clinical literature. Psychopharmacology 212, 1–12 (2010). https://doi.org/10.1007/s00213-010-1932-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-010-1932-6