
Abstract
Introduction
The role of oxidative stress has been well known in neurodegenerative disorders. 3-Nitropropionic acid (3-NP) is a plant-based mycotoxin that produces HD like symptoms in animals. Oxidative stress and nitric oxide mechanisms have been recently proposed in the 3-NP-induced neurotoxicity. Epigallocatechin gallate (EGCG) is one of the major components of green tea, known for its potent antioxidant activity. Besides, neuroprotective effect of EGCG has also been suggested in different experimental models.
Objectives
The present study has been designed to examine possible effect of EGCG against 3-NP induced behavioral, oxidative stress, mitochondrial dysfunction, and striatal damage in rats and its possible interaction with nitric oxide modulators.
Material and methods
Systemic 3-NP (10 mg/kg) administration for 14 days significantly reduced locomotor activity, body weight, grip strength, oxidative defense (raised levels of lipid peroxidation, nitrite concentration, depletion of antioxidant enzyme), and mitochondrial enzymes activity in striatum, cortex, and hippocampal regions of the brain.
Results
Fourteen days of EGCG pretreatment (10, 20, and 40 mg/kg) significantly attenuated behavioral alterations, oxidative damage, mitochondrial complex enzymes dysfunction, and striatal damage in 3-NP-treated animals. l-arginine (50 mg/kg) pretreatment with sub-effective dose of EGCG (20 mg/kg) significantly reversed the protective behavioral, biochemical, cellular, and histological effects of EGCG. However, l-NAME (10 mg/kg) pretreatment with EGCG (20 mg/kg) significantly potentiated the protective effect of EGCG which was significant as compared to their effect per se.
Conclusion
The present study shows that EGCG attenuate 3-NP-induced neurotoxicity, and nitric oxide modulation might be involved in its protective action.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Huntington’s disease (HD) is a neurodegenerative disorder characterized by a progressive degeneration of striatal neurons that leads to both motor and cognitive impairments (Nam et al. 2005). 3-Nitropropionic acid (3-NP) obtains from fungus, irreversibly inhibits succinate dehydrogenase (SDH) enzyme in the tricarboxylic acid cycle and electron transport chain. Systemic 3-NP administration produces striatum-specific lesions, delayed dystonia, and abnormal choreiform movements in rats and humans (Ludolph et al. 1991; Brouillet et al. 1995). 3-NP-induced striatal-specific lesions have been reported to occur by involving secondary excitotoxic mechanisms (Nam et al 2005) possible by indirect activation of glutamate receptors (Zeevalk et al. 1995). Besides, 3-NP toxicity has been reported to affect other regions of the brain such as cerebral cortex, hippocampus, etc. (La Fontaine et al. 2000). Additionally, it has also been reported that oxidative stress plays a key role in 3-NP-induced striatal damage (La Fontaine 2000; Yang et al. 2005). Growing body of evidence suggest that elevated concentration of intracytosol calcium and disruption of transmembrane ion gradients and subsequently production of free radicals are implicated in HD pathology (Deshpandea et al. 2006).
NO is a short-lived bioactive molecule that participates in the physiology and pathophysiology of biological systems in mammals. NO is synthesized in the cytoplasm from l-arginine by nitric oxide synthase (NOS) (Moncada et al. 1992). It has been suggested that excessive NO biosynthesis may be deleterious and causes neurotoxicity in the neurons (Nagai et al. 2002). Studies suggest that excessive concentration of NO plays a major role in the pathogenesis of neurodegenerative diseases such as HD, Alzheimer’s disease, Parkinson’s disease, etc. (Nagai et al. 2002).
Green tea is a popular beverage used worldwide for its various pharmacological effects such as antioxidant, antimutagenic, antiproliferative, and anticarcinogenic properties and neuroprotective activities (Lee et al. 2000). Chemically, green tea contains many polyphenolic compounds, commonly known as catechin. Among them, epigallocatechin gallate (EGCG) is one of the active polyphenols known for its potent antioxidant property (Matsuo et al. 1997), although its cellular mechanism is still unclear. However, reports suggest the involvement of oxidative stress and nitric oxide pathways in its protective actions (Guo et al. 1996; Aucamp et al. 1997). However, to our knowledge, there is no previous study conducted to explore the protective effect of EGCG against 3-NP-induced neurotoxicity in rats.
Therefore, present study has been designed to explore the effect of EGCG and its possible nitric oxide mechanism against 3-NP-induced behavioral, oxidative stress, and mitochondrial and histological alterations in striatum, cortex, and hippocampal regions in rat brain.
Experimental procedure
Animals
Male Wistar rats bred in Central Animal House facility of the Panjab University, Chandigarh and weighing between 250 and 300 g were used. Animals were acclimatized to laboratory conditions prior to experimentation. Animals were kept under standard conditions of light and dark cycle with food and water ad libitum. All the experiments were carried out between 0900 and 1500 hours. The protocol was approved by the Institutional Animal Ethics Committee and carried out in accordance with the Indian National Science Academy Guidelines for the use and care of animals.
Drugs and treatment schedule
The following drugs were used in the present study. 3-NP, l-arginine, and l-NAME (Sigma Chemicals, St. Louis, MO, USA) were diluted with saline (adjust pH 7.4) and administered intraperitoneally to animals. EGCG was suspended in 0.05% w/v sodium carboxy-methyl-cellulose solution and administered by oral route in a constant volume of 0.5 ml/100 g of body weight. Animals were randomly divided into ten groups, 16 animals in each. The study was conducted in different phases. The effect of EGCG was explored in the first phase. In subsequent phases, interactions of EGCG with nitric oxide modulators were explored.
Group 1 was the vehicle-treated group, Group 2 received 3-NP (10 mg/kg), Group 3 received EGCG (40 mg/kg, per oral) per se, Groups 4 to 6 received EGCG (10, 20, and 40 mg/kg) + 3-NP (10 mg/kg), Group 7 received l-arginine (50 mg/kg) + 3-NP (10 mg/kg), Group 8 received l-NAME (10 mg/kg) + 3-NP (10 mg/kg), Group 9 received l-arginine (50 mg/kg) + EGCG (20 mg/kg) + 3-NP (10 mg/kg), and Group 10 received l-NAME (10 mg/kg) + EGCG (20 mg/kg) + 3-NP (10 mg/kg).
Drugs were given administered for 14 days. In the present study, l-arginine and l-NAME administered 1 h prior to EGCG whereas EGCG was administered 1 h prior to 3-NP administration.
TTC staining, striatal lesion volume measurement
On the 15th day, animals were killed for 2,3,5-triphenyltetrazolium chloride (TTC) staining. Brains were quickly removed and placed in ice-cold saline solution. Brains were sectioned at 2-mm intervals. Slices were then subjected in 2% TTC for 20 min at 37°C in the dark and removed and placed in 4% paraformaldehyde, pH 7.4 in 0.1 M phosphate buffer. A lesion volume was measured using computer-based image analysis (Image, NIH).
Measurement of body weight
Animal body weight was recorded on the first and last days of the experimentation. Percent change in body weight was calculated as
Behavioral assessments
Assessment of gross behavioral activity (locomotor activity)
The locomotor activity was assessed by using actophotometer (IMCORP, Ambala, India). The motor activity was detected by infrared beams above the floor of the testing area. Each interruption of a beam on the x- or y-axis generated an electric impulse, which was presented on a digital counter. The apparatus was placed in a darkened light, sound-attenuated, and ventilated testing room. Each animal was observed over a period of 5 min and expressed as counts per 5 min (Kumar et al. 2006).
Rotarod activity
All animals were evaluated for motor coordination and grip by using the rotarod apparatus (Techno, India). The rats were exposed to prior training session before initialization of therapy to acclimate them on rotarod performance. Rats were placed on the rotating rod with a diameter of 7 cm (speed 25 rpm). The cutoff time was 180 s, and each rat performed three separate trials after 5-min gap. The average fall of time was recorded (Kulkarni 1999).
Dissection and homogenization
On day 15, after behavioral assessments, animals were randomized into three groups, for the biochemical, mitochondrial complex, and striatum lesion volume estimation. For the biochemical analysis, animals were killed by decapitation immediately after behavioral assessments. The brains were removed, and the cortex, striatum, and hippocampus were separated by putting on ice. A 10% (w/v) tissue homogenate was prepared in 0.1 M phosphate buffer (pH 7.4). The homogenate was centrifuged at 10,000×g for 15 min. Aliquots of supernatant was separated and used for biochemical estimations.
Measurement of oxidative stress parameters
Measurement of lipid peroxidation
The quantitative measurement of lipid peroxidation was performed according to the method of Wills (1966). The amount of malondialdehyde (MDA), a measure of lipid peroxidation, was measured by reaction with thiobarbituric acid at 532 nm using Perkin Elmer lambda 20 Spectrophotometer (Norwalk, CT, USA). The values were calculated using molar extinction coefficient of chromophore (1.56 × 105 M−1 cm−1) and expressed as percentage of control.
Estimation of nitrite
The accumulation of nitrite in the supernatant, an indicator of the production of nitric oxide (NO), was determined with a colorimetric assay with Greiss reagent (0.1% N-(1-naphthyl) ethylenediame dihydrochloride, 1% sulfanilamide, and 2.5% phosphoric acid) as described by Green et al. (1982). Equal volumes of supernatant and Greiss reagent were mixed; the mixture was incubated for 10 min at room temperature in the dark, and the absorbance at 540 nm was determined with Perkin Elmer lambda 20 spectrophotometer. The concentration of nitrite in the supernatant was determined from a sodium nitrite standard curve and expressed as percentage of control.
Catalase estimation
Catalase activity was assayed by the method of Luck (1971), wherein the breakdown of hydrogen peroxides (H2O2) is measured at 240 nm. Briefly, assay mixture consisted of 3 ml of H2O2 phosphate buffer and 0.05 ml of supernatant of tissue homogenate (10%), and change in absorbance was recorded at 240 nm. The results were expressed as micromole H2O2 decomposed per milligram of protein per minute.
Superoxide dismutase activity
Superoxide dismutase (SOD) activity was assayed according to the method of Kono (1978) wherein the reduction of nitrazobluetetrazolium (NBT), inhibited by the superoxide dismutase, is measured at 560 nm using spectrophotometer. Briefly, the reaction was initiated by the addition of the hydroxylamine hydrochloride to the mixture containing NBT and sample. The results were expressed as unit/mg protein, where one unit of enzyme is defined as the amount of enzyme inhibiting the rate of reaction by 100%.
Protein estimation
The protein content was measured by biuret method using bovine serum albumin as standard (Gornall 1949).
Mitochondrial complex estimation
Isolation of rat brain mitochondria
Rat brain mitochondria were isolated by the method of Berman and Hastings (1999). The brain regions were homogenized in isolation buffer. Homogenate was centrifuged at 13,000×g for 5 min at 4ºC. The pellet was resuspended in isolation buffer with ethylene glycol tetraacetic acid (EGTA) and spun again at 13,000×g for 5 min. The resulting supernatant was transferred to new tubes and topped off with isolation buffer with EGTA and again spun at 13,000×g for 10 min. Pellet containing pure mitochondria was resuspended in isolation buffer without EGTA.
Complex I (NADH dehydrogenase activity)
Complex I was measured spectrophotometrically by the method of King and Howard (1967). The method involves catalytic oxidation of NADH to NAD+ with subsequent reduction of cytochrome C. The reaction mixture contained 0.2 M glycyl glycine buffer pH 8.5, 6 mM NADH in 2 mM glycyl glycine buffer, and 10.5 mM cytochrome C. The reaction was initiated by addition of requisite amount of solubilized mitochondrial sample and followed absorbance change at 550 nm for 2 min.
Complex II (SDH activity)
SDH was measured spectrophotometrically according to King (1967). The method involves oxidation of succinate by an artificial electron acceptor, potassium ferricyanide. The reaction mixture contained 0.2 M phosphate buffer pH 7.8, 1% BSA, 0.6 M succinic acid, and 0.03 M potassium ferricyanide. The reaction was initiated by the addition of mitochondrial sample, and absorbance change was followed at 420 nm for 2 min.
Complex IV (Cytochrome oxidase assay)
Cytochrome oxidase activity was assayed in brain mitochondria according to the method of Sotocassa et al. (1967). The assay mixture contained 0.3 mM reduced cytochrome C in 75 mM phosphate buffer. The reaction was started by the addition of solubilized mitochondrial sample, and the absorbance change was recorded at 550 nm for 2 min.
MTT assay
The MTT assay is based on the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-H-tetrazolium bromide (MTT) by hydrogenase activity in functionally intact mitochondria. The MTT reduction rate was used to assess the activity of the mitochondrial respiratory chain in isolated mitochondria by the method of Liu et al. (1997). Briefly, 100-μl mitochondrial samples were incubated with 10 μl MTT for 3 h at 37°C. The blue formazan crystals were solubilized with dimethylsulfoxide and measured by an ELISA reader at 580-nm filter.
Statistical analysis
The data were analyzed by using ANOVA followed by Tukey’s test. All the values are expressed as mean ± SEM. In all tests, the criterion for statistical significance was P < 0.05.
Results
Effect of EGCG on 3-NP-induced rat striatal degeneration
We first examined the effects of EGCG on neurotoxicity induced by repeated treatment of 3-NP using TTC staining method. 3-NP treatment significantly produced lesions in the striatal regions as compared to vehicle-treated group, where as EGCG treatment significantly reversed these changes as compared to 3-NP-treated group. Further, l-arginine treatment with EGCG (20 mg/kg) reversed their effect as compared to their effect per se. l-NAME (10 mg/kg) treatment with EGCG (20 mg/kg) potentiated their effect as compared to their effect per se (Fig. 1).

Effect of EGCG and nitric oxide modulator on striatal lesions produced by systemic administration of 3-NP
Effect of EGCG on body weight and its modification by nitric oxide modulators in 3-NP-treated rats
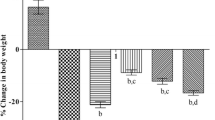
3-NP (10 mg/kg) treatment caused significant decrease in body weight on the 15th day as compared to vehicle-treated group. Further, EGCG (10, 20, and 40 mg/kg, p.o.) treatment significantly attenuated the body weight on the 15th day in 3-NP-treated rats (P < 0.05). l-arginine, nitric oxide precursor (50 mg/kg, per se), and l-NAME, nitric oxide synthase inhibitor (10 mg/kg, per se) did not significantly influence the body weight as compare to the 3-NP or vehicle-treated groups. l-arginine (50 mg/kg) pretreatment with sub-effective dose of EGCG (20 mg/kg) significantly decreased body weight compared to EGCG-treated (20 mg/kg) animals. However, l-NAME pretreatment with EGCG (20 mg/kg) further improved the body weight compared to their effect per se (Fig. 2).

Effect of EGCG and its modification by nitric oxide modulators on body weight in 3-nitropropionic-acid-treated rats. a P < 0.05 versus vehicle-treated, b P < 0.05 versus 3-NP, c P < 0.05 versus [EGCG (10) +3-NP], d P < 0.05 versus [EGCG (20) +3-NP], e P < 0.05 versus [3-NP +l-NAME(10)]-treated group. (One-way ANOVA followed by Tukey’s test)
Effect of EGCG on locomotor activity and its modification by nitric oxide modulators in 3-NP-treated rat
In the present experiment, mean scores for locomotor activity on day 1 for each rat was relatively stable and showed no significant variation. Systemic 3-NP (10 mg/kg) treatment significantly decreased locomotor activity on the 15th day as compared to vehicle-treated group. Fourteen days of EGCG (10, 20, and 40 mg/kg, p.o.) treatment significantly improved locomotor activity as compared to 3-NP-treated rats (Fig. 2). l-arginine (50 mg/kg) pretreatment with sub-effective dose of EGCG (20 mg/kg) significantly reversed the protective effect of EGCG (there was reduction in locomotor activity) in 3-NP-treated rats. However, l-NAME (10 mg/kg) pretreatment with EGCG (20 mg/kg) significantly increased locomotor activity as compared to their effect per se (P < 0.05). EGCG (40 mg/kg, p.o.) per se treatment did not produce any significant effect on locomotor activity as compared to vehicle-treated group (Fig. 3).

Effect of EGCG and its modification by nitric oxide modulators on locomotor activity in 3-NP-treated rats. a P < 0.05 versus vehicle treated, b P < 0.05 versus 3-NP, c P < 0.05 versus [EGCG (10) +3-NP], d P < 0.05 versus [EGCG (20) + 3-NP], e P < 0.05 versus [l-NAME(10) +3-NP]-treated group. (One-way ANOVA followed by Tukey’s test)
Effect of EGCG and its modification by nitric oxide modulators on rotarod activity in 3-NP-treated rats
Systemic 3-NP treatment significantly impaired grip strength performance on day 15th as compared to vehicle-treated group. EGCG (10, 20, and 40 mg/kg, p.o.) treatment significantly improved muscle grip performance compared to control (3-NP)-treated rats (P < 0.05; Fig. 3). l-arginine (50 mg/kg) pretreatment with EGCG (20 mg/kg) impaired grip strength and attenuated the protective the action of EGCG in 3-NP-treated rats. However, l-NAME (10 mg/kg) pretreatment with EGCG (20 mg/kg) significantly improved the grip strength performance as compared to their effect per se. However, EGCG (40 mg/kg, p.o.) per se did not alter grip strength performance as compared to vehicle (P < 0.05; Fig. 4).

Effect of EGCG and its modification by nitric oxide modulators on rotarod activity in 3-NP-treated rats. a P < 0.05 versus vehicle-treated, b P < 0.05 versus 3-NP, c P < 0.05 versus [EGCG (10) +3-NP], d P < 0.05 versus [EGCG (20) + 3-NP], e P < 0.05 versus [3-NP + l-NAME(10)]-treated group. (One-way ANOVA followed by Tukey’s test)
Effect of EGCG on brain oxidative damage (lipid peroxidation, nitrite, SOD, and catalase) and its modification by nitric oxide modulators in 3-NP-treated rats.
Systemic administration of 3-NP caused an increase in lipid peroxidation, nitrite concentration, depleted superoxide dismutase, and catalase enzyme activity in striatum, cortex, and hippocampus regions of the brain as compared to vehicle-treated animals. However, chronic EGCG (10, 20, and 40 mg/kg, p.o.) treatment significantly attenuated MDA, nitrite concentration and restored SOD and catalase enzyme activities in 3-NP-treated animals (P < 0.05; Table 1). l-arginine (50 mg/kg) pretreatment with sub-effective dose of EGCG (20 mg/kg) significantly reversed the protective effect of EGCG in 3-NP-treated group. On the other hand, l-NAME (10 mg/kg) pretreatment with EGCG (20 mg/kg) significantly potentiated the protective effect of EGCG (20) compared to their effect per se. Further, EGCG (40 mg/kg, p.o.), l-arginine (50 mg/kg), and l-NAME (10 mg/kg) per se treatments did not cause any significant change in MDA, nitrite levels, superoxide dismutase, and catalase enzyme activity compared to vehicle (P < 0.05; Table 1).
Effect of EGCG on mitochondrial complexes activity and its modification by nitric oxide modulators in 3-NP-treated rats
Systemic 3-NP (10 mg/kg) administration significantly impaired mitochondrial enzyme complexes (I, II, and IV) activities as compared to vehicle treated rats. Chronic administration of EGCG (10, 20, and 40 mg/kg, p.o.) significantly restored mitochondrial enzyme complex activities as compared to 3-NP-treated group (P < 0.05; Figs. 4, 5, 6). l-arginine (50 mg/kg) pretreatment with sub-effective dose EGCG (20 mg/kg) significantly reversed the protective effect (reduced mitochondrial enzyme complex activity I, II, and IV) of EGCG in 3-NP-treated animals. However, l-NAME (10 mg/kg) pretreatment with EGCG (20 mg/kg) significantly potentiated and restored mitochondrial enzymes complex activities which were significant as compared to their effect per se. EGCG (40 mg/kg, p.o.), l-arginine (50 mg/kg), and l-NAME, (10 mg/kg) per se treatment did not produce any significant effect on the mitochondrial enzyme complexes as compared to the vehicle-treated group (Figs. 5, 6, 7).

Effect of EGCG and its modification by nitric oxide modulators on mitochondrial complex I in 3-NP-treated rats. a P < 0.05 versus vehicle-treated, b P < 0.05 versus 3-NP, c P < 0.05 versus [EGCG (10) +3-NP], d P < 0.05 versus [EGCG (20) + 3-NP], e P < 0.05 versus [3-NP + l-NAME(10)] treated group. (One-way ANOVA followed by Tukey’s test)

Effect of EGCG and its modification by nitric oxide modulators on mitochondrial complex II in 3-NP-treated rats. a P < 0.05 versus vehicle-treated, b P < 0.05 versus 3-NP, c P < 0.05 versus [EGCG (10) +3-NP], d P < 0.05 versus [EGCG (20) + 3-NP], e P < 0.05 versus [3-NP + l-NAME(10)]-treated group. (One-way ANOVA followed by Tukey’s test)

Effect of EGCG and its modification by nitric oxide modulators on mitochondrial complex IV in 3-NP-treated rats. a P < 0.05 versus vehicle-treated, b P < 0.05 versus 3-NP, c P < 0.05 versus [EGCG (10) +3-NP], d P < 0.05 versus [EGCG (20) + 3-NP], e P < 0.05 versus [3-NP + l-NAME(10)]-treated group. (One-way ANOVA followed by Tukey’s test)
Effect of EGCG on MTT assay and its modification by nitric oxide modulators in 3-NP-treated rats
Systemic 3-NP treatment significantly declined number of viable cell as compared to vehicle-treated rats as estimated by MTT assay. Fourteen days systemic treatment with EGCG (10, 20, and 40 mg/kg, p.o.) significantly restored the loss in number viable cell counts as compared to 3-NP-treated group (P < 0.05; Fig. 7). l-arginine (50 mg/kg) pretreatment with EGCG (20 mg/kg) significantly reversed the protective effect of EGCG in 3-NP-treated group. On the other hand, l-NAME (10 mg/kg) pretreatment with EGCG (20 mg/kg) significantly increased viable cell count and potentiated the protective effect of EGCG as compared to their effect per se (P < 0.05; Fig. 8).

Effect of EGCG and its modification by nitric oxide modulators on MTT ability in 3-NP-treated rats. a P < 0.05 versus vehicle-treated, b P < 0.05 versus 3-NP, c P < 0.05 versus [EGCG (10) +3-NP], d P < 0.05 versus [EGCG (20) + 3-NP], e P < 0.05 versus [3-NP + l-NAME(10)]-treated group. (One-way ANOVA followed by Tukey’s test)
Discussion
The present study highlights the therapeutic potential of EGCG against 3-NP-induced histological, behavioral alterations, oxidative damage, and mitochondrial dysfunction. Results demonstrate that: (1) EGCG reversed 3-NP-induced behavioral alterations (improved body weight, locomotor, and rotarod performance), oxidative damage (attenuated lipid peroxidation, nitrite levels, decreased superoxide dismutase, and catalase levels), and restored mitochondrial complex enzyme activities (inhibition of complex I, II, and IV); (2) l-arginine pretreatment reversed the protective effect of EGCG and l-NAME pretreatment significantly potentiate the protective effect of EGCG.
Systemic 3-NP treatment for 14 days significantly caused body weight loss and motor abnormalities (reduced locomotor and grip strength performance), suggesting behavioral toxicity. Supporting to the present investigation, late-stage HD patients also exhibit dysphagia and loss of body weight (Saydoff et al. 2003). Report suggests that 3-NP-induced loss in body weight could be partially due to factors outside the CNS (Saydoff et al. 2003), whereas alterations in locomotor and motor behavioral could be its specific action on striatum (basal ganglia) that control body movement. Besides, studies indicated that abnormal behavioral symptoms in early HD patients are due to primarily either dysfunction of cholinergic interneurons in striatal circuits or cell loss within the lateral striatum, ventral pallidum, and entopedoncular nucleus (Picconi et al. 2006; Massioui et al. 2001).
It seems that 3-NP produces alteration in motor behavioral by influencing striatum. However, the possibility of involvement of other brain areas such as cortex and hippocampus cannot be ignored in 3-NP action (Rodríguez-Martínez et al. 2004; Silva et al. 2007). Intriguingly, systemic treatments with EGCG for 14 days significantly restored the loss in body weight in 3-NP-treated rats. Previous laboratory reports also reconfirm that antioxidant treatments significantly restored the behavioral changes and oxidative defense level in 3-NP-treated animals (Kumar and Kumar 2009; Kumar et al. 2006, 2007). EGCG pretreatment significantly attenuated behavioral alterations (locomotor as well as rotarod performance) following 3-NP administration.
l-Arginine pretreatment significantly reversed the protective effect of EGCG on all behavioral parameters. On the other hand, l-NAME pretreatment caused potentiation of EGCG protective effect, suggesting the involvement of nitric oxide pathway in the protective effect of EGCG. EGCG modulatory effect by nitric oxide modulators explains its neuroprotective effect. It has been shown that EGCG inhibits neuronal NOS (nNOS) in vitro (Lin et al. 1997) and in vivo in hypoxic rats, reducing oxidative stresses (Wei et al. 2004). Previous studies also reported that catechins protect, in vivo, against white matter oxidative damage in childhood-onset hydrocephalus in rats (Etus et al. 2003) and found to be effective in improving learning and memory in senescence-accelerated mice (Unno et al. 2004) including Alzheimer transgenic mice (Kavon Rezai-Zadeh et al. 2008). Neuroprotective effect of EGCG is also confirmed by its ability to cross blood–brain barrier in mammals (Suganuma et al. 1998). In vitro models of HD demonstrate that EGCG can modulate early steps in the aggregation process of an amyloidogenic polyQ-containing protein (Ehrnhoefer et al. 2006). Beneficial activities of EGCG such as radical scavenging, reduction of reactive oxidative species, or chelating of metal ions might also contribute to the decrease of htt aggregation and toxicity in vivo models of HD (Mandel et al. 2005). EGCG might protect neuronal cells by expressing a mutant htt protein from its noxious properties (Ehrnhoefer et al. 2006).
3-NP is a mycotoxin, inhibits succinate dehydrogenase, and causes interruption in mitochondrial electron transport and cellular energy deficit (Nam et al. 2005). It has been reported that systemic administration of 3-NP produce brain lesions that closely replicate the histological, neurochemical, and clinical features of HD (Beal 1993). These lesions may involve secondary excitotoxic oxidative stress and ATP failure mechanisms. It has been shown that excitotoxicity may be linked to free radical generation (Dykens 1994). Evidence suggests the involvement of oxidative stress in 3-NP neurotoxicity that includes production of hydroxyl free radicals, weakness in endogenous antioxidants defense, and increased 3-nitrotyrosine, a marker for peroxynitrite-mediated damage (Binienda 1998; Klivenyi et al. 2000; Matthews et al. 2000). It has also been reported that systemic 3-NP administration produces oxidized proteins in the striatum and cortex as well as massive loss of striatal neurons (La Fontaine 2000). Researchers also confirmed 3-NP-induced lesions and oxidative damage in hippocampus (Rodríguez-Martínez et al. 2004; Silva et al. 2007; Karanian et al. 2006; Burda et al. 2005). Turan and coworkers reported that chemical preconditioning with 3-NP reduces infarct size via a mechanism that may involve increased bioavailability of NO and decreased ONOO– formation (Turan et al. 2006). Previous studies from different groups reported that 3-NP significantly induced oxidative damage and impaired antioxidant defense enzymes in the brain (Kumar and Kumar 2009; Kumar et al. 2006, 2007; Túnez et al. 2006, 2007; Túnez and Santamaría 2009; Pérez-De La Cruz et al. 2009; Garcia et al. 2008)
Antioxidant drugs strategies (Curcumin, resveratrol; Kumar et al. 2006, 2007) as well as overexpression of genes involved in attenuating oxidative stress showed a significant neuroprotective effects against 3-NP neurotoxicity (Beal et al. 1995). In the present study, 3-NP significantly induced oxidative damage (increased lipid peroxidation, nitrite concentration, and depleted superoxide and catalase levels) in the striatum, cortex, and hippocampal regions. These changes significantly restored by EGCG pretreatment and modulated by nitric oxide modulators. This explains that nitric oxide modulation could be involved in the neuroprotective action of EGCG in addition to its antioxidant effect. EGCG protects against NO stress-induced neuronal damage after ischemia by acting as an antioxidant (Buttemeyer et al. 2003). Reports also suggest that EGCG exerted a protective effect against iron-induced oxidative stress in the rat brain and reduced lipid peroxidation in synaptosomes (Guo et al. 1996). EGCG increased catalase activity and attenuated nitrite concentration (Arimoto-Kobayashi et al. 2003), suggesting that catechins directly produce antioxidant action as well as indirectly influence the activity of other antioxidant enzymes. Besides, a number of in vitro and in vivo studies have shown the antioxidant and antiapoptotic effect of EGCG (Morley et al. 2005; Xie et al. 2004; Mandel et al. 2005).
It has been demonstrated that EGCG can scavenge superoxide and hydroxyl radicals (Guo et al. 1996), peroxyl radicals, NO (Kelly et al. 2001), carbon-center free radicals, singlet oxygen, lipid free radicals (Zhao et al. 2001), and peroxynitrite by preventing nitration of tyrosine. Besides, catechins exert their antioxidant effects through ultra rapid electron transfer from catechins to ROS-induced radical sites on DNA (Anderson et al. 2001). The inhibition of calcium influx by catechins may, therefore, be an important mechanism by which catechins prevent neuronal damage. NO produced by inducible nitric oxide synthase (iNOS) and nNOS plays an important biological role in pathophysiological processes including neurotoxicity (Moncada 1992). The modulation of the various NOS isoforms could be involved in the neuroprotective effects of EGCG. It has been shown that EGCG inhibits iNOS, nNOS in vitro (Lin et al. 1997), and in vivo in hypoxic rats, reducing oxidative stress (Wei et al. 2004). There are substantial evidences indicating that EGCG protective effect partially may be due to its NO scavenging and NOS inhibiting activity (Nagai et al. 2002; Tedeschi et al. 2004). The nNOS isoform of NOS produces toxic effect through NO. Therefore, EGCG inhibition of nNOS may be one of its neuroprotective mechanisms. EGCG inhibits nNOS activity after stimulation with lipopolysaccharide and interferon γ in mouse peritoneal cells (Chan et al. 1997) and iNOS mRNA expression after treatment with LPS, IFN (Chan et al. 1997; Paquay et al. 2000), interleukin (IL)-1 and tumor necrosis factor α (TNF α) (Tedeschi et al. 2004) in vitro.
It has been suggested that EGCG can elicit antioxidant action by involving several ways such as: (a) free radical scavenging activity and reactive nitrogen species (Guo et al. 1996) (b) blockade of iNOS and nNOS induction (Chan et al. 1997). These mechanisms may contribute to its potent antioxidant and putative neuroprotective actions as shown in Fig. 9.

Possible mechanism of action of EGCG against 3-NP induced neurotoxicity in rats
3-NP induces damage to cell-signalling pathways in the brain and impairs ATP in neuronal mitochondria, which leads to cell apoptosis. Mitochondrial respiratory chain complexes that produce ATP for cellular functioning damage by 3-NP induced neurotoxicity both in vivo and in vitro. In the present study, 3-NP significantly altered mitochondrial complex enzymes activities in the striatum, cortex, and hippocampal regions of brain which was restored the functional activities by EGCG treatment. Further, l-arginine pretreatment significantly reversed the protective effect of EGCG whereas l-NAME pretreatment potentiated the protective effect of EGCG. Previous report suggests that EGCG improve functional activity of mitochondrial complexes in ischemia rat model (Sutherland et al. 2005). EGCG significantly attenuates the impairment in citrate synthase activity induced by ischemia (Sutherland et al. 2005). EGCG prevents mitochondrial damage in isoproterenol-induced cardiac toxicity in rats (Devika et al. 2008). Therefore, EGCG restores mitochondrial complex activity as well as prevents the leakage of important mitochondrial matrix components into the extracellular space, thus, preserving normal ATP function and halt cell death.
3-NP treatment further caused selectively lesion in the striatum as measured by TTC staining. One of the main indicators of neuronal cell death is excitotoxicity. Excitotoxin-induced cell death is derived from the disturbance of intracellular Ca2+ homeostasis. Recent reports indicated that 3-NP-caused cellular ATP exhaustion that is coupled to intracellular Ca2+ elevation. 3-NP-induced intracellular Ca2+ elevation might be mediated via either through voltage dependent Ca2+ channel following depolarization with Na+ influx or NMDA and non-NMDA receptor activation (Deshpande et al. 2006). EGCG treatment significantly attenuated these striatal lesion volumes in 3-NP-treated group. Previous reports also suggest that antioxidant like ginseng significantly decreases striatal lesion volume in 3-NP-treated rats (Kim et al. 2005). On the other hand, l-NMAE significantly potentiated the protective effect of EGCG as compared to their effect per se, suggesting the involvement of nitric oxide mechanism in the protective effect of EGCG.
Present study highlights and extends the neuroprotective effects of EGCG. EGCG reversed the behavioral, histological and cellular alterations against 3-NP toxicity. Study further provides evidence that NOS inhibition might play a pivotal role in neuroprotective effect of EGCG.
References
Anderson RF, Fisher LJ, Hara Y, Harris T, Mak WB, Melton LD (2001) Green tea catechins partially protect DNA from (-) OH radical induced strand breaks and base damage through fast chemical repair of DNA radicals. Carcinogenesis 22:1189–1193
Arimoto-Kobayashi S, Inada N, Sato Y, Sugiyama C, Okamoto K, Hayatsu H, Negishi T (2003) Inhibitory effects of (-)-epigallocatechin gallate on the mutation, DNA strand cleavage, and DNA adduct formation by heterocyclic amines. J Agric Food Chem 51:5150–5153
Aucamp J, Gaspar A, Hara Y, Apostolides Z (1997) Inhibition of xanthine oxidase by catechins from tea (Camellia sinensis). Antican Res 17:4381–4386
Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT (1993) Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci 13:4181–4192
Beal MF, Ferrante RJ, Henshaw R, Matthews RT, Chan PH, Kowall NW, Epstein CJ, Schulz JB (1995) 3-Nitropropionic acid neurotoxicity is attenuated in copper/zinc superoxide dismutase transgenic mice. J Neurochem 65:919–922
Berman SB, Hastings TG (1999) Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson’s disease. J Neurochem 73:1127–1137
Binienda Z, Simmons C, Hussain S, Slikker W Jr, Ali SF (1998) Effect of acute exposure to 3-nitropropionic acid on activities of endogenous antioxidants in the rat brain. Neurosci Lett 251:173–176
Brouillet E, Hantraye P (1995) Effects of chronic MPTP and 3-nitropropionic acid in nonhuman primates. Curr Opin Neurol 8:469–473
Burda J, Matiasová M, Gottlieb M, Danielisová V, Némethová M, Garcia L, Salinas M, Burda R (2005) Evidence for a role of second pathophysiological stress in prevention of delayed neuronal death in the hippocampal CA1 region. Neurochem Res 30:1397–1405
Büttemeyer R, Philipp AW, Schlenzka L, Mall JW, Beissenhirtz M, Lisdat F (2003) Epigallocatechin gallate can significantly decrease free oxygen radicals in the reperfusion injury in vivo. Transplant Proc 35:3116–3120
Chan MM, Fong D, Ho CT, Huang HI (1997) Inhibition of inducible nitric oxide synthase gene expression and enzyme activity by epigallocatechin gallate, a natural product from green tea. Biochem Pharmacol 54:1281–1286
Deshpande SB, Hida H, Takei-Io N, Masuda T, Baba H, Nishino H (2006) Involvement of nitric oxide in 3-nitropropionic acid-induced striatal toxicity in rats. Brain Res 1108:205–215
Devika PT, Stanely MPP (2008) (-) Epigallocatechin-gallate (EGCG) prevents mitochondrial damage in isoproterenol-induced cardiac toxicity in albino Wistar rats: a transmission electron microscopic and in vitro study. Pharmacol Res 57:351–357
Dykens JA (1994) Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated Ca and Na: implications for neurodegeneration. J Neurochem 63:584–591
Ehrnhoefer DE, Duennwald M, Markovic P, Wacker JL, Engemann S, Roark M, Legleiter J, Marsh JL, Thompson LM, Lindquist S, Muchowski PJ, Wanker EE (2006) Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington's disease models. Hum Mol Genet 15:2743–2751
El Massioui N, Ouary S, Cheruel F, Hantraye P, Brouillet E (2001) Perseverative behavior underlying attentional set-shifting deficits in rats chronically treated with the neurotoxin 3-nitropropionic acid. Exp Neurol 172:172–181
Etus V, Altug T, Belce A, Ceylan S (2003) Green tea polyphenol (-) - epigallocatechin gallate prevents oxidative damage on periventricular white matter of infantile rats with hydrocephalus. J Exp Med 200:203–209
Garcia E, Limon D, Perez-De La Cruz V, Giordano M, Diaz-Muñoz M, Maldonado PD, Herrera-Mundo MN, Pedraza-Chaverri J, Santamaria A (2008) Lipid peroxidation, mitochondrial dysfunction and neurochemical and behavioral deficits in different neurotoxic models: protective role of S-allylcysteine. Free Radic Res 42:892–902
Gornall AG, Bardawill CJ, David MM (1949) Determination of serum proteins by means of the biuret reaction. J Biol Chem 177:751–756
Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannebaum SR (1982) Analysis of nitrate, nitrite, and [15 N] nitrate in biological fluids. Ann Biochem 126:131–138
Guo Q, Zhao B, Li M, Shen S, Xin W (1996) Studies on protective mechanisms of four components of green tea polyphenols against lipid peroxidation in synaptosomes. Biochem Biophys Acta 1304:210–222
Karanian DA, Baude AS, Brown QB, Parsons CG, Bahr BA (2006) 3-Nitropropionic acid toxicity in hippocampus: protection through N-methyl-D-aspartate receptor antagonism. Hippocampus 16:834–42
Kelly MR, Geigerman CM, Loo G (2001) Epigallocatechin gallate protects U937 cells against nitric oxide-induced cell cycle arrest and apoptosis. J Cell Biochem 81:647–658
Kim JH, Kim S, Yoon IS, Lee JH, Jang BJ, Jeong SM, Lee BH, Han JS, Oh S, Kim HC, Park TK, Rhim H, Nah SY (2005) Protective effects of ginseng saponins on 3-nitropropionic acid-induced striatal degeneration in rats. Neuropharmacology 48:743–756
King TE (1967) Preparation of succinate dehydrogenase and reconstitution of succinate oxidase. Methods Enzymol 10:322–331
King TE, Howard RL (1967) Preparations and properties of soluble NADH dehydrogenases from cardiac muscle. Methods Enzymol 10:275–284
Klivenyi P, Andreassen OA, Ferrante RJ, Dedeoglu A, Mueller G, Lancelot E, Bogdanov M, Andersen JK, Jiang D, Beal MF (2000) Mice deficient in cellular glutathione peroxidase show increased vulnerability to malonate, 3-nitropropionic acid, and 1-methyl-4-phenyl- 1, 2, 5, 6-tetrahydropyridine. J Neurosci 20:1–7
Kono Y (1978) Generation of superoxide radical during auto-oxidation of hydroxylamine and an assay of superoxide dismutase. Arch Biochem Biophysics 186:189–195
Kulkarni SK (1999) Handbook of Experimental Pharmacology, 3rd Edition. Vallabh Parkashan, New Delhi, India
Kumar P, Kumar A (2009) Possible role of sertraline against 3-nitropropionic acid induced behavioral, oxidative stress and mitochondrial dysfunctions in rat brain. Prog Neuropsychopharmacol Biol Psychiatry 33:100–108
Kumar P, Padi SS, Naidu PS, Kumar A (2006) Effect of resveratrol on 3-nitropropionic acid-induced biochemical and behavioral changes: possible neuroprotective mechanisms. Behav Pharmacol 17:485–492
Kumar P, Padi SS, Naidu PS, Kumar A (2007) Cyclooxygenase inhibition attenuates 3-nitropropionic acid-induced neurotoxicity in rats: possible antioxidant mechanisms. Fundam Clin Pharmacol 21:297–306
La Fontaine M, Geddes J, Banks A, Butterfield DA (2000) Effect of exogenous and endogenous antioxidants on 3-nitropropionic acid induced oxidative stress and striatal lesions: insights into Huntington’s disease. J Neurochem 75:1709–1715
Lee SR, Kim SP, Kim JE (2000) Protective effect of topiramate against hippocampal neuronal damage after transient global ischemia in the gerbils. Neurosci Lett 281:184–187
Lin YL, Lin JK (1997) (-)-Epigallocatechin-3-gallate blocks the induction of nitric oxide synthase by down-regulating lipopolysaccharide-induced activity of transcription factor nuclear factor-kB. Mol Pharmacol 52:465–472
Liu Y, Peterson DA, Kimura H, Schubert D (1997) Mechanisms of cellular 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolinium bromide (MTT) reduction. J Neurochem 69(2):581–593
Luck H (1971) Catalase. In: Bergmeyer HU (ed) Methods of enzymatic analysis. Academic, New York, pp 885–893
Ludolph AC, He F, Spencer PS, Hammerstad J, Sabri M (1991) 3-Nitropropionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can J Neurol Sci 18:492–498
Mandel SA, Avramovich-Tirosh Y, Reznichenko L, Zheng H, Weinreb O, Amit T, Youdim MB (2005) Multifunctional activities of green tea catechins in neuroprotection. Modulation of cell survival genes, iron-dependent oxidative stress and PKC signaling pathway. Neurosignals 14:46–60
Matsuo N, Yamada K, Shoji K, Mori M, Sugano M (1997) Effect of tea polyphenols on histamine release from rat basophilic leukemia (RBL-2H3) cells: the structure-inhibitory activity relationship. Allergy 52:58–64
Matthews RT, Yang L, Jenkins BG, Ferrante RJ, Rosen BR, Kaddurah-Daouk R, Beal MF (2000) Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington’s disease. J Neurosci 18:156–163
Moncada S, Palmer RMJ, Higgs DA (1992) Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol Rev 43:109–142
Morley N, Clifford T, Salter L, Campbell S, Gould D, Curnow A (2005) The green tea polyphenol (-)-epigallocatechin gallate and green tea can protect human cellular DNA from ultraviolet and visible radiation-induced damage. Photodermatol Photoimmunol Photomed 21:15–22
Nagai K, Jiang MH, Hada J, Nagata T, Yajima Y, Yamamoto S, Nishizaki T (2002) (-)-Epigallocatechin gallate protects against NO stress induced neuronal damage after ischemia by acting as an anti-oxidant. Brain Res 956:319–322
Nam E, Lee SM, Koh SE, Joo WS, Maeng S, Im HI, Kim YS (2005) Melatonin protects against neuronal damage induced by 3-nitropropionic acid in rat striatum. Brain Res 1046:90–96
Paquay JB, Haenen GR, Stender G, Wiseman SA, Tijburg LB, Bast A (2000) Protection against nitric oxide toxicity by tea. J Agric Food Chem 48:5768–5772
Pérez-De La Cruz V, Elinos-Calderón D, Robledo-Arratia Y, Medina-Campos ON, Pedraza-Chaverrí J, Ali SF, Santamaría A (2009) Targeting oxidative/nitrergic stress ameliorates motor impairment, and attenuates synaptic mitochondrial dysfunction and lipid peroxidation in two models of Huntington's disease. Behav Brain Res 199:210–217
Picconi B, Passino E, Sgobio C, Bonsi P, Barone I, Ghiglieri V, Pisani A, Bernardi G, Ammassari-Teule M, Calabresi P (2006) Plastic and behavioral abnormalities in experimental Huntington’s disease: a crucial role for cholinergic interneurons. Neurobiol Dis 22:143–152
Rezai-Zadeh K, Arendash GW, Hou H, Fernandez F, Jensen M, Runfeldt M, Shytle RD, Tan J (2008) Green tea epigallocatechin-3-gallate (EGCG) reduces beta-amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer transgenic mice. Brain Res 1214:177–187
Rodríguez-Martínez E, Rugerio-Vargas C, Rodríguez AI, Borgonio-Pérez G, Rivas-Arancibia S (2004) Antioxidant effects of taurine, vitamin C, and vitamin E on oxidative damage in hippocampus caused by the administration of 3-nitropropionic acid in rats. Int J Neurosci 114:1133–1145
Saydoff JA, Liu LS, Garcia RA, Hu Z, Li D, von Borstel RW (2003) Oral uridine pro-drug PN401 decreases neurodegeneration, behavioral impairment, weight loss and mortality in the 3-nitropropionic acid mitochondrial toxin model of Huntington’s disease. Brain Res 994:44–54
Silva RH, Abílio VC, Kameda SR, Takatsu-Coleman AL, Carvalho RC, Ribeiro Rde A, Tufik S, Frussa-Filho R (2007) Effects of 3-nitropropionic acid administration on memory and hippocampal lipid peroxidation in sleep-deprived mice. Prog Neuropsychopharmacol Biol Psychiatry 31:65–70
Sottocasa GL, Kuylenstierna B, Ernster L, Bergstrand A (1967) An electron-transport system associated with the outer membrane of liver mitochondria. A biochemical and morphological study. J Cell Biol 32:415–428
Suganuma M, Okabe S, Oniyama M, Tada Y, Ito H, Fujiki H (1998) Wide distribution of [3H] (-)-epigallocatechin gallate, a cancer preventive tea polyphenol, in mouse tissue. Carcinogenesis 19:1771–1776
Sutherland BA, Shaw OM, Clarkson AN, Jackson DN, Sammut IA, Appleton I (2005) Neuroprotective effects of (-)-epigallocatechin gallate following hypoxia–ischemia-induced brain damage: novel mechanisms of action. Faseb J 19:258–260
Tedeschi E, Menegazzi M, Yao Y, Suzuki H, Forstermann U, Kleinert H (2004) Green tea inhibits human inducible nitric-oxide synthase expression by down-regulating signal transducer and activator of transcription-1alpha activation. Mol Pharmacol 65:111–120
Túnez I, Santamaría A (2009) Model of Huntington’s disease induced with 3-nitropropionic acid. Rev Neurol 48:430–434
Túnez I, Montilla P, del Carmen Muñoz M, Medina FJ, Drucker-Colín R (2006) Effect of transcranial magnetic stimulation on oxidative stress induced by 3-nitropropionic acid in cortical synaptosomes. Neurosci Res 56:91–95
Túnez I, Feijóo M, Collado JA, Medina FJ, Peña J, Muñoz Mdel C, Jimena I, Franco F, Rueda I, Muntané J, Montilla P (2007) Effect of testosterone on oxidative stress and cell damage induced by 3-nitropropionic acid in striatum of ovariectomized rats. Life Sci 80:1221–1227
Turan N, Csonka C, Csont T, Giricz Z, Fodor G, Bencsik P, Gyöngyösi M, Cakici I, Ferdinandy P (2006) The role of peroxynitrite in chemical preconditioning with 3-nitropropionic acid in rat hearts. Cardiovasc Res 70:384–390
Unno K, Takabayashi F, Oku N (2004) Improvement in brain function and oxidative damage of aged senescence-accelerated mice by green tea catechins. Intl Congress Series 1260:409–412
Wei IH, Wu YC, Wen CY, Shieh JY (2004) Green tea polyphenol (-) - epigallocatechin gallate attenuates the neuronal NADPH-d/nNOS expression in the nodose ganglion of acute hypoxic rats. Brain Res 999:73–80
Wills ED (1966) Mechanism of lipid peroxide formation in animal. Biochem J 99:667–676
Xie D, Liu G, Zhu G, Wu W, Ge S (2004) (-)-Epigallocatechin-3-gallate protects cultured spiral ganglion cells from H2O2-induced oxidizing damage. Acta Otolaryngol 124:464–470
Yang L, Calingasan NY, Chen J, Ley JJ, Becker DA, Beal MF (2005) A novel azulenyl nitrone antioxidant protects against MPTP and 3-nitropropionic acid neurotoxicities. Exp Neurol 191:86–93
Zeevalk GD, Derr-Yellin E, Nicklas WJ (1995) NMDA receptor involvement in toxicity to dopamine neurons in vitro caused by the succinate dehydrogenase inhibitor 3-nitropropionic acid. J Neurochem 64:455–458
Zhao B, Guo Q, Xin W (2001) Free radical scavenging by green tea polyphenols. Methods Enzymol 335:217–231
Acknowledgment
Authors gratefully acknowledged the financial support of Indian Council of Medical Research (ICMR), New Delhi for carrying out this work. The Senior Research Fellowship (Puneet Kumar) of the Indian Council of Medical Research (ICMR), New Delhi, is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kumar, P., Kumar, A. Protective effects of epigallocatechin gallate following 3-nitropropionic acid-induced brain damage: possible nitric oxide mechanisms. Psychopharmacology 207, 257–270 (2009). https://doi.org/10.1007/s00213-009-1652-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-009-1652-y