
Abstract
Rationale
Antipsychotic-induced parkinsonism (AIP) is a severe adverse affect of neuroleptic treatment. Interindividual heterogeneity in AIP development and severity is associated with risk factors such as antipsychotic drug type, old age, and female gender. There is evidence for genetic predisposition to develop AIP but the variants that confer susceptibility or protection are mostly unknown.
Objective
To identify genes related to AIP susceptibility, we performed a pharmacogenomic genome-wide association study (GWAS) for AIP severity.
Methods
Three hundred ninety-seven American schizophrenia patients who participated in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE)-GWAS project were included in our analysis. Patients had been randomized to treatment with antipsychotic monotherapy for periods ranging from 2 weeks to 18 months during phase 1 of the CATIE trial. They were regularly assessed for AIP severity using the modified Simpson–Angus Scale (SAS). For statistical analysis, patients were dichotomized as cases (average SAS mean global score > 0.3 during CATIE phase 1, N = 199) or controls (average SAS mean global score 0, N = 198).
Results
Using logistic regression and controlling for population stratification, age, gender, SAS score at baseline, and concomitant use of anticholinergic drugs, we identified several single-nucleotide polymorphisms associated with AIP severity. Although none reached the GWAS significance level of P < 4.2 × 10−7, some promising candidate genes for further research on genetic predisposition to AIP were identified including EPF1, NOVA1, and FIGN.
Conclusions
Our finding may contribute to understanding of the pathophysiology of AIP as well as to a priori identification of patients vulnerable for development of AIP.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The use of antipsychotic, neuroleptic drugs is associated with the development of extrapyramidal symptoms (EPS) which may be acute and reversible (such as dystonia, parkinsonism, and akathisia) or long lasting and chronic (tardive dyskinesia and dystonia; Blanchet 2003; Hansen et al. 1997). EPS are a major problem in schizophrenia treatment due to their negative effect on adherence to treatment, patient distress, social stigma, and reduced quality of life (Lindenmayer et al. 2004; Haddad and Dursun 2008). Antipsychotic-induced parkinsonism (AIP) is the most common manifestation of EPS (Rochon et al. 2005; Tenback et al. 2006). Clinically, AIP is very similar to idiopathic Parkinson's disease (PD). It is characterized by bradykinesia, tremor, rigidity, and stooped posture. Other manifestations are gait disturbance, salivation, and seborrheic dermatitis (Hansen et al. 1997; Hirose 2006; Haddad and Dursun 2008). AIP is thought to be caused by blockade of dopamine receptors in the nigrostriatal pathway, although additional hypotheses have been suggested (Casey 2004). It has been shown that early EPS, including parkinsonism, are predictors of tardive dyskinesia (Tenback et al. 2006), but the effect of EPS on antipsychotic treatment outcome is not clear (Caligiuri and Lohr 1997).
AIP prevalence data vary widely among studies, ranging from 15% to more than 50% of antipsychotic-treated patients (Hansen et al. 1997; Hirose 2006). The substantial heterogeneity may stem from interstudy differences in medication regimens, patient demographic background data, and variable phenotype definitions. Well-documented clinical and demographic risk factors for AIP are the use of high-potency neuroleptics, old age, and female gender (Ebadi and Srinivasan 1995; Caligiuri and Peavy 2000). Atypical, second-generation antipsychotics (SGAs) are generally considered less likely to cause EPS than typical, first-generation drugs (FGA; Geddes et al. 2000; Park et al. 2005), although EPS risk is not negligible with SGA (Modestin et al. 2008). Moreover, the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) did not show a difference between SGA and the typical antipsychotic, perphenazine, with regard to acute EPS prevalence (Lieberman et al. 2005).
AIP is considered to be an acute side effect. According to data from the 1960s which relate to FGA, 50% of cases manifest AIP within the first month of drug administration and 90% during the first 72 days (Ayd 1961). Other researchers observed that majority of patients develop AIP within 20 days (Freyhan 1959) while Medinar et al. (1962) reported AIP within the first week of treatment. Improvement and recovery of AIP symptoms within 2 months was reported in two thirds of patients (Stephen and Williamson 1984). However, AIP is also observed as a late-onset manifestation (Lerner et al. 2007).
In addition to the epidemiological risk factors, genetic factors may contribute to interindividual differences in AIP susceptibility (Basile et al. 2002; Arranz and de Leon 2007; Greenbaum et al. 2009). Using the candidate gene approach, several polymorphisms within genes encoding receptors for dopamine and serotonin have been studied for association with AIP, but findings are not conclusive (Gunes et al. 2008; Kaiser et al. 2002; Nakazono et al. 2005; Güzey et al. 2007; Dolzan et al. 2007; Al Hadithy et al. 2008). Associations have been reported for the VNTR polymorphism in DAT1 (Güzey et al. 2007), Taq1, and 141CIns/Del variants in DRD2 (Guzey et al. 2007; Al Hadithy et al. 2008), and the HTR2C polymorphism Cys23Ser (Gunes et al. 2008;Al Hadithy et al. 2008). Genes associated with PD such as Alpha-synuclein, LRRK2, Parkin, Pink1, DJ-1, and UCHL1 have not been specifically studied for association with AIP thus far (Lesage and Brice 2009).
Recently, our group reported association of the RGS2 gene with AIP in a sample of 115 Jewish schizophrenia patients treated with antipsychotics for 2 weeks (Greenbaum et al. 2007); this finding was replicated in a further study of 184 US patients (Greenbaum et al. 2009). A protective effect of the functional 3´UTR single-nucleotide polymorphism (SNP), rs4606, was of particular interest (Greenbaum et al. 2009). Identifying genetic risk factors for AIP may not only improve current understanding of its pathophysiology but also allow prediction of AIP risk among schizophrenia patients prior to antipsychotic treatment.
Genome-wide association studies (GWASs) are a well-established tool in the search for common genetic variations in complex disorders including psychiatric and neurological diseases (Cichon et al. 2009). Several pharmacogenetic GWASs have been published recently, some of them with impressive success (review by Crowley et al. 2009). In contrast to candidate gene-based methods, the genome-wide pharmacogenomic approach allows unbiased, “hypothesis-free” detection of DNA variants associated with the phenotype of interest. Here, we describe the first case-control, pharmacogenomic GWAS for AIP severity. This study employs phenotype and genotype data from the CATIE project (Lieberman et al. 2005). We performed a secondary analysis of the data that aimed to identify genetic variants associated with AIP severity. To the best of our knowledge, this is the first GWAS of AIP reported thus far.
Materials and methods
Phenotype and genotype data access
We used the phenotypic and genotypic data from the CATIE study (Lieberman et al. 2005) and CATIE-GWAS (Sullivan et al. 2008) to conduct a secondary analysis of AIP. Phenotype and genotype data were obtained from the National Institute of Mental Health Center for Collaborative Genetic Studies on Mental Disorders (www.nimhgenetics.org), following signature of a distribution agreement, as required.
Antipsychotic-induced parkinsonism GWAS—sample description
Subjects for the current AIP study were participants in the original CATIE case-control GWAS for schizophrenia described in detail by Sullivan et al. (2008). All were diagnosed with schizophrenia according to Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) structured clinical interview (performed by CATIE personnel), 18–67 years old and male or female of mixed ancestry who were treated with antipsychotics drugs, and provided written informed consent to participate in the study. Patients were excluded from participation if they were diagnosed with schizoaffective disorder or mental retardation, had a history of treatment resistance or serious adverse effects of antipsychotic treatment, and were pregnant or breastfeeding (Stroup et al. 2003).
Antipsychotic-induced parkinsonism GWAS design and pharmacological intervention
The CATIE study, which took place in the USA between January 2001 and December 2004, was a multiphase, randomized, controlled study assessing the response of schizophrenia patients to antipsychotic medication (Lieberman et al. 2005; Stroup et al. 2003). The study included 1,460 subjects, all diagnosed with schizophrenia according to DSM-IV criteria. During CATIE phase 1, following randomization, participants were treated with one of five antipsychotic drugs: perphenazine, olanzapine, quetiapine, risperidone, and ziprasidone. Since some of the participants received antipsychotic treatment before joining the study and being randomized, overlap with prior antipsychotic drug treatment was allowed for the first 28 days of participation. Patients, diagnosed with tardive dyskinesia at baseline, were randomized only to treatment with the four SGA and not perphenazine (phase 1A). For simplicity, the term “CATIE phase 1” (which includes phase 1A) is used in this paper.
Patients were treated according to an antipsychotic drug treatment protocol in the context of a double-blind design for a maximum of 18 months. Treatment duration and discontinuation were determined by judgment of the CATIE clinicians. Maximal participation in phase 1 was 18 months. If patients discontinued phase 1 before end point, they were offered to continue participation in the CATIE trial as part of phase 2 or 3 and received alternative antipsychotic treatment.
Participants were assessed at baseline, after 1 month, after 3 months, and then every 3 months up to 18 months. Assessments included several clinical efficacy scales and adverse effect scales, mainly weight gain and EPS. We excluded patients whose participation in CATIE phase 1 was shorter than 14 days.
Antipsychotic-induced parkinsonism assessment
Patients participating in the CATIE study were assessed for AIP with a modified Simpson–Angus scale (SAS). The modified SAS scale is based on the original SAS (Simpson and Angus 1970) and includes six items, each rated for severity from 0–4: gait, arm dropping, shoulder shaking, elbow rigidity, fixation of position or wrist rigidity, and tremor. It does not include four items which appear in the original version: leg pendulousness, head dropping, glabellar tap, and salivation. The mean score is obtained by adding the items and dividing by six. SAS is a common tool to assess EPS and parkinsonism among antipsychotic-treated patients; the modified scale does not cover akinesia and bradykinesia manifestations. To establish a robust and reproducible phenotype reflecting development of parkinsonism after treatment with antipsychotics, a dichotomized AIP severity phenotype was defined based on the average of all SAS mean global scores (SAS-MGS) for a particular patient during CATIE phase 1 (excluding baseline measurement). Since an SAS threshold of 0.3 for parkinsonism is commonly accepted (Janno et al. 2005), cases were defined as individuals whose average SAS-MGS during phase 1 (not including baseline measurement) was 0.3 and above while the average SAS-MGS of controls was 0 for the entire period of phase 1. Individuals with an intermediate average SAS-MGS (0 < SAS-MGS < 0.3) were not included in the analysis. This phenotype definition was used to overcome potential bias caused by extreme ratings or discrepancies between evaluators.
Genotyping was performed by Perlegen Sciences using two different chips—Affymetrix 500K (500,568 SNPs) and the Perlegen custom 164K chip (164,871 SNPs). In total, 665,439 SNPs were genotyped. However, 157,048 SNPs failed the Perlegen quality control process (described by Sullivan et al. 2008). Of the remaining 508,286 SNPs, 13,030 SNPs were removed due to minor allele frequency < 0.01 or missingness > 0.05362. In total, 495,172 SNPs were included in the analysis set. Seven hundred forty-one individuals were available for AIP-GWAS analysis (after exclusion of duplicates and samples with missingness > 0.2). Of this available data pool, GWAS genotypes of 397 individuals were included in our case-control analysis.
Population stratification
Since cases and controls are of mixed ancestry (129 African Americans, 257 white, 11 mixed), we used principal component analysis (Price et al. 2006; Reich et al. 2008) to correct for possible effects of population stratification. This kind of correction is specific to a candidate marker's variation in frequency across ancestral populations, minimizing spurious associations while maximizing power to detect true associations (Price et al. 2006). Three principal components, which were computed using HelixTree software (http://www.goldenhelix.com) and accounted for a large portion of the variation in genotypes, were chosen. These three principal components were included as covariates in the logistic regression model.
Single-marker analyses
All SNPs were tested for association with the dichotomized case-control AIP phenotype using logistic regression analysis. Based on prior clinical data (see Introduction) and univariate analyses, we identified seven potential covariates to be checked for inclusion in our regression model: (1) age, (2) gender, (3) type of antipsychotic drug prescribed (perphenazine, olanzapine, quetiapine, risperidone, or ziprasidone), (4) average dose of antipsychotic drug (adjusted to chlorpromazine units), (5) concomitant treatment with anticholinergic drugs during phase 1, (6) days of CATIE phase 1 participation (varies from 15 to 601), and (7) SAS score at baseline. To select covariates, we checked for association of these variables with the dependent variable (average SAS-MGS during CATIE phase 1, not including baseline) using Mann–Whitney or chi-square tests. Only significant variables (P < 0.05) were included in the regression model. These analyses were done using SPSS 15.0 (SPSS Inc., Chicago, IL, USA).
To perform GWAS with the AIP phenotype, we used PLINK software version 0.9 (Purcell et al. 2007). We considered a P value of 4.2 × 10−7 as significant at the genome-wide level (Liu et al. 2009; Freimer and Sabatti 2004; Lencz et al. 2007). For gene annotation, we used University of California Santa Cruz Genome Browser (http://genome.cse.ucsc.edu/) and NCBI databases (http://www.ncbi.nlm.nih.gov).
Results
Sample description
Out of 741 cases genotyped originally for the CATIE-GWAS, 397 individuals were used for AIP-GWAS analysis, 199 affected (average SAS-MGS 0.3 and above) and 198 unaffected (SAS-MGS score of 0). This sample consists of 284 males and 113 females aged 41.3 ± 11.6 (18–66) years. Two hundred fifty-seven are of white ancestry; 129 are of African American ancestry, and 11 are mixed. At baseline, 76.32% were treated with antipsychotic drugs, and 24.94% received concomitant anticholinergic treatment during phase 1. These 397 individuals participated in the CATIE study for 247 ± 196 (15–601) days and were treated during phase 1 with perphenazine (19.9%), olanzapine (22.4%), quetiapine (21.9%), risperidone (22.9%), and ziprasidone (12.8%). Additional details, including a comparison of cases and controls on the background variables, appear in Table 1.
Selection of covariates
As shown in Table 1, four covariates were found to be associated with average SAS-MGS during CATIE phase 1: (1) age (P < 0.001; Mann–Whitney test), (2) gender (P = 0.02; chi-square test), (3) concomitant treatment with anticholinergic drugs during phase 1 (P < 0.001; chi-square test), and (4) SAS score at base line (P < 0.001; Mann–Whitney test). These four covariates were included in the regression model. Days of participation, drug type, and mean drug doses in chlorpromazine units were not significantly associated with SAS-MGS and were not included in the regression model.
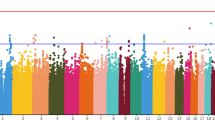
Results of single-marker association tests
The top results of our case-control GWAS for association with AIP severity included 15 SNPs that were significant at P < 1 × 10−4 (Table 2). The most significant association (rs12476047, P = 3.13 × 10−6) falls short of the GWAS significance threshold. This SNP (chromosome 2) is located more than 146 kilobases (kb) from the nearest known gene, fidgetin (FIGN). Of the top 15 SNPs, four are located within gene introns: Early B-cell Factor 1 (EBF1), Rap Guanine Nucleotide Exchange Factor (PARGEF5), Zinc Finger Protein, Multitype 2 (ZFPM2), and Bromodomain Adjacent to Zinc Finger Domain, 2B (BAZ2B). Another SNP is located 177 bases away from the Glutaredoxin Cysteine Rich 2 (GRXCR2) gene. None of these genes has been previously reported to be associated with AIP, schizophrenia, or PD. Two adjacent intergenic chromosome 14 SNPs that were associated with AIP severity (rs8006700, P = 2.31 × 10−5; rs1950420, P = 5.1 × 10−5) are located 95 kb and 74 kb (respectively) from the Neuro-Oncological Ventral Antigen 1 (NOVA1) gene.
In addition, we separately focused on four previously reported AIP candidate genes (DAT1, DRD2, HTR2C, and RGS2) and six candidate genes for idiopathic PD (Alpha-synuclein, LRRK2, Parkin, Pink1, DJ-1, and UCHL1) and analyzed their relation to AIP severity in our sample. These genes were selected based on literature review (see Introduction). Since most of reported associated variants were not genotyped in the current study platform, and in order to study the candidate genes association systematically, we analyzed all the SNPs genotyped within these genes. None of them was found significantly associated with AIP severity after correction for number of SNPs analyzed within each gene (Supplementary Table 1).
Discussion
We have performed a genome-wide, case-control, pharmacogenomic screen for genetic variants associated with AIP severity based on a secondary analysis of publicly available genotype and phenotype data from the CATIE study (Lieberman et al. 2005; Sullivan et al. 2008). Our analysis included 397 schizophrenia patients treated for at least 2 weeks with one antipsychotic drug and assessed regularly for AIP. Our dichotomized phenotype was defined on the basis of the average of SAS mean global score measurements during CATIE phase 1 (not including baseline measurement), using an extreme distribution of phenotype-analysis approach. Use of the average score of multiple clinical measurements of SAS-MGS during the phase 1 time period rather than a single measurement (e.g., the highest score) to determine individual AIP score is in keeping with the prospective nature of the CATIE study in which patients were followed for up to 18 months. Average scores are less prone to bias due to occasional outlying scores that may result from interindividual differences in AIP evaluation, exceptional increases in drug doses, and changes in patient adherence to treatment during follow-up. Moreover, since AIP development is dose dependent and all patients are expected to eventually develop AIP if high-enough doses are prescribed (Hirose 2006), we believe that relying on average SAS-MGS measurements taken over several months of follow-up is an appropriate strategy. Second, we used the “best responders” (who did not develop any sign of AIP during the follow-up despite chronic treatment with antipsychotics) as controls while patients with the highest SAS-MGS scores (0.3 as a cutoff) were defined as cases. Focusing on the extremes of a sample distribution is regarded as one of the most advantageous strategies in conducting pharmacogenomic GWASs (Crowley et al. 2009). To ensure that differences in individual SAS at baseline would not affect AIP scores during the study (the majority of patients were treated with antipsychotics before entering the study), we controlled for this covariate in the logistic regression model.
Methodological limitations of this GWAS for AIP severity include the fact that five different antipsychotic drugs were prescribed, each with a different propensity to induce AIP (one FGA and four SGA). In addition, the doses were not uniform but adjusted individually. Thus, one may argue that AIP severity differences could stem from difference in drug allocation and/or higher doses between the case and control groups rather than genetic predisposition. However, in agreement with the findings of the original CATIE report (Lieberman et al. 2005), we did not observe association of drug type or average dose (standardized to chlorpromazine unit) during phase 1 with AIP severity (see Table 1). On the other hand, there was a statistically significant difference between cases and controls in concomitant use of anticholinergic medication (see Table 1). To overcome this possible confounder, the concomitant use of anticholinergic agents during phase 1 was included as a dichotomous covariate in our logistic regression model. A further point to be noted is that there are more males than females in the AIP group; this is contradictory to textbook knowledge that females are more susceptible to parkinsonism induced by antipsychotic drugs. However, in the overall CATIE sample, 74% of the participants available for genotyping were men; therefore, the core sample was not representative in terms of gender distribution.
In this study, AIP was assessed using the modified SAS. The original SAS is a ten-item scale commonly used to assess AIP in both research and clinical contexts (Janno et al. 2005). However, this scale has been criticized for over-emphasizing rigidity items as well as for differences in sensitivity between SAS and DSM-IV case definitions of neuroleptic-induced parkinsonism (Janno et al. 2004). In the CATIE study, a modified version of SAS was used (including six items). Although the number of items in our SAS version is six instead of ten (as in the original version), we use the widely accepted SAS mean global score of 0.3 as a cutoff point for the existence of parkinsonism (Simpson and Angus 1970) since this score reflects a mean and not a total score. In addition, and in accordance with our “extremes of distribution” approach, the threshold of 0.3 and above approximately represents the upper third of the CATIE phase 1 average SAS-MGS while 0 approximately represents the lower third of the sample.
As reviewed by Crowley et al. (2009), the number of pharmacogenomic GWASs reported in the literature studying association with drug-induced phenotypes is gradually increasing. These studies have a relatively small sample size compared with disease-oriented GWASs (which include thousands of participants). In spite of this limitation, some important and impressive pharmacogenomic findings have been reported mainly concerning relatively rare phenotypes (e.g., Link et al. 2008; Kindmark et al. 2008). With relatively small sample sizes, it is easier to find susceptibility variants for rare side effects (resembling monogenetic heritability) than variants associated with common drug-induced phenotypes (Crowley et al. 2009). AIP is a common adverse effect, and it is reasonable to assume that each AIP-associated variant has only a small effect on the emergence of the phenotype. We assume that the relatively small sample size we used to study this common adverse effect is the main reason for our failure to find AIP-associated SNPs that reach genome-wide significance.
Nevertheless, some interesting candidate genes for further research and replication trials were found. One attractive AIP candidate gene is EBF1 (rs891903, intron 6, P = 4.06 × 10−5) which encodes a transcription factor that controls neurogenesis in the CNS (Garcia-Dominguez et al. 2003) and is implicated in the development of nigrostriatal neurons (Lobo et al. 2008). A recent study by Yin et al. (2009) demonstrated that EBF1 plays a regulatory role in the development of dopaminergic neurons and is critical for the migration of mesodiencephalic dopaminergic neurons to the substantia nigra (Yin et al. 2009). The RAPGEF5 gene (rs7804311, intron 9, P = 5.64 × 10−5), also known as MR-GEF, encodes a guanine nucleotide-exchange factor which takes part in signaling pathways related to telencephalic neurogenesis (Bithell et al. 2003).
Most of our top SNPs are intergenic rather than located within annotated genes. Their distance to the nearest gene ranges from 177 base pairs (bp) to more than a million kb (see Table 2). For example, our top AIP severity-associated SNP (rs12476047) is located 146 kb away from the FIGN gene, which encodes the fidgetin protein, a member of the AAA family of ATPase that functions as a chaperone (Yang et al. 2006). This gene is involved in developmental processes in several body organs (Cox et al. 2000). Intergenic variants may play an important role in regulation of nearby gene expression, as enhancers, repressors, or transcription-factor binding sites. Moreover, recent studies have demonstrated the importance and prevalence of intergenic transcription, extensive transcription of non protein coding DNA regions outside annotated genes that may have regulatory role (Martens et al. 2005; Khaitovich et al. 2006). Biologically speaking, a plausible candidate gene for AIP severity in our study is NOVA1; two intergenic AIP severity-associated SNPs, rs8006700 and rs1950420, are respectively located 95 and 74 kb away from the gene (Table 2). NOVA1 encodes a neuronal-specific RNA-binding protein, which serves as an antigen recognized by the antisera of patients with the rare paraneoplastic opsoclonus–myoclonus ataxia (POMA; Buckanovich et al. 1996; Ruggiu et al. 2009). POMA affects motor neurons in the brain stem, cerebellum, and spinal cord, and is associated with several types of cancer (Buckanovich et al. 1996).
The underlying pathophysiology of AIP is unclear but is probably mediated by decreased dopaminergic transmission along the nigrostriatal pathway (Blanchet 2003). It is well established that dopamine D2 receptor occupancy by antipsychotics in the nigrostriatal pathway is related to parkinsonism, and all the clinically effective antipsychotics drugs block this receptor (Casey 2004; Miyamoto et al. 2005). Occupancy of more than 80% of D2 receptors by typical antipsychotics substantially increases the risk of AIP (Hirose 2006) while atypical antipsychotic D2 receptor occupancy is usually lower and depends on the specific drug (Miyamoto et al. 2005). Other hypotheses of AIP mechanisms focus on differences in the dissociation rate of typical versus atypical drugs from D2 receptors and/or the contribution of serotonin receptors blockade (Casey 2004). Thus, genetic variants may influence susceptibility to AIP by more than one biological mechanism.
Our findings may also be relevant to the general context of schizophrenia susceptibility genes not limited to AIP. Studies in the preantipsychotic medication era as well as modern studies on antipsychotic-naïve patients clearly point to the presence of dyskinesia and parkinsonism among schizophrenia patients (Koning et al. 2008; Whitty et al. 2009). For example, Caligiuri and colleagues found that 29% of antipsychotic-naïve patients suffered from rigidity and 37% from tremor (Caligiuri et al. 1993). Parkinsonism was documented in 15% of 142 chronically ill, antipsychotic-naïve Indian patients (McCreadie et al. 1996). Additional studies have supported these findings (Whitty et al. 2009; Chakos et al. 1992). Thus, it is possible that abnormal movements may be an intrinsic endophenotype of schizophrenia and not only a side effect of antipsychotic medication (Koning et al. 2008; Bombin et al. 2005). Koning et al. (2008) suggested that parkinsonism may represent an intrinsic part of the disease process, involving impairment in the nigrostriatal pathway. If parkinsonism and dyskinesia are indeed a trait feature of schizophrenia, and related to the genetic background of schizophrenia, then it is possible that schizophrenia and movement disorders share common risk or protective genetic variants. In a meta-analysis, parkinsonism and dyskinesia were found to be significantly more prevalent in healthy first-degree relatives of schizophrenia patients as compared with healthy controls (Koning et al. 2008). Although it is not clear whether EPS are associated with poorer or better pharmacological treatment outcomes (Chakos et al. 1992; Chatterjee et al. 1995), it is possible that susceptibility genes for AIP are risk factors for a heritable schizophrenia endophenotype reflecting a dopaminergic disturbance in the basal ganglia (Caligiuri and Lohr 1997). A possible future research direction is to study our top AIP candidate genes in the context of the genetics of schizophrenia or idiopathic PD.
The implications of EPS (and AIP in particular) for the quality of daily life of antipsychotic-treated patients may be substantial, and patients may suffer from social stigma and distress (Haddad and Dursun 2008). AIP can result in weakness, muscle aching, impaired ability to perform occupational and social tasks due to impaired dexterity, and, in severe cases, lead to falls and injury (Haddad and Dursun 2008). Thus, a priori prediction of AIP susceptibility is an important clinical need for better management of vulnerable patients, maintaining low drug doses, early treatment with anticholinergic agents, and preference for SGA. In addition, indentifying susceptibility or protective genetic variants associated with AIP may contribute to our basic understanding of underlying pathophysiology.
References
Al Hadithy AF, Wilffert B, Stewart RE, Looman NM, Bruggeman R, Brouwers JR, Matroos GE, van Os J, Hoek HW, van Harten PN (2008) Pharmacogenetics of parkinsonism, rigidity, rest tremor, and bradykinesia in African–Caribbean inpatients: differences in association with dopamine and serotonin receptors. Am J Med Genet B Neuropsychiatr Genet 147B(6):890–897
Arranz MJ, de Leon J (2007) Pharmacogenetics and pharmacogenomics of schizophrenia: a review of last decade of research. Mol Psychiatry 12(8):707–747
Ayd F (1961) A survey of drug induced extrapyramidal reaction. JAMA 175:1054–1060
Basile VS, Masellis M, Potkin SG, Kennedy JL (2002) Pharmacogenomics in schizophrenia: the quest for individualized therapy. Hum Mol Genet 11(20):2517–2530
Bithell A, Alberta J, Hornby F, Stiles CD, Williams BP (2003) Expression of the guanine nucleotide exchange factor, mr-gef, is regulated during the differentiation of specific subsets of telencephalic neurons. Brain Res Dev Brain Res 146(1–2):107–118
Blanchet PJ (2003) Antipsychotic drug-induced movement disorders. Can J Neurol Sci Suppl 1:S101–S107
Bombin I, Arango C, Buchanan RW (2005) Significance and meaning of neurological signs in schizophrenia: two decades later. Schizophr Bull 31(4):962–977
Buckanovich RJ, Yang YY, Darnell RB (1996) The onconeural antigen Nova-1 is a neuron-specific RNA-binding protein, the activity of which is inhibited by paraneoplastic antibodies. J Neurosci 16(3):1114–1122
Caligiuri MP, Lohr JB (1997) Instrumental motor predictors of neuroleptic-induced parkinsonism in newly medicated schizophrenia patients. J Neuropsychiatry Clin Neurosci 9(4):562–567
Caligiuri MP, Peavy G (2000) An instrumental study of the relationship between extrapyramidal signs and psychosis in Alzheimer's disease. J Neuropsychiatry Clin Neurosci 12(1):34–39
Caligiuri MP, Lohr JB, Jeste DV (1993) Parkinsonism in neuroleptic–naive schizophrenic patients. Am J Psychiatry 150(9):1343–1348
Casey DE (2004) Pathophysiology of antipsychotics drug-induced movement disorders. J Clin Psychiatry 65(supp):25–28
Chakos MH, Mayerhoff DI, Loebel AD, Alvir JM, Lieberman JA (1992) Incidence and correlates of acute extrapyramidal symptoms in first episode of schizophrenia. Psychopharmacol Bull 28(1):81–86
Chatterjee A, Chakos M, Koreen A, Geisler S, Sheitman B, Woerner M, Kane JM, Alvir J, Lieberman JA (1995) Prevalence and clinical correlates of extrapyramidal signs and spontaneous dyskinesia in never-medicated schizophrenic patients. Am J Psychiatry 152(12):1724–1729
Cichon S, Craddock N, Daly M, Faraone SV, Gejman PV, Kelsoe J, Lehner T, Levinson DF, Moran A, Sklar P, Sullivan PF, Psychiatric GWAS Consortium Coordinating Committee (2009)Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry 166(5):540–556
Cox GA, Mahaffey CL, Nystuen A, Letts VA, Frankel WN (2000) The mouse fidgetin gene defines a new role for AAA family proteins in mammalian development. Nat Genet 26(2):198–202
Crowley JJ, Sullivan PF, McLeod HL (2009) Pharmacogenomic genome-wide association studies: lessons learned thus far. Pharmacogenomics 10(2):161–163
Dolzan V, Plesnicar BK, Serretti A, Mandelli L, Zalar B, Koprivsek J, Breskvar K (2007) Polymorphisms in dopamine receptor DRD1 and DRD2 genes and psychopathological and extrapyramidal symptoms in patients on long-term antipsychotic treatment. Am J Med Genet B Neuropsychiatr Genet 144B(6):809–815
Ebadi M, Srinivasan SK (1995) Pathogenesis, prevention, and treatment of neuroleptic-induced movement disorders. Pharmacol Rev 47(4):575–604
Freimer N, Sabatti C (2004) The use of pedigree, sib-pair and association studies of common diseases for genetic mapping and epidemiology. Nat Genet 36(10):1045–1051
Freyhan FA (1959) Therapeutic implications of differential effects of new phenothiazine compounds. Am J Psychiatry 115:577–585
Garcia-Dominguez M, Poquet C, Garel S, Charnay P (2003) Ebf gene function is required for coupling neuronal differentiation and cell cycle exit. Development 130(24):6013–6025
Geddes J, Freemantle N, Harrison P, Bebbington P (2000) Atypical antipsychotics in the treatment of schizophrenia: systematic overview and meta-regression analysis. BMJ 321(7273):1371–1376
Greenbaum L, Strous RD, Kanyas K, Merbl Y, Horowitz A, Karni O, Katz E, Kotler M, Olender T, Deshpande SN, Lancet D, Ben-Asher E, Lerer B (2007) Association of the RGS2 gene with extrapyramidal symptoms induced by treatment with antipsychotic medication. Pharmacogenet Genomics 17(7):519–528
Greenbaum L, Smith RC, Rigbi A, Strous R, Teltsh O, Kanyas K, Korner M, Lancet D, Ben-Asher E, Lerer B (2009) Further evidence for association of the RGS2 gene with antipsychotic-induced parkinsonism: protective role of a functional polymorphism in the 3′-untranslated region. Pharmacogenomics J 9(2):103–110
Gunes A, Dahl ML, Spina E, Scordo MG (2008) Further evidence for the association between 5-HT2C receptor gene polymorphisms and extrapyramidal side effects in male schizophrenic patients. Eur J Clin Pharmacol 64(5):477–482
Güzey C, Scordo MG, Spina E, Landsem VM, Spigset O (2007) Antipsychotic-induced extrapyramidal symptoms in patients with schizophrenia: associations with dopamine and serotonin receptor and transporter polymorphisms. Eur J Clin Pharmacol 63(3):233–241
Haddad PM, Dursun SM (2008) Neurological complications of psychiatric drugs: clinical features and management. Hum Psychopharmacol 23(Suppl 1):15–26
Hansen TE, Casey DE, Hoffman WF (1997) Neuroleptic intolerance. Schizophr Bull 23(4):567–582
Hirose G (2006) Drug induced parkinsonism: a review. J Neurol 253(Suppl 3):iii22–iii24
Janno S, Holi M, Tuisku K, Wahlbeck K (2004) Prevalence of neuroleptic-induced movement disorders in chronic schizophrenia inpatients. Am J Psychiatry 161(1):160–163
Janno S, Holi MM, Tuisku K, Wahlbeck K (2005) Validity of Simpson–Angus Scale (SAS) in a naturalistic schizophrenia population. BMC Neurol 5(1):5
Kaiser R, Tremblay PB, Klufmöller F, Roots I, Brockmöller J (2002) Relationship between adverse effects of antipsychotic treatment and dopamine D(2) receptor polymorphisms in patients with schizophrenia. Mol Psychiatry 7(7):695–705
Khaitovich P, Kelso J, Franz H, Visagie J, Giger T, Joerchel S, Petzold E, Green RE, Lachmann M, Pääbo S (2006) Functionality of intergenic transcription: an evolutionary comparison. PLoS Genet 2(10):e171
Kindmark A, Jawaid A, Harbron CG, Barratt BJ, Bengtsson OF, Andersson TB, Carlsson S, Cederbrant KE, Gibson NJ, Armstrong M, Lagerström-Fermér ME, Dellsén A, Brown EM, Thornton M, Dukes C, Jenkins SC, Firth MA, Harrod GO, Pinel TH, Billing-Clason SM, Cardon LR, March RE (2008) Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenomics J 8(3):186–195
Koning JP, Tenback DE, van Os J, Aleman A, Kahn RS, van Harten PN. (2008) Dyskinesia and parkinsonism in antipsychotic-naive patients with schizophrenia, first-degree relatives and healthy controls: a meta-analysis. Schizophr Bull. [Epub ahead of print]
Lencz T, Morgan TV, Athanasiou M, Dain B, Reed CR, Kane JM, Kucherlapati R, Malhotra AK (2007) Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry 12(6):572–580
Lesage S, Brice A (2009) Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 18(R1):R48–R59
Lerner V, Libov I, Kaptsan A, Miodownik C, Dwolatzky T, Levine J (2007) The prevalence of neuroleptic drug-induced tardive movement subsyndromes among schizophrenic and schizoaffective patients residing in the southern region of Israel. Isr J Psychiatry Relat Sci 44(1):20–28
Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK (2005) Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 353(12):1209–1223
Lindenmayer JP, Eerdekens E, Berry SA, Eerdekens M (2004) Safety and efficacy of long-acting risperidone in schizophrenia: a 12-week, multicenter, open-label study in stable patients switched from typical and atypical oral antipsychotics. J Clin Psychiatry 65(8):1084–1089
Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, Gut I, Lathrop M, Collins R (2008) SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 359(8):789–799
Liu YZ, Pei YF, Guo YF, Wang L, Liu XG, Yan H, Xiong DH, Zhang YP, Levy S, Li J, Haddock CK, Papasian CJ, Xu Q, Ma JZ, Payne TJ, Recker RR, Li MD, Deng HW (2009) Genome-wide association analyses suggested a novel mechanism for smoking behavior regulated by IL15. Mol Psychiatry. 14:668–680
Lobo MK, Yeh C, Yang XW (2008) Pivotal role of early B-cell factor 1 in development of striatonigral medium spiny neurons in the matrix compartment. J Neurosci Res 86(10):2134–2146
Martens JA, Wu PY, Winston F (2005) Regulation of an intergenic transcript controls adjacent gene transcription in Saccharomyces cerevisiae. Genes Dev 19(22):2695–2704
McCreadie RG, Thara R, Kamath S, Padmavathy R, Latha S, Mathrubootham N, Menon MS (1996) Abnormal movements in never-medicated Indian patients with schizophrenia. Br J Psychiatry 168(2):221–226
Medinar C, Kramer MD, Kurland AA (1962) Biperiden in the treatment of phenothiazine-induced extrapyramidal reactions. JAMA 182:1127–1128
Miyamoto S, Duncan GE, Marx CE, Lieberman JA (2005) Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry 10(1):79–104
Modestin J, Wehrli MV, Stephan PL, Agarwalla P (2008) Evolution of neuroleptic-induced extrapyramidal syndromes under long-term neuroleptic treatment. Schizophr Res 100(1–3):97–107
Nakazono Y, Abe H, Murakami H, Koyabu N, Isaka Y, Nemoto Y, Murata S, Tsutsumi Y, Ohtani H, Sawada Y (2005) Association between neuroleptic drug-induced extrapyramidal symptoms and dopamine D2-receptor polymorphisms in Japanese schizophrenic patients. Int J Clin Pharmacol Ther 43(4):163–171
Park S, Ross-Degnan D, Adams AS, Sabin J, Kanavos P, Soumerai SB (2005) Effect of switching antipsychotics on antiparkinsonian medication use in schizophrenia: population-based study. Br J Psychiatry 187:137–142
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38(8):904–909
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575
Reich D, Price AL, Patterson N (2008) Principal component analysis of genetic data. Nat Genet 40(5):646–649
Rochon PA, Stukel TA, Sykora K, Gill S, Garfinkel S, Anderson GM, Normand SL, Mamdani M, Lee PE, Li P, Bronskill SE, Marras C, Gurwitz JH (2005) Atypical antipsychotics and parkinsonism. Arch Intern Med 165(16):1882–1888
Ruggiu M, Herbst R, Kim N, Jevsek M, Fak JJ, Mann MA, Fischbach G, Burden SJ, Darnell RB (2009) Rescuing Z+agrin splicing in Nova null mice restores synapse formation and unmasks a physiologic defect in motor neuron firing. Proc Natl Acad Sci USA 106(9):3513–3518
Simpson G, Angus MP (1970) Scale for assessment extrapyramidal side effects. Acta Psychiatr Scand 212:11–19
Stephen PJ, Williamson J (1984) Drug-induced parkinsonism in the elderly. Lancet 2(8411):1082–1083
Stroup TS, McEvoy JP, Swartz MS, Byerly MJ, Glick ID, Canive JM, McGee MF, Simpson GM, Stevens MC, Lieberman JA (2003) The National Institute of Mental Health Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) project: schizophrenia trial design and protocol development. Schizophr Bull 29(1):15–31
Sullivan PF, Lin D, Tzeng JY, van den Oord E, Perkins D, Stroup TS, Wagner M, Lee S, Wright FA, Zou F, Liu W, Downing AM, Lieberman J, Close SL (2008) Genomewide association for schizophrenia in the CATIE study: results of stage 1. Mol Psychiatry 13(6):570–584
Tenback DE, van Harten PN, Slooff CJ, van Os J (2006) Evidence that early extrapyramidal symptoms predict later tardive dyskinesia: a prospective analysis of 10,000 patients in the European Schizophrenia Outpatient Health Outcomes (SOHO) study. Am J Psychiatry 163(8):1438–1440
Whitty PF, Owoeye O, Waddington JL (2009) Neurological signs and involuntary movements in schizophrenia: intrinsic to and informative on systems pathobiology. Schizophr Bull 35(2):415–424
Yang Y, Mahaffey CL, Bérubé N, Frankel WN (2006) Interaction between fidgetin and protein kinase A-anchoring protein AKAP95 is critical for palatogenesis in the mouse. J Biol Chem 281(31):22352–22359
Yin M, Liu S, Yin Y, Li S, Li Z, Wu X, Zhang B, Ang SL, Ding Y, Zhou J (2009) Ventral mesencephalon-enriched genes that regulate the development of dopaminergic neurons in vivo. J Neurosci 29(16):5170–5182
Acknowledgement
The principal investigators of the CATIE (Clinical Antipsychotic Trials of Intervention Effectiveness) trial were Jeffrey A. Lieberman, M.D., T. Scott Stroup, M.D., M.P.H., and Joseph P. McEvoy, M.D. The CATIE trial was funded by a grant from the National Institute of Mental Health (N01 MH900001) along with MH074027 (PI PF Sullivan). Genotyping was funded by Eli Lilly and Company.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ana Alkelai and Lior Greenbaum contributed equally to this work.
Electronic supplementary materials
Below is the link to the electronic supplementary material.
Supplementary Table 1
(DOC 56 kb)
Rights and permissions
About this article
Cite this article
Alkelai, A., Greenbaum, L., Rigbi, A. et al. Genome-wide association study of antipsychotic-induced parkinsonism severity among schizophrenia patients. Psychopharmacology 206, 491–499 (2009). https://doi.org/10.1007/s00213-009-1627-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-009-1627-z