
Abstract
Rationale
Mesolimbic dopamine (DA) is a critical component of the brain circuitry regulating behavioral activation and effort-related processes. Research involving choice tasks has shown that rats with impaired DA transmission reallocate their instrumental behavior away from food-reinforced tasks with high response requirements and instead select less effortful food-seeking behaviors.
Objective
Previous work showed that adenosine A2A antagonism can reverse the effects of the DA antagonist haloperidol in an operant task that assesses effort-related choice. The present work used a T-maze choice procedure to assess the effects of adenosine A2A and A1 antagonism.
Materials and methods
With this task, the two arms of the maze have different reinforcement densities (four vs. two food pellets), and a vertical 44 cm barrier is positioned in the arm with the higher density, presenting the animal with an effort-related challenge. Untreated rats strongly prefer the arm with the high density of food reward and climb the barrier in order to obtain the food.
Results
Haloperidol produced a dose-related (0.05–0.15 mg/kg i.p.) reduction in the number of trials in which the rats chose the high-barrier arm. Co-administration of the adenosine A2A receptor antagonist MSX-3 (0.75, 1.5, and 3.0 mg/kg i.p.), but not the A1 antagonist 8-cyclopentyl-1,3-dipropylxanthine (0.75, 1.5, and 3.0 mg/kg i.p.), reversed the effects of haloperidol on effort-related choice and latency.
Conclusions
Adenosine A2A and D2 receptors interact to regulate effort-related decision making, which may have implications for the treatment of psychiatric symptoms such as psychomotor slowing or anergia that can be observed in depression, parkinsonism, and other disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Motivational stimuli often have activating effects, and goal-directed behavior frequently is characterized by a high degree of activity, vigor, or persistence in work output (Salamone and Correa 2002). These activational aspects of motivation are adaptive because they enable organisms to overcome work-related response costs that separate them from significant stimuli (Salamone et al. 1997, 2007; Salamone and Correa 2002; Van den Bos et al. 2006). Conversely, lack of behavioral activation can be maladaptive; in humans, symptoms such as psychomotor slowing, anergia, and fatigue are fundamental aspects of depression, as well as other psychiatric and neurological disorders (Demyttenaere et al. 2005; Salamone et al. 2006, 2007; Yurgelun-Todd et al. 2007; Capuron et al. 2007; Majer et al. 2008). Brain dopamine (DA), particularly in the nucleus accumbens, appears to be one of the critical components of the brain circuitry controlling effort-related behavioral processes and behavioral activation (Salamone et al. 1997, 2005, 2007; Barbano and Cador 2007; Phillips et al. 2007; Robbins and Everitt 2007). Nucleus accumbens DA depletions make rats very sensitive to ratio requirements in operant lever-pressing schedules (Sokolowski and Salamone 1998; Correa et al. 2002; Mingote et al. 2005). Moreover, DA-depleted rats show alterations in response allocation in tasks that measure effort-related choice behavior (Salamone et al. 2007). Some studies in this area have used a concurrent fixed ratio 5 (FR5)/chow feeding procedure to study effort-related choice (Salamone et al. 1991, 2002, 2007). With this task, rats have the option of responding on a FR5 lever-pressing schedule for a highly preferred food (high carbohydrate food pellets) or approaching and consuming a less preferred food (standard rodent chow) that is freely available in the chamber. Low-to-moderate doses of DA antagonists that act on either D1 or D2 family receptors suppress lever pressing for food but actually increase chow intake (Salamone et al. 1991, 2002; Cousins et al. 1994; Koch et al. 2000; Sink et al. 2008). Nucleus accumbens is the DA terminal region most closely associated with these effects of impaired DA transmission (Cousins et al. 1993; Sokolowski and Salamone 1998; Koch et al. 2000; Nowend et al. 2001). Furthermore, the effects of interference with DA transmission were shown to differ substantially from those produced by pre-feeding to reduce food motivation (Salamone et al. 1991) and by appetite-suppressant drugs with various neurochemical profiles, including amphetamine (Cousins et al. 1994), fenfluramine (Salamone et al. 2002), and cannabinoid CB1 antagonists (Sink et al. 2008).
Additional studies in this area have employed a T-maze task to assess effort-related choice behavior (Salamone et al. 1994; Cousins et al. 1996; Walton et al. 2003; Denk et al. 2005; Schweimer et al. 2005; Floresco and Ghods-Sharifi 2007). In this task, two arms of the maze can have different reinforcement densities (e.g., four vs. two food pellets, or four vs. zero), and under some conditions, a vertical barrier can be placed in the arm with the higher reward density to vary task difficulty. When there is no barrier present, untreated rats preferred the high reward density arm, and neither haloperidol nor accumbens DA depletion altered arm preference when no barrier was in the maze (Salamone et al. 1994). Under conditions in which the arm with four food pellets was partially blocked with the barrier, but the other arm contained no pellets (i.e., the only way to get food was to climb the barrier), rats with accumbens DA depletions were relatively slow but still chose the high-density arm, climbed the barrier, and consumed the food (Cousins et al. 1996). Nevertheless, DA manipulations dramatically altered choice behavior when the high-density arm (four pellets) had the barrier in place, and the arm without the barrier had an alternative source of food (two pellets). Under these conditions, rats treated with DA antagonists or accumbens DA depletions showed decreased choice of the high-density arm and increased choice of the low-density arm (Cousins et al. 1996; Salamone et al. 1994; Denk et al. 2005).
Other brain areas and transmitters in addition to nucleus accumbens DA also are involved in effort-related processes (e.g., prefrontal cortex, amygdala, and ventral pallidal γ-aminobutyric acid; see Walton et al. 2006; Denk et al. 2005; Schweimer et al. 2005; Floresco and Ghods-Sharifi 2007; Salamone et al. 2007; Farrar et al. 2008), and recent studies have focused upon the purine nucleoside adenosine (Farrar et al. 2007; Font et al. 2008; Mingote et al. 2008). Adenosine A2A receptors are heavily concentrated in striatal areas, including the caudate/putamen and nucleus accumbens (Schiffmann et al. 1991; DeMet and Chicz-DeMet, 2002; Ferré et al. 2004). There is considerable evidence of a functional interaction between striatal and accumbens DA D2 receptors and adenosine A2A receptors (Fink et al. 1992; Ferré 1997; Hillion et al. 2002; Fuxe et al. 2003). This interaction often has been investigated in relation to neostriatal motor functions involved in parkinsonism (Ferré et al. 1997, 2001, 2008; Hauber and Munkel 1997; Svenningsson et al. 1999; Hauber et al. 2001; Wardas et al. 2001; Morelli and Pinna 2002; Correa et al. 2004; Pinna et al. 2005; Ishiwari et al. 2007; Salamone et al. 2008a,b). However, researchers also have identified functions of adenosine A2A receptor transmission related to cognition (Takahashi et al. 2008) and aspects of motivation (O’Neill and Brown 2006; Farrar et al. 2007; Font et al. 2008; Mingote et al. 2008). Farrar et al. (2007) demonstrated that systemic injections of the adenosine A2A antagonist MSX-3 reversed the haloperidol-induced shift in choice behavior seen in rats responding on the operant concurrent choice task.
The present work was undertaken to examine the role of DA/adenosine A2A receptor interactions in effort-related choice behavior by assessing the ability of an adenosine A2A receptor antagonist to reverse the behavioral impairment in T-maze performance induced by the DA antagonist haloperidol. Unlike the concurrent choice task, which studies response allocation between free operant lever pressing and chow intake (e.g., Farrar et al. 2007), the T-maze task allows for assessment of discrete choices on a trial-by-trial basis. For these studies, one arm of the maze contained a high density of food reinforcement (four pellets), and the other arm contained a low density of food reinforcement (two pellets). Response costs were different in the two arms due to the presence of a large vertical barrier located in the high-density arm of the maze. Experiment 1 studied the effects of different doses of haloperidol (Veh, 0.05, 0.10, and 0.15 mg/kg i.p.) on T-maze performance in order to identify the dose of haloperidol to be used for the subsequent reversal studies. Experiment 2A assessed the ability of the adenosine A2A antagonist MSX-3 (0.75–3.0 mg/kg i.p.) to reverse the effects of 0.15 mg/kg haloperidol on T-maze performance. In order to compare the effects of antagonists that act on different adenosine receptors, experiment 3A studied the ability of the adenosine A1 antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX; 0.75–3.0 mg/kg i.p.) to reverse the effects of 0.15 mg/kg haloperidol. Additional control studies (2B and 3B) assessed the effects of injections of the high doses of MSX-3 and DPCPX on T-maze performance in the absence of haloperidol.
Materials and methods
Subjects
Adult male, drug-naive, Sprague–Dawley rats (Harlan Sprague–Dawley, Indianapolis, IN, USA) were housed in a colony maintained at 23°C at with 12-h light/dark cycles (lights on at 0700 hours). The rats (N = 20) weighed 290–340 g at the beginning of the study and were food-deprived to 85% of their free-feeding body weight for the experiment. Rats were fed supplemental chow to maintain the 85% free-feeding body weight throughout the study with water available ad libitum in the home cages. Animal protocols were approved by the University of Connecticut Institutional Animal Care and Use Committee and followed NIH guidelines.
Pharmacological agents and selection of doses
Haloperidol (Sigma Chemical, St. Louis, MO, USA) was dissolved in a 0.3% tartaric acid solution (pH = 4.0); this 0.3% tartaric acid solution also was used as the vehicle control for the haloperidol injections. The adenosine A2A antagonist used was MSX-3 ((E)-phosphoric acid mono-[3-[8-[2-(3-methoxyphenyl)vinyl]-7-methyl-2,6-dioxo-1-prop-2-ynyl-1,2,6,7-tetrahydropurin-3-yl] propyl] ester disodium salt). MSX-3 was synthesized at the laboratory of Dr. Christa Müller at the Pharmazeutisches Institut, Universität Bonn, in Bonn, Germany. For the preparation of the drug solution, MSX-3 (free acid) was dissolved in 0.9% saline, and pH was adjusted by titrating with microliter quantities of 1.0 N NaOH until the solid drug was in solution. The final pH was usually 7.5 ± 0.2 and was not allowed to exceed 7.8. MSX-3 is a pro-drug that is cleaved in vivo into the pharmacologically active adenosine antagonist MSX-2 (Hockemeyer et al. 2004). MSX-2 has a 100-fold binding selectivity for A2A vs. A1 receptors (Solinas et al. 2005). DPCPX was obtained from Tocris and was dissolved in a 20% ethanol vehicle; this compound is approximately 1,000-fold selective for A1 receptors relative to A2A receptors (Fredholm and Lindström 1999).
Doses of haloperidol used for the dose–response study (experiment 1) were based upon previously performed research (Salamone et al. 1994) and on pilot studies. The results of experiment 1 were used to select the dose of haloperidol employed in the reversal studies (i.e., 0.15 mg/kg i.p.). Doses of MSX-3 for the reversal study were based upon previous research (Farrar et al. 2007) and unpublished pilot data. The dose range for DPCPX that was used was based upon doses listed in published behavioral studies involving i.p. administration in rats (Prediger et al. 2005; Aubel et al. 2007; Maione et al. 2007; Lobato et al. 2008).
Apparatus and testing procedures
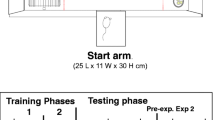
Food-deprived rats were trained in the T-maze apparatus. The start arm consisted of an enclosed Plexiglas box (29 × 21 × 21 cm) with a wire mesh floor grid. The test arm of each side of the maze was a box 99 × 32 × 59 cm. The test arm and back walls of the maze were made of Plexiglas, and the floor was wire mesh. The doorway from the start arm to the maze was a stainless steel guillotine door. The high-density arm provided four food pellets (45 mg each, Bioserve, Frenchtown, NJ, USA) and the low-density arm provided two food pellets. Pellets were located in small glass dishes placed against the far walls of the maze arms. Half the rats had the high-density arm with the barrier consistently located on the left side, while half the rats had the high-density arm and barrier on the right side. Rats were trained in several different phases. All rats received 1 week of initial training, which allowed them free access to both arms of the T-maze upon exiting the start arm. During initial training, no barrier was present, and rats were allowed to consume all pellets in both high- and low-density arms of the maze before being returned to the start arm. Upon completion of this initial training, rats were then trained to select between the high- and low-density arms, with no barrier in place. For these and all subsequent procedures, the rat was removed after entering one arm of the T-maze and consuming the pellets in that arm, and 30 trials were conducted each day. After this phase, rats were then trained with a barrier placed in the high-density arm, halfway between the start box and the food; rats were initially trained for 1 week with a small plastic barrier (11.3 cm) and then for several weeks with a medium-sized (23.4 cm) wire mesh barrier. Upon successful completion of the medium barrier training (i.e., >90% choice of the barrier arm out of 30 trials), a final wire mesh barrier (44 cm) was introduced halfway between the start arm door and the food dish. Rats were trained on the high-barrier choice procedure until they selected the high-density arm greater than 90% of trials per session. After successful completion of high-barrier training, drug testing commenced. For all drug studies, including the baseline days between drug treatments, the high barrier was present in the high-density arm, and no barrier was present in the low-density arm.
Experimental design
All experiments used a within group design, with all rats receiving their i.p. drug treatments in the study in a randomly varied order (one treatment per week; no treatment sequence repeated across different animals in the experiment). Baseline training sessions with no drug treatments were conducted four additional days per week.
Experiment 1: Effect of haloperidol on T-maze performance
Rats were trained before drug testing as described above. Rats (n = 5) received i.p. injections of tartaric acid vehicle, 0.05 mg/kg haloperidol, 0.1 mg/kg haloperidol, and 0.15 mg/kg haloperidol (all injections 50 min before testing). All rats were tested for 30 trials. The observer recorded the number of high- and low-density choices, as well as the response latency (start door opening to food dish area).
Experiment 2: Ability of MSX-3 to reverse the effects of haloperidol
Trained rats (n = 9) received the flowing treatments in experiment 2A: tartaric acid vehicle (50 min before testing) plus saline vehicle i.p. (20 min before testing), 0.15 mg/kg haloperidol i.p. (50 min before testing) plus saline vehicle i.p. (20 min before testing), 0.15 mg/kg haloperidol i.p. (50 min before testing) plus 0.75 mg/kg MSX-3 i.p. (20 min before testing), 0.15 mg/kg haloperidol i.p. (50 min before testing) plus 1.5 mg/kg MSX-3 i.p. (20 min before testing), and 0.15 mg/kg haloperidol i.p. (50 min before testing) plus 3.0 mg/kg MSX-3 i.p. (20 min before testing) and were tested for 30 trials. The observer recorded the number of high- and low-density choices, as well as the response latency (start door opening to food dish area). For experiment 2B, a group of rats (n = 5; the same rats that had completed experiment 1) received injections of either saline or 3.0 mg/kg MSX-3 (i.p.; 20 min before testing).
Experiment 3: Ability of DPCPX to reverse the effects of haloperidol
An additional group of trained rats (n = 6) received the flowing treatments in experiment 3A: tartaric acid vehicle (50 min before testing) plus saline vehicle i.p. (20 min before testing), 0.15 mg/kg haloperidol i.p. (50 min before testing) plus saline vehicle i.p. (20 min before testing), 0.15 mg/kg haloperidol i.p. (50 min before testing) plus 0.75 mg/kg DPCPX i.p. (20 min before testing), 0.15 mg/kg haloperidol i.p. (50 min before testing) plus 1.5 mg/kg DPCPX i.p. (20 min before testing), and 0.15 mg/kg haloperidol i.p. (50 min before testing) plus 3.0 mg/kg DPCPX i.p. (20 min before testing), and all rats were tested for 30 trials. As with experiment 2, the observer recorded the number of high- and low-density choices and the response latency (start door opening to food dish area). For experiment 3B, another group of rats (n = 6; the same rats that had completed experiment 3) received injections of either saline or 3.0 mg/kg DPCPX (i.p.; 20 min before testing).
Statistical analyses
In these experiments, there were no differences between animals that had the high-density arm to the left as opposed to those trained on the right, so these data were combined for further analyses. The total number of high-density arm selections (i.e., barrier crossings) was analyzed with repeated measures analysis of variance (ANOVA); selections of the low-density arm were not statistically analyzed because no animals failed to make a choice, and thus they are simply the mirror image of the high-density arm data. In experiment 1, repeated measures ANOVA was conducted on four treatment levels. For experiments 2A and 3A, repeated measures ANOVA was conducted for each of the five treatment levels (tartaric acid vehicle plus saline vehicle, 0. 15 mg/kg haloperidol plus saline vehicle, and 0.15 mg/kg haloperidol plus each dose of MSX-3 or DPCPX). Paired comparisons were performed using non-orthogonal planned comparisons that employed the overall error term (Keppel 1991); for experiment 1, the three haloperidol conditions were compared with vehicle, and for experiments 2 and 3, the data for the haloperidol plus vehicle treatment condition were contrasted with the other four treatments. The t test was used for analyses of experiments 2B and 3B.
Results
Experiment 1: Effects of haloperidol
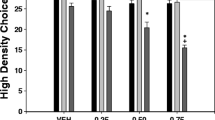
The data for selection of the high-density arm (i.e., crossing the barrier) for experiment 1 are shown in Fig. 1. Repeated measures ANOVA indicated that there was an overall significant effect of haloperidol treatment [F(3,12) = 31.46, p < 0.001]. Planned comparisons revealed that haloperidol produced a significant decrease in high-density arm selection compared to vehicle-treated control rats at both the 0.10 and 0.15 mg/kg dose (p < 0.01). Furthermore, haloperidol induced a significant increase in the selection of the low reinforcement density arm of the T-Maze (data not shown), and all haloperidol-treated rats consumed every pellet that was present in their chosen arm on each trial. There were no trials in which vehicle or haloperidol-treated rats failed to choose one of the two arms of the maze.

Effect of i.p. administration of the DA antagonist haloperidol on arm choice in the maze. Mean (±SEM) number of barrier arm choices after treatment with vehicle or various doses of haloperidol are shown (**p < 0.01, different from vehicle). Regression analysis revealed that there was a significant linear relation between dose and arm choice [F(1,18) = 92.7, p < 0.001; r 2 = 0.84]
Experiment 2: Reversal with MSX-3
The data on high-density arm selection (i.e., barrier crossings) for rats treated with MSX-3 and haloperidol are shown in Fig. 2a. Repeated measures ANOVA indicated that there was an overall significant effect of drug treatment on arm choice [F(4, 32) = 37.64, p < 0.001]. Haloperidol-vehicle treated rats showed a significant decrease in the selection of the number of barrier crossings as compared to vehicle–vehicle-treated rats (non-orthogonal planned comparisons; p < 0.001). Co-administration of MSX-3 with haloperidol produced a significant dose-related increase in selection of the high-density arm with the barrier relative to haloperidol plus vehicle-treated rats (planned comparisons; p < 0.01). Mean run latencies for haloperidol and MSX-3 co-administration are shown in Fig. 2b. Repeated measures ANOVA indicated an overall significant effect of drug treatment on run latency [F(4,32) = 4.59, p < 0.01]. Administration of haloperidol caused a significant increase in response latency as compared to vehicle–vehicle-treated rats (non-orthogonal planned comparisons; p < 0.001). Additionally, non-orthogonal planned comparisons indicated that a significant decrease in latency compared to haloperidol–vehicle-treated rats occurred upon co-administration of MSX-3 in a dose-dependent manner (0.75 mg/kg MSX-3, p < 0.001; 1.5 and 3.0 mg/kg MSX-3, p < 0.05). Experiment 2B assessed the effects of the high dose of MSX-3 on T-maze performance in the absence of haloperidol (Table 1). There was no significant effect on arm choice produced by MSX-3 (t = 1.0, df = 4, n.s.) and also no significant effect on latency (t = 2.4, df = 4, n.s.). However, four of the five animals did show slight decreases in latency after MSX-3 injection relative to saline injection.

a Effects of the adenosine A2A antagonist MSX-3 on T-maze arm choice in rats co-administered haloperidol. Mean (±SEM) number of barrier arm choices after treatment with vehicle or haloperidol plus various doses of MSX-3 are shown. Veh/Veh (vehicle plus vehicle), HAL/Veh (0.15 mg/kg haloperidol plus vehicle), HAL/0.75 M (0.15 mg/kg haloperidol plus 0.75 mg/kg MSX-3), HAL/1.5 M (0.15 mg/kg haloperidol plus 1.5 mg/kg MSX-3), HAL/3.0 M (0.15 mg/kg haloperidol plus 3.0 mg/kg MSX-3). # p < 0.01, different from vehicle/vehicle, planned comparison; **p < 0.01, different from vehicle plus haloperidol, planned comparison. b Effects of the adenosine A2A antagonist MSX-3 on run latency in rats co-administered haloperidol. Mean (+SEM) run latency (i.e., average across 30 trials, expressed in seconds) after treatment with vehicle or haloperidol plus various doses of MSX-3 are shown. Veh/Veh (vehicle plus vehicle), HAL/Veh (0.1 mg/kg haloperidol plus vehicle), HAL/0.75 M (0.1 mg/kg haloperidol plus 0.75 mg/kg MSX-3), HAL/1.5 M (0.1 mg/kg haloperidol plus 1.5 mg/kg MSX-3), HAL/3.0 M (0.1 mg/kg haloperidol plus 3.0 mg/kg MSX-3). # p < 0.01, different from vehicle/vehicle, planned comparison; **p < 0.01, different from vehicle plus haloperidol, planned comparison
Experiment 3: DPCPX plus haloperidol
The mean (+SEM) number of high-density arm selections (i.e., barrier crossings) for rats treated with DPCPX and haloperidol are shown in Fig. 3a. Repeated measures ANOVA indicated that there was an overall significant effect of drug treatment on arm choice [F(4,20) = 38.1, p < 0.001]. Haloperidol plus vehicle-treated rats showed a significant decrease in the selection of the high-density arm as compared to vehicle–vehicle-treated rats (planned comparisons, p < 0.001). However, co-administration of DPCPX with haloperidol did not reverse the effects of haloperidol; in fact, treatment with 3.0 mg/kg DPCPX plus haloperidol significantly reduced selection of the barrier arm relative to haloperidol plus vehicle (p < 0.01). Mean run latencies for haloperidol and DPCPX co-administration are shown in Fig. 3b. Repeated measures ANOVA indicated an overall significant effect of drug treatment on run latency [F(4,20) = 4.27, p < 0.05]. The planned comparison between haloperidol plus vehicle and vehicle–vehicle control approached significance (p = 0.065); however, subsequent non-parametric analysis with the Wilcoxon test did show a significant difference (p < 0.05). In addition, non-orthogonal planned comparisons indicated that there was a significant increase in latency compared to the haloperidol plus vehicle treatment condition in animals administered haloperidol plus 3.0 mg/kg DPCPX. In experiment 3B (Table 1), DPCPX without haloperidol had no significant effect on arm choice (t = 1.2, df = 5, n.s.) or on latency (t = 2.4, df = 5, n.s.). Five of the six animals showed slightly longer latencies after injections of DPCPX than they did after injections of vehicle.

a Effects of the adenosine A1 antagonist DPCPX on T-maze arm choice in rats co-administered haloperidol. Mean (±SEM) number of barrier arm choices after treatment with vehicle or haloperidol plus various doses of DPCPX are shown. Veh/Veh (vehicle plus vehicle), HAL/Veh (0.15 mg/kg haloperidol plus vehicle), HAL/0.75D (0.15 mg/kg haloperidol plus 0.75 mg/kg DPCPX), HAL/1.5D (0.15 mg/kg haloperidol plus 1.5 mg/kg DPCPX), and HAL/3.0D (0.15 mg/kg haloperidol plus 3.0 mg/kg DPCPX). # p < 0.01, different from vehicle/vehicle, planned comparison; *p < 0.05, different from vehicle plus haloperidol, planned comparison. b Effects of the adenosine A1 antagonist DPCPX on run latency in rats co-administered haloperidol. Mean (±SEM) run latency (i.e., average across 30 trials, expressed in sec) after treatment with vehicle or haloperidol plus various doses of DPCPX are shown. Veh/Veh (vehicle plus vehicle), HAL/Veh (0.1 mg/kg haloperidol plus vehicle), HAL/0.75D (0.1 mg/kg haloperidol plus 0.75 mg/kg DPCPX), HAL/1.5D (0.1 mg/kg haloperidol plus 1.5 mg/kg MSX-3), and HAL/3.0D (0.1 mg/kg haloperidol plus 3.0 mg/kg DPCPX). ## p < 0.05, different from vehicle/vehicle, Wilcoxon test; *p < 0.05, different from vehicle plus haloperidol, planned comparison
Discussion
Consistent with previous findings, administration of the DA antagonist haloperidol produced a significant decrease in selection of the high reward density T-maze arm that contained the barrier (Salamone et al. 1994; Cousins et al. 1996; Denk et al. 2005). Correspondingly, haloperidol administration also increased the selection of the low-density T-maze arm. Thus, despite drug-induced decreases in the selection of the high-density arm with the barrier, rats that received haloperidol injections were able to engage in food-motivated behaviors by selecting an alternative route of food selection (i.e., the low-density arm with no barrier). The shift from selection of the high-barrier arm to the no-barrier arm with the lower density of reinforcement occurred in a dose-dependent manner, with the greatest effects being seen at 0.15 mg/kg. Previous research has shown that neither haloperidol nor accumbens DA depletions affected arm choice between four and two food pellets when there was no barrier present (Salamone et al. 1994). This finding indicates that haloperidol was not affecting discrimination of the density of reward (see also Martin-Iverson et al. 1987) or the memory for which arm had the higher density of food present. The present findings with the T-maze are analogous to the results that have been reported to occur with administration of DA antagonists to rats responding on the operant FR5/chow feeding concurrent choice task (Salamone et al. 1991, 2002; Sink et al. 2008). Taken together, these observations support the hypothesis that administration of low doses of DA antagonists can affect choice behavior by making animals more sensitive to the work requirements of a task (Salamone and Correa 2002; Salamone et al. 1991, 2007; Kelley et al. 2005; Baldo and Kelley 2007).
In experiment 2, co-administration of the adenosine A2A antagonist MSX-3 with haloperidol reversed the effect of the DA D2 receptor antagonist. MSX-3 significantly increased selection of the barrier (i.e., high density) arm in haloperidol-treated rats. Furthermore, the highest dose of MSX-3 completely reversed the effects of DA D2 receptor antagonism induced by haloperidol; rats that received haloperidol plus 3.0 mg/kg MSX-3 selected the barrier arm roughly the same number of times as they did when they were treated with vehicle control injections. MSX-3 also was capable of reversing the increase in response latencies induced by the DA antagonist. MSX-3 administered in the absence of haloperidol did not have any behavioral effects on maze performance, except that a few animals actually ran slightly faster in the maze. These results from the present study demonstrate that adenosine A2A receptor antagonism can restore the alterations in effort-related choice that are induced by haloperidol. This observation is consistent with recent studies showing that MSX-3 could reverse the effects of haloperidol and eticlopride in rats tested on the operant FR5/chow feeding concurrent choice procedure (Farrar et al. 2007; Worden et al. 2008).
Although previous studies have examine the ability of MSX-3 to reverse the effects of haloperidol using other tasks (e.g., Farrar et al. 2007), the present study also assessed the effects of the adenosine A1 antagonist DPCPX. Injections of DPCPX (0.75–3.0 mg/kg i.p.) failed to reverse the effects of haloperidol in experiment 3. In fact, co-administration of DPCPX with haloperidol tended to reduce selection of the barrier arm even more than haloperidol plus vehicle and also tended to produce further increases in latency, which could reflect some type of motor or motivational impairment. Although the 0.75–3.0 mg/kg doses of DPCPX were ineffective at reversing the actions of haloperidol in the present study, this i.p. dose range of DPCPX has been shown to be effective in studies with rats that recorded behaviors related to nociception, depression, memory, and other processes (Prediger and Takahashi 2005; Aubel et al. 2007; Maione et al. 2007; Lobato et al. 2008). The present results are consistent with previous studies showing differences between the behavioral effects of adenosine A1 and A2A receptor antagonists (Marston et al. 1998; Mandryk et al. 2005; Prediger et al. 2005). More specifically, the present data suggest that there are differential actions of adenosine A1 and A2A receptor antagonists in terms of how these drugs interact with the DA D2 antagonism produced by haloperidol. The neurochemical basis of this differential interaction is not absolutely clear; however, it is possible that it is related to findings showing that adenosine A1 and A2A receptors are localized on different populations of cells in striatal areas, including the nucleus accumbens (Ferré 1997). Adenosine A2A receptors tend to be co-localized on striatal and accumbens medium spiny neurons with DA D2 receptors, and these receptors appear to interact in a manner related to the development of heterodimers or convergence on to the same signal transduction mechanisms (Fink et al. 1992; Ferré 1997; Svenningsson et al. 1999; Pinna et al. 1999; Hillion et al. 2002; Fuxe et al. 2003). In contrast, adenosine A1 receptors are more likely to be co-localized with DA D1 receptors (Ferré 1997). Although the present results could reflect independent actions of MSX-3 and haloperidol, it is reasonable to suggest that MSX-3 was so effective in reversing the actions of haloperidol on T-maze performance because of the direct interaction between adenosine A2A and DA D2 receptors located on the same medium spiny neurons. Although DPCPX failed to reverse the effects of haloperidol in the T-maze, it is possible that DPCPX would be able to reverse an alteration in T-maze performance if it were induced by a D1 antagonist.
The present findings support the hypothesis that DA and adenosine systems in the brain, possibly in nucleus accumbens, interact in the regulation of effort-related functions (Salamone et al. 2007; Farrar et al. 2007; Font et al. 2008; Mingote et al. 2008). Additional studies should investigate the effects of local administration of MSX-3 into the nucleus accumbens and other brain sites (e.g., Ishiwari et al. 2007; Font et al. 2008) in order to characterize more specifically the brain locus at which adenosine and DA receptors are interacting to influence T-maze performance. Identification of the brain systems involved in regulating behavioral activation and effort-based choice in animals serve to highlight the overlap between activational aspects of motivation and quantitative features of motor control (Salamone et al. 2007). Moreover, this research may provide important clues regarding the neural mechanisms involved in clinical psychopathologies related to psychomotor slowing, fatigue, or anergia in depression and parkinsonism (Salamone et al. 2006, 2007; Farrar et al. 2007; Capuron et al. 2007). For example, it is possible that A2A receptor antagonists could be used to treat these energy-related disorders in humans (Salamone et al. 2007) or to reverse the motivational effects of D2 antagonists that are used clinically to treat psychoses.
References
Aubel B, Kayser V, Farré A, Hamon M, Bourgoin S (2007) Evidence for adenosine- and serotonin-mediated antihyperalgesic effects of cizolirtine in rats suffering from diabetic neuropathy. Neuropharmacology 52:487–496, doi:10.1016/j.neuropharm.2006.08.017
Baldo BA, Kelley AE (2007) Discrete neurochemical coding of distinguishable motivational processes: insights from nucleus accumbens control of feeding. Psychopharmacology (Berl) 191:439–459, doi:10-1007/s00213-007-0741-z
Barbano MF, Cador M (2007) Opioids for hedonic experience and dopamine to get ready for it. Psychopharmacology 191:497–506, doi:10.1007/s00213-006-0521-1
Capuron L, Pagnoni G, Demetrashvili MF, Lawson DH, Fornwalt FB, Woolwine B, Berns GS, Gregory S, Nemeroff CB, Miller AH (2007) Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology 32:2384–2392, doi:10.1038/sj.npp.1301362
Correa M, Carlson BB, Wisniecki A, Salamone JD (2002) Nucleus accumbens dopamine and work requirements on interval schedules. Behav Brain Res 137:179–187, doi:10.1016/S0166-4328(02)00292-9
Correa M, Wisniecki A, Betz A, Dobson DR, O'Neill MF, O'Neill MJ, Salamone JD (2004) The adenosine A2A antagonist KF17837 reverses the locomotor suppression and tremulous jaw movements induced by haloperidol in rats: possible relevance to parkinsonism. Behav Brain Res 148:47–54, doi:10.1016/S0166-4328(03)00178-5
Cousins MS, Sokolowski JD, Salamone JD (1993) Different effects of nucleus accumbens and ventrolateral striatal dopamine depletions on instrumental response selection in the rat. Pharmacol Biochem Behav 46:953–951, doi:10.1016/0091-3057(93)90226-J
Cousins MS, Wei W, Salamone JD (1994) Pharmacological characterization of performance on a concurrent lever pressing/feeding choice procedure: effects of dopamine antagonist, cholinomimetic, sedative and stimulant drugs. Psychopharmacology 116:529–537, doi:10.1007/BF02247489
Cousins MS, Atherton A, Turner L, Salamone JD (1996) Nucleus accumbens dopamine depletions alter relative response allocation in a T-maze cost/benefit task. Behav Brain Res 74:189–197, doi:10.1016/0166-4328(95)00151-4
DeMet EM, Chicz-DeMet A (2002) Localization of adenosine A2A-receptors in rat brain with [3H]ZM-241385. Naunyn Schmiedebergs Arch Pharmacol 366:478–481, doi:10.1007/s00210-002-0613-3
Demyttenaere K, De Fruyt J, Stahl SM (2005) The many faces of fatigue in major depressive disorder. Int J Neuropsychopharmacol 8:93–105, doi:10.1017/S1461145704004729
Denk F, Walton ME, Jennings KA, Sharp T, Rushworth MF, Bannerman DM (2005) Differential involvement of serotonin and dopamine systems in cost–enefit decisions about delay or effort. Psychopharmacology 179:587–596, doi:10.1007/s00213-004-2059-4
Farrar AM, Pereira M, Velasco F, Hockemeyer J, Muller CE, Salamone JD (2007) Adenosine A(2A) receptor antagonism reverses the effects of dopamine receptor antagonism on instrumental output and effort-related choice in the rat: implications for studies of psychomotor slowing. Psychopharmacology 191:579–586, doi:10.1007/s00213-006-0554-5
Farrar AM, Font L, Pereira M, Mingote SM, Bunce JG, Chrobak JJ, Salamone JD (2008) Forebrain circuitry involved in effort-related choice: injections of the GABAA agonist muscimol into ventral pallidum alters response allocation in food-seeking behavior. Neuroscience 152:321–330, doi:10.1016/j.neuroscience.2007.12.034
Ferré S (1997) Adenosine-dopamine interactions in the ventral striatum. Implications for the treatment of schizophrenia. Psychopharmacology 133:107–120, doi:10.1007/s002130050380
Ferré S, Fredholm BB, Morelli M, Popoli P, Fuxe K (1997) Adenosine–dopamine receptor–receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci 20:482–487, doi:10.1016/S0166-2236(97)01096-5
Ferré S, Popoli P, Giménez-Llort L, Rimondini R, Müller CE, Strömberg I, Ögren SO, Fuxe K (2001) Adenosine/dopamine interaction: implications for the treatment of Parkinson's disease. Parkinsonism Relat Disord 7(3):235–241, doi:10.1016/S1353-8020(00)00063-8
Ferré S, Ciruela F, Canals M, Marcellino D, Burgueno J, Casado V, Hillion J, Torvinen M, Fanelli F, Benedetti Pd P, Goldberg SR, Bouvier M, Fuxe K, Agnati LF, Lluis C, Franco R, Woods A (2004) Adenosine A2A-dopamine D2 receptor-receptor heteromers. Targets for neuro-psychiatric disorders. Parkinsonism Relat Disord 10:265–271, doi:10.1016/j.parkreldis.2004.02.014
Ferre S, Ciruela F, Borycz J, Solinas M, Quarta D, Antoniou K, Quiroz C, Justinova Z, Lluis C, Franco R, Goldberg SR (2008) Adenosine A1–A2A receptor heteromers: new targets for caffeine in the brain. Front Biosci 13:2391–2399, doi:10.2741/2852
Fink JS, Weaver DR, Rivkees SA, Peterfreund RA, Pollack AE, Adler EM, Reppert SM (1992) Molecular cloning of the rat A2A adenosine receptor: selective co-expression with D2 dopamine receptors in rat striatum. Brain Res Mol Brain Res 14:186–195, doi:10.1016/0169-328X(92)90173-9
Floresco SB, Ghods-Sharifi S (2007) Amygdala-prefrontal cortical circuitry regulates effort-based decision making. Cereb Cortex 17:251–260, doi:10.1093/cercor/bhj143
Font L, Mingote S, Farrar AM, Pereira M, Worden L, Stopper C, Port RG, Salamone JD (2008) Intra-accumbens injections of the adenosine A(2A) agonist CGS 21680 affect effort-related choice behavior in rats. Psychopharmacology 199:515–526, doi:10.1007/s00213-008-1174-z
Fredholm BB, Lindström K (1999) Autoradiographic comparison of the potency of several structurally unrelated adenosine receptor antagonists at adenosine A1 and A(2A) receptors. Eur J Pharmacol 380:197–202
Fuxe K, Agnati LF, Jacobsen K, Hillion J, Canals M, Torvinen M, Tinner-Staines B, Staines W, Rosin D, Terasmaa A, Popoli P, Leo G, Vergoni V, Lluis C, Ciruela F, Franco R, Ferré S (2003) Receptor heteromerization in adenosine A2A receptor signaling: relevance for striatal function and Parkinson’s disease. Neurology 61:S19–S23
Hauber W, Munkel M (1997) Motor depressant effects mediated by dopamine D2 and adenosine A2A receptors in the nucleus accumbens and the caudate-putamen. Eur J Pharmacol 323:127–131, doi:10.1016/S0014-2999(97)00040-X
Hauber W, Neuscheler P, Nagel J, Müller CE (2001) Catalepsy induced by a blockade of dopamine D1 or D2 receptors was reversed by a concomitant blockade of adenosine A2A receptors in the caudate putamen of rats. Eur J Neurosci 14:1287–1293, doi:10.1046/j.0953-816x.2001.01759.x
Hillion J, Canals M, Torvinen M, Casado V, Scott R, Terasmaa A, Hansson A, Watson S, Olah ME, Mallol J, Canela EI, Zoli M, Agnati LF, Ibanez CF, Lluis C, Franco R, Ferré S, Fuxe K (2002) Coaggregation, cointernalization, and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J Biol Chem 277:18091–18097, doi:10.1074/jbc.M107731200
Hockemeyer J, Burbiel JC, Müller CE (2004) Multigram-scale syntheses, stability, and photoreactions of A2A adenosine receptor antagonists with 8-styrylxanthine structure: potential drugs for Parkinson’s disease. J Org Chem 69:3308–3318, doi:10.1021/jo0358574
Ishiwari K, Madson LJ, Farrar AM, Mingote SM, Valenta JP, DiGianvittorio MD, Frank LE, Correa M, Hockemeyer J, Muller C, Salamone JD (2007) Injections of the selective adenosine A2A antagonist MSX-3 into the nucleus accumbens core attenuate the locomotor suppression induced by haloperidol in rats. Behav Brain Res 178:190–199, doi:10.1016/j.bbr.2006.12.020
Kelley AE, Baldo BA, Pratt WE, Will MJ (2005) Corticostriatal-hypothalamic circuitry and food motivation: integration of energy, action and reward. Physiol Behav 86:773–795, doi:10.1016/j.physbeh.2005.08.066
Keppel G (1991) Design and analysis: a researcher’s handbook. Prentice-Hall, Englewood Cliffs, NJ
Koch M, Schmid A, Schnitzler HU (2000) Role of muscles accumbens dopamine D1 and D2 receptors in instrumental and Pavlovian paradigms of conditioned reward. Psychopharmacology 152:67–73, doi:10.1007/s002130000505
Lobato KR, Binfaré RW, Budni J, Rosa AO, Santos AR, Rodrigues AL (2008) Involvement of the adenosine A1 and A2A receptors in the antidepressant-like effect of zinc in the forced swimming test. Prog Neuropsychopharmacol Biol Psychiatry 32:994–999, doi:10.1016/j.pnpbp.2008.01.012
Maione S, de Novellis V, Cappellacci L, Palazzo E, Vita D, Luongo L, Stella L, Franchetti P, Marabese I, Rossi F, Grifantini M (2007) The antinociceptive effect of 2-chloro-2′-C-methyl-N6-cyclopentyladenosine (2′-Me-CCPA), a highly selective adenosine A1 receptor agonist, in the rat. Pain 131:281–292, doi:10.1016/j.pain.2007.01.013
Majer M, Welberg LA, Capuron L, Pagnoni G, Raison CL, Miller AH (2008) IFN-alpha-induced motor slowing is associated with increased depression and fatigue in patients with chronic hepatitis C. Brain Behav Immun 22:870–880, doi:10.1016/j.bbi.2007.12.009
Mandryk M, Fidecka S, Poleszak E, Malec D (2005) Participation of adenosine system in the ketamine-induced motor activity in mice. Pharmacol Reports 57:55–60
Marston HM, Finlayson K, Maemoto T, Olverman HJ, Akahane A, Sharkey J, Butcher SP (1998) Pharmacological characterization of a simple behavioral response mediated selectively by central adenosine A1 receptors, using in vivo and in vitro techniques. J Pharmacol Exp Ther 285:1023–1030
Martin-Iverson MT, Wilke D, Fibiger HC (1987) Effect of haloperidol and d-amphetamine on perceived quantity of food and tones. Psychopharmacology 93:374–381
Mingote S, Weber SM, Ishiwari K, Correa M, Salamone JD (2005) Ratio and time requirements on operant schedules: effort-related effects of nucleus accumbens dopamine depletions. Eur J Neurosci 21:1749–1757
Mingote S, Font L, Farrar AM, Vontell R, Worden L, Stopper CM, Port RG, Sink KS, Bunce JG, Chrobak JJ, Salamone JD (2008) Nucleus accumbens adenosine A2A receptors regulate exertion of effort by acting on the ventral striatopallidal pathway. J Neurosci 28:9037–9046
Morelli M, Pinna A (2002) Interaction between dopamine and adenosine A2A receptors as a basis for the treatment of Parkinson’s disease. Neurol Sci 22:71–72, doi:10.1007/s100720170052
Nowend KL, Arizzi M, Carlson BB, Salamone JD (2001) D1 or D2 antagonism in nucleus accumbens core or dorsomedial shell suppresses lever pressing for food but leads to compensatory increases in chow consumption. Pharmacol Biochem Behav 69:373–382, doi:10.1016/S0091-3057(01)00524-X
O'Neill M, Brown VJ (2006) The effect of the adenosine A2A antagonist KW-6002 on motor and motivational processes in the rat. Psychopharmacology 184:46–55, doi:10.1007/s00213-005-0240-z
Phillips PE, Walton ME, Jhou TC (2007) Calculating utility: preclinical evidence for cost–benefit analysis by mesolimbic dopamine. Psychopharmacology 191:483–495, doi:10.1007/s00213-006-0626-6
Pinna A, Wardas J, Cozzolino A, Morelli M (1999) Involvement of adenosine A2A receptors in the induction of c-fos expression by clozapine and haloperidol. Neuropsychopharmacol 20:44–51, doi:10.1016/S0893-133X(98)00051-7
Pinna A, Wardas J, Simola N, Morelli M (2005) New therapies for the treatment of Parkinson’s disease: adenosine A2A receptor antagonists. Life Sci 77:3259–3267, doi:10.1016/j.lfs.2005.04.029
Prediger RD, Takahashi RN (2005) Modulation of short-term social memory in rats by adenosine A1 and A(2A) receptors. Neurosci Lett 376:160–165, doi:10.1016/j.neulet.2004.11.049
Prediger RD, Fernandes D, Takahashi RN (2005) Blockade of adenosine A2A receptors reverses short-term social memory impairments in spontaneously hypertensive rats. Behav Brain Res 159:197–205, doi:10.1016/j.bbr.2004.10.017
Robbins TW, Everitt BJ (2007) A role for mesencephalic dopamine in activation: commentary on Berridge (2006). Psychopharmacology 191:433–437, doi:10.1007/s00213-006-0528-7
Salamone JD, Correa M (2002) Motivational views of reinforcement: implications for understanding the behavioral functions of nucleus accumbens dopamine. Behav Brain Res 137:3–25, doi:10.1016/S0166-4328(02)00282-6
Salamone JD, Steinpreis RE, McCullough LD, Smith P, Grebel D, Mahan K (1991) Haloperidol and nucleus accumbens dopamine depletion suppress lever pressing for food but increase free food consumption in a novel food choice procedure. Psychopharmacology 104:515–521, doi:10.1007/BF02245659
Salamone JD, Cousins MS, Bucher S (1994) Anhedonia or anergia? Effects of haloperidol and nucleus accumbens dopamine depletion on instrumental response selection in a T-maze cost/benefit procedure. Behav Brain Res 65:221–229, doi:10.1016/0166-4328(94)90108-2
Salamone JD, Cousins MS, Snyder BJ (1997) Behavioral functions of nucleus accumbens dopamine: empirical and conceptual problems with the anhedonia hypothesis. Neurosci Biobehav Rev 21:341–359, doi:10.1016/S0149-7634(96)00017-6
Salamone JD, Arizzi M, Sandoval MD, Cervone KM, Aberman JE (2002) Dopamine antagonsts alter response allocation but do not suppress appetite for food in rats: Contrast between the effects of SKF 83566, raclopride and fenfluramine on a concurrent choice task. Psychopharmacology 160:371–380, doi:10.1007/s00213-001-0994-x
Salamone JD, Correa M, Mingote SM, Weber SM (2005) Beyond the reward hypothesis: alternative functions of nucleus accumbens dopamine. Curr Opin Pharmacol 5:34–41, doi:10.1016/j.coph.2004.09.004
Salamone JD, Correa M, Mingote SM, Weber SM, Farrar AM (2006) Nucleus accumbens dopamine and the forebrain circuitry involved in behavioral activation and effort-related decision making: implications for understanding anergia and psychomotor slowing in depression. Curr Psychiatr Rev 2:267–280, doi:10.2174/157340006776875914
Salamone JD, Correa M, Farrar A, Mingote SM (2007) Effort-related functions of nucleus accumbens dopamine and associated forebrain circuits. Psychopharmacology 191:461–482, doi:10.1007/s00213-006-0668-9
Salamone JD, Betz AJ, Ishiwari K, Felsted J, Madson L, Mirante B, Clark K, Font L, Korbey S, Sager TN, Hockemeyer J, Muller CE (2008a) Tremorolytic effects of adenosine A2A antagonists: implications for parkinsonism. Front Biosci 13:3594–3605, doi:10.2741/2952
Salamone JD, Ishiwari K, Betz AJ, Farrar AM, Mingote SM, Font L, Hockemeyer J, Müller CE, Correa M (2008b) Dopamine/adenosine interactions related to locomotion and tremor in animal models: Possible relevance to parkinsonism. Parkinsonism Relat Disord 14:S130–S134, doi:10.1016/j.parkreldis.2008.04.017
Schiffmann SN, Jacobs O, Vanderhaeghen JJ (1991) Striatal restricted adenosine A2A receptor (RDC8) is expressed by enkephalin but not by substance P neurons: an in situ hybridization histochemistry study. J Neurochem 57:1062–1071, doi:10.1111/j.1471-4159.1991.tb08257.x
Schweimer J, Saft S, Hauber W (2005) Involvement of catecholamine neurotransmission in the rat anterior cingulate in effort-related decision making. Behav Neurosci 119:1687–1692, doi:10.1037/0735-7044.119.6.1687
Sink KS, Vemuri VK, Olszewska T, Makriyannis A, Salamone JD (2008) Cannabinoid CB1 antagonists and dopamine antagonists produce different effects on a task involving response allocation and effort-related choice in food-seeking behavior. Psychopharmacology 196:565–574, doi:10.1007/s00213-007-0988-4
Sokolowski JD, Salamone JD (1998) The role of nucleus accumbens dopamine in lever pressing and response allocation: Effects of 6-OHDA injected into core and dorsomedial shell. Pharmacol Biochem Behav 59:557–566, doi:10.1016/S0091-3057(97)00544-3
Solinas M, Ferré S, Antoniou K, Quarta D, Justinova Z, Hockemeyer J, Pappas LA, Segal PN, Wertheim C, Müller CE, Goldberg SR (2005) Involvement of adenosine A1 receptors in the discriminative-stimulus effects of caffeine in rats. Psychopharmacology 179:576–586
Svenningsson P, Le Moine C, Fisone G, Fredholm BB (1999) Distribution, biochemistry and function of striatal adenosine A2A receptors. Prog Neurobiol 59:355–396, doi:10.1016/S0301-0082(99)00011-8
Takahashi RN, Pamplona FA, Prediger RD (2008) Adenosine receptor antagonists for cognitive dysfunction: a review of animal studies. Front Biosci 13:2614–2632, doi:10.2741/2870
Van den Bos R, van der Harst J, Jonkman S, Schilders M, Spruijt B (2006) Rats assess costs and benefits according to an internal standard. Behav Brain Res 171:350–354, doi:10.1016/j.bbr.2006.03.035
Walton ME, Bannerman DM, Alterescu K, Rushworth MF (2003) Functional specialization within medial frontal cortex of the anterior cingulate for evaluating effort-related decisions. J Neurosci 23:6475–6479
Walton ME, Kennerley SW, Bannerman DM, Phillips PE, Rushworth MF (2006) Weighing up the benefits of work: behavioral and neural analyses of effort-related decision making. Neural Netw 19:1302–1314, doi:10.1016/j.neunet.2006.03.005
Wardas J, Konieczny J, Lorenc-Koci E (2001) SCH 58261, an A2A adenosine receptor antagonist, counteracts parkinsonian-like muscle rigidity in rats. Synapse 41:160–171, doi:10.1002/syn.1070
Worden LT, Shariari M, Farrar AM, Sink KS, Hockemeyer J, Muller C, Salamone JD (2009) The adenosine A2A antagonist MSX-3 reverses the effort-related effects of dopamine blockade: differential interaction with D1 and D2 family antagonists. Psychopharmacology (in press)
Yurgelun-Todd DA, Sava S, Dahlgren MK (2007) Mood disorders. Neuroimaging Clin N Am 17:511–521, doi:10.1016/j.nic.2007.08.001
Acknowledgments
This work was supported by a grant to J.S. from the National Institute of Mental Health (MH078023). Many thanks to Dr. Merce Correa for her valuable comments and suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mott, A.M., Nunes, E.J., Collins, L.E. et al. The adenosine A2A antagonist MSX-3 reverses the effects of the dopamine antagonist haloperidol on effort-related decision making in a T-maze cost/benefit procedure. Psychopharmacology 204, 103–112 (2009). https://doi.org/10.1007/s00213-008-1441-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-008-1441-z