
Abstract
Rationale
Antagonism at serotonin 5-HT2A and 5-HT2C receptors modulates cortical and striatal dopamine (DA) release and may underlie some aspects of the clinical efficacy of ‘atypical’ antipsychotic compounds. However, it is not known whether 5-HT2A/2C receptor-mediated modulation of DA release can be quantified with non-invasive neurochemical imaging, as would be required for investigation of these processes in man.
Objective
The objective of the study was to perform a feasibility study in the rat in order to determine whether 5-HT2A/2C modulation of DA release can be observed using positron emission tomography (PET) imaging.
Materials and methods
Rats were administered with either vehicle, a combined 5-HT2A/2C antagonist (ketanserin, 3 mg/kg i.p.), or the more selective 5-HT2C antagonist SB 206,553 (10 mg/kg i.p.) 30 min before administration of the PET DA D2 receptor radiotracer [11C]raclopride (∼11 MBq) and were then scanned for 60 min using a quad-high-density avalanche chamber small animal tomograph. Using the same technique, modulation of amphetamine (4 mg/kg)-induced decreases in [11C]raclopride binding by 5-HT2A antagonism (SR 46349B, 0.2 mg/kg i.v.) was also determined.
Results
Consistent with the increase in DA release measured by others using microdialysis, 5-HT2C antagonism markedly reduced striatal [11C]raclopride binding (p < 0.003), while amphetamine-induced reductions in striatal [11C]raclopride binding (p < 0.001) were attenuated by 5-HT2A antagonist administration (p = 0.04).
Conclusions
These results inform the feasibility of monitoring 5-HT2A/2C receptor-mediated modulation of DA systems in man using PET and, more generally, demonstrate that D2 radiotracer PET imaging may be used to monitor the efficacy of new DA modulators in attenuating stimulated DA release.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Serotonergic projections from the midbrain raphe nuclei exert a profound modulatory influence over dopamine (DA) release in the striatum (see Alex and Pehek 2007 and Kapur and Remington 1996). As pharmacological manipulation of these serotonergic inputs may provide a useful avenue in treatment of schizophrenia, substance abuse, and obsessive compulsive and affective disorders (Kapur and Remington 1996; Moresco et al. 2007; Rothman and Baumann 2006), interaction between 5-HT and DA systems is an important area for translational research. Although influences of serotonergic systems on striatal DA release have been comprehensively determined in experimental animals using invasive techniques (e.g., microdialysis), the extent to which these findings may be investigated non-invasively in man is unclear.
Many ‘atypical’ antipsychotic drugs show high binding affinity at 5-HT2A and 5-HT2C receptors (Ichikawa and Meltzer 1999; Meltzer et al. 1989; 2003; Roth et al. 1992), and antidepressants such as mirtazepine have 5-HT2C affinity (Chanrion et al. 2008). Modulation of striatal DA release by 5-HT2A and 5-HT2C compounds has been extensively investigated in animals using microdialysis to measure changes in extracellular DA concentrations. Results have shown that 5-HT2A receptor antagonists do not alter striatal DA levels when administered under baseline conditions (De Deuwaerdère and Spampinato 1999; Gobert et al. 2000; Schmidt and Fadayel 1996; Sorensen et al. 1993) but markedly attenuate increases in DA release stimulated, for example, by amphetamine administration (Auclair et al. 2004a, b; De Deuwaerdère and Spampinato 1999; Lucas and Spampinato 2000; Porras et al. 2002; Schmidt et al. 1992). Accordingly, while 5-HT2A receptor activation has little effect on basal DA release, stimulated DA release is facilitated (Gobert and Millan 1999; Ichikawa and Meltzer 1995; Kuroki et al. 2003; Lucas and Spampinato 2000).
The role of 5-HT2C receptors in regulation of DA release is somewhat oppositional to that of the 5-HT2A subtype. 5-HT2C receptors are constitutionally active and exert tonic inhibitory control over DA release from both mesolimbic and nigrostriatal neurons (De Deuwaerdère et al. 2004). Thus, basal (tonic) DA cell firing and terminal DA release is enhanced by 5-HT2C antagonists/inverse agonists and inhibited by 5-HT2C agonists (Alex et al. 2005; De Deuwaerdère et al. 2004; De Deuwaerdère and Spampinato 1999; Di Giovanni et al. 1999; 2000; Di Matteo et al. 1998; 1999; Gobert et al. 2000; Navailles et al. 2006; Porras et al. 2002).
The aforementioned studies in experimental animals have provided an important foundation for predicting 5-HT2A and 5-HT2C influences on striatal DA release. In man, such neurochemical changes may be investigated using positron emission tomography (PET) imaging with DA D2 receptor radiotracers such as [11C]raclopride that are sensitive to changes in endogenous neurotransmitter. A decrease in D2 radiotracer binding caused by ‘competition’ between the D2 radiotracer and free DA for D2 receptor binding can be interpreted as an increase in synaptic DA (Laruelle 2000). However, the extent to which changes in DA detected using microdialysis and that measured using PET is unclear, as relationships between alterations in extracellular DA levels (as determined using microdialysis) and changes in PET D2 radiotracer binding may be complex (see Laruelle 2000 for review). It is therefore important to determine whether 5-HT2A and 5-HT2C modulation of striatal DA release might be quantified using in vivo PET in small animals before undertaking comparable clinical investigations of the interaction of these systems in man.
Some research suggests that 5-HT2A and 5-HT2C modulation of striatal DA release can be detected using PET, although 5-HT receptor subtype-selective compounds have not yet been investigated. Administration of the hallucinogenic drug psilocybin, an agonist at 5-HT1A and 5-HT2A receptors (McKenna et al. 1990), significantly decreases [11C]raclopride binding in man (Vollenweider et al. 1999), indicating increased DA release. In baboons, decreases in striatal [11C]raclopride binding have also been shown following administration of the 5-HT receptor antagonists altanserin (Dewey et al. 1995) and ketanserin (Tsukada et al. 1999). However, both these compounds have higher affinity at 5-HT2A than 5-HT2C receptors [altanserin: 20-fold (Tan et al. 1999); ketanserin: 14-fold (Glennon et al. 2002)], which, on the basis of the microdialysis studies cited above (Di Matteo et al. 1999; Gobert et al. 2000; Porras et al. 2002), would be expected to produce opposing effects; if 5-HT2A antagonism predominates, this may be hypothesized to reduce increases in DA release elicited by 5-HT2C antagonism. Indeed, although the microdialysis studies carried out in parallel with the PET studies detailed above demonstrated increases in striatal DA following altanserin or ketanserin administration (Dewey et al. 1995; Tsukada et al. 1999), other microdialysis investigations have reported no effect of ketanserin on baseline DA levels (Nash 1990; Ng et al. 1999).
We therefore investigated the modulatory influence of 5-HT2A and 5-HT2C antagonists on striatal DA levels in rats in vivo using [11C]raclopride PET. Specifically, we wished to compare the effects of the mixed 5-HT2A/2C antagonist ketanserin with that of more selective 5-HT2A and 5-HT2C compounds. We used the selective 5-HT2A antagonist SR 46349B (Rinaldi-Carmona et al. 1992), previously shown to reduce amphetamine-stimulated DA release using microdialysis (Auclair et al. 2004a; Porras et al. 2002). For investigation of the effects of 5-HT2C antagonism, we used SB 206,553, which has 160-fold selectivity for 5-HT2C versus 5-HT2A sites (Forbes et al. 1995) but cannot discriminate between 5-HT2B and 5-HT2C receptors (Kennett et al. 1996). SB 206,553 is particularly able to enhance DA release in vivo (De Deuwaerdère et al. 2004; Gobert et al. 2000). It should be noted that many 5-HT2A and 5-HT2C ‘antagonists’, including those employed here, actually have inverse agonist properties (Berg et al. 1999; 2005; De Deuwaerdère et al. 2004; Herrick-Davis et al. 2000; Weiner et al. 2001).
Materials and methods
A total of 32 adult male Sprague–Dawley rats (Harlan Olac, UK; body weight mean ± SD = 306 ± 22 g) were group-housed in standard conditions. All investigations were carried out in accordance with the UK Animals (Scientific Procedures) Act, 1986 and associated guidelines.
[11C]Raclopride PET scan protocol
Rats were anesthetized with isoflurane and a mixture of N2O and O2. To ensure correct head position, rats were positioned in a stereotaxic frame (made in-house) and placed in a quad-high-density avalanche chamber (HIDAC) small animal tomograph (Oxford Positron Systems). Each PET scan used a small aliquot of [11C]raclopride from a radiosynthesis intended for human PET, prepared routinely according to the method of Farde et al. (1988). [11C]Raclopride was administered via a previously catheterized lateral tail vein. The mean ± SD injectate was 10.9 ± 0.97 MBq, with an associated stable content of 0.9 ± 0.7 nmol/kg. Using our previously measured in vivo ED50 value of 17 nmol/kg, receptor occupancy due to co-injected stable compound was estimated as ∼5% (Hume et al. 1998). Immediately following administration of [11C]raclopride, emission data were acquired in list mode for 60 min, after which rats were euthanized via an intravenous injection of sodium pentobarbitone. Data for experiments 1 and 2 (see below) was collected over different time periods, between which scanner performance varied. We therefore used date-matched control animals for each experiment.
To reconstruct scan sinograms, list-mode emission data were binned into 0.5-mm isotropic voxels using filtered back-projection (Hamming filter, 0.6 cut-off). This resulted in a spatial resolution of ∼0.5 mm full-width at half-maximum (Myers and Hume 2002). Image volumes were then transferred into ANALYZE software for volume of interest (VOI) analysis (Robb and Hanson 1991). To sample [11C]raclopride binding in the striatum, a VOI template based on stereotaxic coordinates (Hume et al. 2001) was projected onto each scan volume. Data were sampled from the dorsal striatum (2 × 140 voxels), ventral striatum (2 × 12 voxels), and cerebellum (764 voxels). While the microdialysis studies cited in the “Introduction” measure DA release in the nucleus accumbens, by using PET, we are unable to separate the nucleus accumbens from the olfactory tubercle, so we use the term ‘ventral striatum’ to describe this brain area.
The total binding/non-specific binding ratio in each VOI was calculated from tissue: cerebellum ratios, which assumes that the reference tissue (i.e., cerebellum) has a negligible number of D2 receptors and that the non-specifically bound radiotracer shows similar behavior in the VOI and the reference region. Although data from the quad-HIDAC scanner was in list mode, allowing rebinning into dynamic time–activity curves, the count statistics do not allow measurement of tissue concentrations in VOI with low signals such as the cerebellum (Hirani et al. 2003; Hume et al. 2001). To increase count statistics, VOI analysis was limited to a single 40-min time frame, beginning 20 min after [11C]raclopride injection. Previous studies have shown that [11C]raclopride takes ∼20 min to reach dynamic equilibrium in isoflurane-anesthetized rats and that the striatum/cerebellum ratio remains unchanged from 20–60 min after [11C]raclopride injection (Hume et al. 1996). Ratio data acquired in the 20–60-min time frame correlates well with individual binding potential measurements derived from time–activity curves (Houston et al. 2004), and we have repeatedly demonstrated the usefulness of this approach (Hirani et al. 2003; Houston et al. 2004; Hume et al. 2001; Le Masurier et al. 2004).
Specific binding ratio (SBR) was defined as ([11C]raclopride VOI/[11C]raclopride cerebellum ratio) − 1. Levene’s test was used to confirm equality of variance in data, and the effects of pharmacological manipulations versus control on dorsal and ventral striatal [11C]raclopride SBR were determined using two-tailed independent sample t tests. The threshold for statistical significance was set at an α level of 0.05, and statistical analysis was performed using SPSS software for Windows (SPSS Inc. Version 14.0).
Experiment 1: Effect of 5HT2A and 5-HT2C antagonism on DA release
Rats were administered with either vehicle (control; 0.9% NaCl, n = 5), the mixed 5-HT2A/2C antagonist ketanserin tartate (Tocris Bioscience, UK, 3 mg/kg i.p. n = 6), or the 5-HT2C/2B inverse agonist SB 206,553 (Tocris Bioscience, UK; 10 mg/kg i.p. n = 5). These doses were chosen as 3 mg/kg ketanserin reduces [11C]raclopride binding in monkeys using PET (Tsukada et al. 1999), and a 10-mg/kg dose of SB 206,553 markedly increases DA release in the rat striatum (De Deuwaerdère et al. 2004). All drugs were administered 30 min before the start of the scan. In order to minimize any confounding effects of subsequent anesthesia, all drugs were administered to anesthetized animals, which were not recovered before the scan.
Experiment 2: Effect of 5HT2A antagonism on amphetamine-stimulated DA release
Rats were administered with either vehicle (0.9% NaCl, n = 6) or 4 mg/kg amphetamine sulphate (Sigma UK, n = 5) i.p. 30 min before the scan. To examine the effects of 5-HT2A antagonism on amphetamine-stimulated DA release, a separate group of rats was administered with the 5-HT2A antagonist SR 46349B (Sanofi-Synthelabo, France; 0.2 mg/kg i.v.) 5 min before amphetamine (n = 5). This 0.2 mg/kg i.v. dose of SR 46349B was shown to totally occupy 5-HT2A receptors in ex vivo biodistribution studies (Shah et al. 1998). Amphetamine was injected at a dose of 4 mg/kg as we have previously demonstrated marked reductions in striatal [11C]raclopride SBR at this dose using PET (Houston et al. 2004). All drugs were administered to anesthetized animals, which were not recovered before the scan.
Results
Figures 1 and 3 illustrate the images that were obtained in the control or drug-treated animals using the quad-HIDAC system. Coronal, horizontal, and sagittal slices are illustrated at the level of the striatum. Although each scan was analyzed separately, ‘parametric’ images are shown for illustration purposes. The volumes contain data acquired at steady state (20–60 min following raclopride injection) and were obtained by normalizing each dataset through dividing individual images by corresponding individual cerebellar VOI data and then calculating the average volume (using ANALYZE AVW, image algebra). Bilateral striata are clearly noticeable. The extra-cerebral bilateral hotspots on the horizontal and sagittal planes represent non-specific (i.e., non-saturable) binding to the lachrymal glands.

Mean parametric maps calculated from individual scan data using ANALYZE software (Robb and Hanson 1991). The top row shows the localization of the VOI in representative coronal, horizontal, and sagittal slices (DStrR, DStrL: right and left dorsal striatum; VStrR, VStrL: right and left ventral striatum; cblm: cerebellum). The colored images show [11C]raclopride average distribution volume ratios (VOI/cerebellum) sampled at steady state (20–60 min following [11C]raclopride injection) in animals treated with vehicle i.p. (0.9% NaCl; n = 5), 3 mg/kg ketanserin i.p. (n = 6), or 10 mg/kg SB 206,553 i.p. (n = 5). For each group, the same coronal, horizontal, and sagittal slices are presented. The extra-cerebral bilateral hotspots rostral to the striata represent non-specific binding in lachrymal (Harderian) glands (see Hume et al. 1996)
Experiment 1: Effect of 5HT2A and 5-HT2C antagonism on baseline DA release
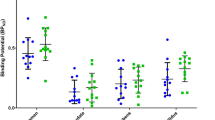
Figure 1 shows the mean parametric images obtained in rats administered with vehicle, 3 mg/kg ketanserin, or 10 mg/kg SB 206,553 30 min before [11C]raclopride injection. The reduction in specific signal in the striata of rats treated with SB 206,553 is clearly visible. The values associated with this effect are presented in Table 1. As illustrated graphically in Fig. 2, ketanserin administration had no effect on [11C]raclopride binding in the dorsal striatum (2%; t (9) = 1.058; p = 0.357, ns); the mean SBR was 3.16 ± 0.09 in control (vehicle-treated) rats and 3.11 ± 0.12 in rats administered with 3 mg/kg ketanserin. In contrast, in the ventral portion of the striatum, the mean SBR was 2.39 ± 0.08 in control (vehicle-treated) rats and 2.09 ± 0.08 in ketanserin-treated rats, leading to a significant reduction in [11C]raclopride binding (12%; t (9) = 3.473; p = 0.007). Due to concerns regarding the extent to which the dorsal and ventral striatum can be dissociated, we additionally examined data obtained when the dorsal and ventral striatal SBR values were combined. These values were calculated by adjusting for the number of voxels in each VOI and then calculating the mean SBR. Under this analysis, no significant effect of ketanserin administration on [11C]raclopride binding in the striatum was detected (t (9) = 0.544; p = 0.232).

Mean [11C]raclopride specific binding ratios (SBRs) in the dorsal and ventral striatum [11C]raclopride binding in rats that were administered with either vehicle i.p. (0.9% NaCl; n = 5), 3 mg/kg ketanserin i.p. (n = 6), or 10 mg/kg SB 206,553 i.p. (n = 5). Thirty minutes following drug administration, rats were injected with [11C]raclopride i.v. and scanned using a quad-HIDAC small animal tomograph. SBRs are (VOI/cerebellum) − 1 radioactivity concentration ratios for scan data sampled at steady state (20–60 min following [11C]raclopride injection). p values are calculated using independent-samples t tests and, * indicates statistical significance (p < 0.05)
Administration of the 5-HT2B/2C inverse agonist SB 206,553 produced highly significant reductions in [11C]raclopride SBR in both the dorsal (t (8) = 4.149; p = 0.003) and ventral striatum (t (8) = 6.004; p < 0.001), as SB 206,553-treated rats showed a SBR of 2.79 ± 0.11 and 1.93 ± 0.03 in the dorsal and ventral striatum, respectively. The percent reductions in SBR following SB 206,553 administration were 12% in the dorsal striatum and 19% in the ventral striatum.
Experiment 2: Effect of 5-HT2A antagonism on amphetamine-stimulated DA release
Figure 3 shows selected coronal slices at the level of the striatum from 3D ‘parametric’ image volumes obtained in rats administered with vehicle alone, 4 mg/kg amphetamine, or 4 mg/kg amphetamine plus 0.2 mg/kg SR 43649B 30 min before [11C]raclopride injection. The reduction in [11C]raclopride binding that was detected following amphetamine administration can be clearly seen by visual inspection of the images in Fig. 3. Values that were obtained in experiment 2 are presented in Table 2. Analysis showed that administration of amphetamine alone produced a highly significant reduction in [11C]raclopride binding in the dorsal (34%; t (9) = 6.261; p < 0.001) and ventral striatum (32%;t (9) = 6.337; p < 0.001). The SBR values in the dorsal and ventral striatum were 2.87 ± 0.12 and 1.90 ± 0.04 in vehicle-treated (control) animals, respectively, and 1.89 ± 0.09 and 1.29 ± 0.09 in amphetamine-alone-treated animals, respectively. These results are illustrated graphically in Fig. 4.

Mean parametric maps calculated from individual scan data using ANALYZE software (Robb and Hanson 1991). The top row shows the localization of the VOI in representative coronal, horizontal, and sagittal slices (DStrR, DStrL: right and left dorsal striatum; VStrR, VStrL: right and left ventral striatum; cblm: cerebellum). The colored images show [11C]raclopride average distribution volume ratios (VOI/cerebellum) sampled at steady state (20–60 min following [11C]raclopride injection) in animals treated with vehicle i.p. (0.9% NaCl; n = 6), 4 mg/kg i.p. amphetamine (Amph; n = 5) or 4 mg/kg i.p. amphetamine preceded by 0.2 mg/kg i.v. SR 46349B (n = 5). For each group, the same coronal, horizontal, and sagittal slices are presented. The extra-cerebral bilateral hotspots rostral to the striata represent non-specific binding in lachrymal (Harderian) glands (see Hume et al. 1996)

Mean [11C]raclopride specific binding ratios (SBRs) in the dorsal and ventral striatum [11C]raclopride binding in rats that were administered either with vehicle i.p. (0.9% NaCl; n = 6), 4 mg/kg i.p. amphetamine (n = 5), or 4 mg/kg i.p. amphetamine preceded by 0.2 mg/kg i.v. SR 46349B (n = 5). Thirty minutes following drug administration, rats were injected with [11C]raclopride i.v. and scanned using a quad-HIDAC small animal tomograph. SBRs are (VOI/cerebellum) − 1 radioactivity concentration ratios for scan data sampled at steady state (20–60 min following [11C]raclopride injection). p values are calculated using independent-samples t tests, and * indicates statistical significance relative to vehicle, while § indicates statistical significance relative to amphetamine (p < 0.05)
When animals were pre-treated with SR 46349B prior to amphetamine administration, the SBR in the dorsal and ventral striatum was also significantly less than that of vehicle-treated animals (dorsal striatum: 24%; t (9) = 4.655; p = 0.001; ventral striatum: 23%; t (9) = 4.907; p = 0.001). Pre-treatment with the 5-HT2A antagonist SR 46349B attenuated the amphetamine-induced reduction in [11C]raclopride binding in the dorsal striatum; the mean SBR in the SR 46349B plus amphetamine group was 2.18 ± 0.07 (13% increase from amphetamine-alone values, t (8) = −2.454; p = 0.040). No significant effects of SR 46349B pre-treatment were found in the ventral striatum (12%; t (8) = −1.279; p = 0.237, ns), although when dorsal and ventral striatal values were combined, a significant attenuation of the amphetamine response was observed following SR 26349B administration (t (8) = 2.542; p = 0.035).
Discussion
Using PET in the rat, we have clearly demonstrated decreases in [11C]raclopride binding following 5-HT2C antagonism and an attenuation of amphetamine-induced decreases in [11C]raclopride binding following 5-HT2A antagonism. These data are in accordance with the opposing effects of 5-HT2A and 5-HT2C receptor antagonists on DA release as previously obtained with microdialysis (Auclair et al. 2004a; De Deuwaerdère et al. 2004; Di Giovanni et al. 1999; 2000; Di Matteo et al. 1998; 1999; Gobert et al. 2000; Lucas and Spampinato 2000; Porras et al. 2002) and provide evidence that such interactions can be observed non-invasively using [11C]raclopride PET imaging.
We first examined the effects of the mixed 5-HT2A/2C antagonist ketanserin on striatal [11C]raclopride binding, as this compound is suitable for human administration. Ketanserin significantly reduced [11C]raclopride binding in the rat ventral but not dorsal striatum, and no changes were detected when the striatum was analyzed as a whole. At the same 3-mg/kg dose, ketanserin has previously been shown to markedly reduce striatal [11C]raclopride binding in the rhesus monkey dorsal striatum as shown with PET (Tsukada et al. 1999). It is unclear why ketanserin produced significant decreases in [11C]raclopride binding in the dorsal striatum in the study of Tsukada et al. (1999), but not in the present investigation. One possibility is that there may be species differences in dose sensitivity or 5-HT2A/2C receptor distribution. Indeed, when microdialysis is used to measure changes in dorsal extracellular DA following ketanserin administration, increases in DA levels are observed in monkeys (Tsukada et al. 1999), but not rats (Nash 1990; Ng et al. 1999).
Administration of the 5-HT2B/2C antagonist SB 206,553 decreased [11C]raclopride binding in both the dorsal and ventral striatum, which is in close accordance with the increases in DA release that have been detected with microdialysis following administration of this compound (Alex et al. 2005; De Deuwaerdère et al. 2004; De Deuwaerdère and Spampinato 1999; Di Giovanni et al. 1999; 2000; Gobert et al. 2000; Navailles et al. 2006; Porras et al. 2002). This effect is likely to be mediated by the5-HT2C receptor; although SB 206,553 also has significant 5-HT2B affinity, 5-HT2B expression is low in the brain, and selective 5-HT2B receptor antagonists do not increase DA release (Duxon et al. 1997; Gobert et al. 2000).
We also observed a significant attenuation of amphetamine-induced decreases in [11C]raclopride binding in the striatum following administration of the 5-HT2A antagonist SR 46349B. These results parallel those obtained with microdialysis studies of dopamine release (Porras et al. 2002; Auclair et al. 2004a), but are of a smaller magnitude, perhaps because of the large dose of amphetamine we employed. Despite the small effect size, to our knowledge, this is the first PET study to show attenuation of an amphetamine-induced decrease in [11C]raclopride binding by a compound that does not compete with amphetamine at the receptor level (e.g., Villemagne et al. 1999) or reduces DA release via tyrosine depletion (e.g., Le Masurier et al. 2004). A significant limitation of this study is that we did not investigate the effects of SR 46349B alone on [11C]raclopride binding. However, we predict that 5-HT2A antagonism alone would have no effect; microdialysis studies have repeatedly reported no effect of 5-HT2A antagonism on dopamine release (De Deuwaerdère and Spampinato 1999; Gobert et al. 2000; Schmidt and Fadayel 1996; Sorensen et al. 1993) or DA neuron firing rate (Olijslagers et al. 2004). Nonetheless, this lack of effect on DA release should also be confirmed using [11C]raclopride PET, as should the effects of a 5-HT2A agonist, before proceeding to human studies.
As this initial investigation was intended as a feasibility study to determine whether 5-HT2A/2C modulation of DA release can be observed with [11C]raclopride PET, we used high doses of ketanserin, SB 206,553, and SR 46349B to maximize the probability of observing significant experimental effects. We also employed a relatively high dose of amphetamine (4 mg/kg) to provide a large initial effect size for potential reversal, although lower doses of amphetamine also produce decreases in [11C]raclopride binding (Houston et al. 2004). This work may now be extended to examining the dose–response relationships between 5-HT2A/2C compounds and percentage change in [11C]raclopride binding; in particular, it is important to confirm whether these effects are observed at 5-HT2A/2C occupancy levels comparable to those achieved clinically.
5-HT2C receptors are localized on GABAergic interneurons in the ventral tegmental area (VTA) and substantia nigra (Bubar and Cunningham 2007; Eberle-Wang et al. 1997), from which position they modulate dopamine neuron activity (Di Giovanni et al. 2001; Prisco et al. 1994). Administration of SB 206,553, the 5-HT2C antagonist employed in the present study, enhances the basal firing rate of DA neurons (Di Giovanni et al. 1999; Gobert et al. 2000; Porras et al. 2002) and can transform the firing pattern of mesolimbic DA neurons into burst mode (Gobert et al. 2000). The data presented here demonstrate that the resulting increases in striatal DA release can be detected using [11C]raclopride PET.
The mechanism by which 5-HT2A receptor activation may decrease stimulated DA release is less clear. The ability of SR 46349B to decrease amphetamine-stimulated DA release does not occur when either amphetamine or SR 46349B are focally injected into the striatum but requires systemic amphetamine administration and 5-HT2A inhibition in the VTA (Auclair et al. 2004a). The precise mechanism remains to be established but has been hypothesized to involve an indirect mechanism arising through amphetamine-induced noradrenaline release in the prefrontal cortex, which in turn regulates glutamatergic afferents to the VTA, at which site 5-HT2A modulation occurs (Auclair et al. 2004a,b; Tassin 2008).
Ability to quantify 5-HT receptor modulation of dopamine release in man may be particularly useful in examining the clinical mechanism of action of antipsychotic and antidepressant drugs. Most atypical antipsychotics possess inverse agonist activity at the 5-HT2C receptor (Berg et al. 1999; Herrick-Davis et al. 2000) and many antidepressants, including mianserin, mirtazepine, and agomelatine, are also 5-HT2C inverse agonists (Chanrion et al. 2008). 5-HT2A affinity has been suggested to contribute toward antipsychotic efficacy and low extra-pyramidal side effects (Meltzer et al. 1989; 2003; Ichikawa and Meltzer 1999). Indeed, SR 46349B has previously reached clinical trial to assess antipsychotic efficacy (Meltzer et al. 2004), as has the selective 5-HT2A antagonist MDL100907 (Potkin et al. 2001).
As we have demonstrated that changes in DA release in the striatum produced 5-HT2A/2C antagonism can be imaged in vivo using PET in the rat, these investigations may now be extended to examine the dose–response profile of antidepressant and antipsychotic compounds with 5-HT2A/C (but not D2) affinity. As the therapeutic effects of these compounds are also likely to be associated with 5-HT2A/2C-mediated DA release in the prefrontal cortex, this work should be extended to PET studies with high affinity D2 radiotracers such as [11C]FLB457, which may be sensitive to changes in cortical DA levels (Montgomery et al. 2007). These studies will guide future human PET studies investigating the association between 5-HT2A/2C antagonism, DA release, and clinical profile of antidepressant and antipsychotic compounds and demonstrate that [11C]raclopride PET may be used to monitor the efficacy of new pharmacotherapies using a functional response in addition to direct ligand–drug interactions.
In conclusion, our studies demonstrate and justify the use of [11C]raclopride PET in man to monitor the interactions of 5-HT and DA systems in vivo following selective pharmacological manipulations of 5-HT2A and 5-HT2C targets.
Abbreviations
- SB 206,553:
-
5-methyl-1-(3-pyridylcarbamoyl)-1,2,3,5-tetrahydropyrrolo[2,3-f]indole hydrochloride
- SR 46349B:
-
(1(Z)-[2-(dimethylamino)ethoxyimino]-1(2-fluorophenyl)-3-(4-hydroxyphenyl)-2(E)-propene)
References
Ahmad R, Opacka-Juffry J, Houston G, Hirani E, Hume S (2005) The effect of 5-HT2A antagonists on amphetamine-evoked dopamine release in rats, measured by positron emission tomography. Mol Imaging Biol 7(2):152
Alex KD, Pehek EA (2007) Pharmacologic mechanisms of serotonergic regulation of dopamine neurotransmission. Pharmacol Ther 113(2):296–320
Alex KD, Yavanian GJ, McFarlane HG, Pluto CP, Pehek EA (2005) Modulation of dopamine release by striatal 5-HT2C receptors. Synapse 55(4):242–251
Auclair A, Blanc G, Glowinski J, Tassin JP (2004a) Role of serotonin 2A receptors in the d-amphetamine-induced release of dopamine: comparison with previous data on alpha1b-adrenergic receptors. J Neurochem 91(2):318–326
Auclair A, Drouin C, Cotecchia S, Glowinski J, Tassin JP (2004b) 5-HT2A and alpha1b-adrenergic receptors entirely mediate dopamine release, locomotor response and behavioural sensitization to opiates and psychostimulants. Eur J Neurosci 20(11):3073–3084
Berg KA, Stout BD, Cropper JD, Maayani S, Clarke WP (1999) Novel actions of inverse agonists on 5-HT2C receptor systems. Mol Pharmacol 55(5):863–872
Berg KA, Harvey JA, Spampinato U, Clarke WP (2005) Physiological relevance of constitutive activity of 5-HT2A and 5-HT2C receptors. Trends Pharmacol Sci 26(12):625–630
Bubar MJ, Cunningham KA (2007) Distribution of serotonin 5-HT2C receptors in the ventral tegmental area. Neuroscience 146(1):286–297
Chanrion B, Mannoury la Cour C, Gavarini S, Seimandi M, Vincent L, Pujol JF, Bockaert J, Marin P, Millan MJ (2008) Inverse agonist and neutral antagonist actions of antidepressants at recombinant and native 5-HT2C receptors: differential modulation of cell surface expression and signal transduction. Mol Pharmacol 73:748–757
De Deurwaerdère P, Spampinato U (1999) Role of 5-HT2A and 5-HT2C receptor subtypes in the control of accumbal and striatal dopamine release elicited in vivo by dorsal raphe nucleus electrical stimulation. J Neurochem 73:1033–1042
De Deuwaerdère P, Navailles S, Berg KA, Clarke WP, Spampinato U (2004) Constitutive activity of the serotonin2C receptor inhibits in vivo dopamine release in the rat striatum and nucleus accumbens. J Neurosci 24:3235–3241
Dewey SL, Smith GS, Logan J, Alexoff D, Ding YS, King P, Pappas N, Brodie JD (1995) Serotonergic modulation of striatal dopamine measured with positron emission tomography (PET) and in vivo microdialysis. J Neurosci 15(1 Pt 2):821–829
Di Giovanni G, De Deurwaerdère P, Di Mascio M, Di Matteo V, Esposito E, Spampinato U (1999) Selective blockade of serotonin-2C/2B receptors enhances mesolimbic and mesostriatal dopaminergic function: a combined in vivo electrophysiological and microdialysis study. Neuroscience 91:587–597
Di Giovanni G, Di Matteo V, Di Mascio M, Esposito E (2000) Preferential modulation of mesolimbic vs. nigrostriatal dopaminergic function by serotonin2C/2B receptor agonists: a combined in vivo electrophysiological and microdialysis study. Synapse 35:53–61
Di Giovanni G, Di Matteo V, La Grutta V, Esposito E (2001) m-Chlorophenylpiperazine excites non-dopaminergic neurons in the rat substantia nigra and ventral tegmental area by activating serotonin-2C receptors. Neuroscience 103(1):111–116
Di Matteo V, Di Giovanni G, Di Mascio M, Esposito E (1998) Selective blockade of serotonin2C/2B receptors enhances dopamine release in the rat nucleus accumbens. Neuropharmacology 37:265–272
Di Matteo V, Di Giovanni G, Di Mascio M, Esposito E (1999) SB242084, a selective serotonin2c receptor antagonist, increases dopaminergic transmission in the mesolimbic system. Neuropharmacology 38:1195–1205
Duxon MS, Flanigan TP, Reavley AC, Baxter GS, Blackburn TP, Fone KCF (1997) Evidence for expression of the 5-hydroxytryptamine-2B receptor protein in the rat central nervous system. Neuroscience 76:323–329
Eberle-Wang K, Mikeladze Z, Uryu K, Chesselet MF (1997) Pattern of expression of the serotonin2C receptor messenger RNA in the basal ganglia of adult rats. J Comp Neurol 384(2):233–247
Egerton A, Grasby PM (2007) Direct and indirect dopamine challenges for PET imaging. J Psychopharmacol 21(7):MB01
Farde L, Wiesel FA, Jansson P, Uppfeldt G, Wahlen A, Sedvall G (1988) An open label trial of raclopride in acute schizophrenia. Confirmation of D2-dopamine receptor occupancy by PET. Psychopharmacology (Berl) 94:1–7
Forbes IT, Ham P, Booth DH, Martin RT, Thompson M, Baxter GS, Blackburn TP, Glen A, Kennett GA, Wood MD (1995) 5-Methyl-1-(3-pyridylcarbamoyl)-1,2,3,5 tetrahydropyrrolo[2,3-f]indole: a novel 5-HT2C/5-HT2B receptor antagonist with improved affinity, selectivity, and oral activity. J Med Chem 38(14):2524–2530
Glennon RA, Metwally K, Dukat M, Ismaiel AM, De los Angeles J, Herndon J, Teitler M, Khorana N (2002) Ketanserin and spiperone as templates for novel serotonin 5-HT(2A) antagonists. Curr Top Med Chem 2(6):539–558
Gobert A, Millan MJ (1999) Serotonin (5-HT)2A receptor activation enhances dialysate levels of dopamine and noradrenaline, but not 5-HT, in the frontal cortex of freely-moving rats. Neuropharmacology 38(2):315–317
Gobert A, Rivet J-M, Lejeune F, Newman-Tancredi A, Adhumeau-Auclair A, Nicolas JP (2000) Serotonin2C receptors tonically suppress the activity of mesocortical dopaminergic and adrenergic, but not serotonergic, pathways: a combined dialysis and electrophysiological analysis in the rat. Synapse 36:205–221
Herrick-Davis K, Grinde E, Teitler M (2000) Inverse agonist activity of atypical antipsychotic drugs at human 5-hydroxytryptamine2C receptors. J Pharmacol Exp Ther 295(1):226–232
Hirani E, Sharp T, Sprakes M, Grasby P, Hume S (2003) Fenfluramine evokes 5-HT2A receptor-mediated responses but does not displace [11C]MDL 100907: small animal PET and gene expression studies. Synapse 50(3):251–260
Houston GC, Hume SP, Hirani E, Goggi JL, Grasby PM (2004) Temporal characterisation of amphetamine-induced dopamine release assessed with [11C]raclopride in anaesthetised rodents. Synapse 51(3):206–212
Hume SP, Lammertsma AA, Myers R, Rajeswaran S, Bloomfield PM, Ashworth S, Fricker RA, Torres EM, Watson J, Jones T (1996) The potential of high-resolution positron emission tomography to monitor striatal dopaminergic function in rat models of disease. J Neurosci Methods 67:103–112
Hume SP, Gunn RN, Jones T (1998) Pharmacological constraints associated with positron emission tomographic scanning of small laboratory animals. Eur J Nucl Med 25:173–176
Hume S, Hirani E, Opacka-Juffry J, Myers R, Townsend C, Pike V, Grasby P (2001) Effect of 5-HT on binding of [(11)C] WAY 100635 to 5-HT(IA) receptors in rat brain, assessed using in vivo microdialysis and PET after fenfluramine. Synapse 41:150–159
Ichikawa J, Meltzer HY (1995) DOI, a 5-HT2A/2C receptor agonist, potentiates amphetamine-induced dopamine release in rat striatum. Brain Res 698(1–2):204–208
Ichikawa J, Meltzer HY (1999) Relationship between dopaminergic and serotonergic neuronal activity in the frontal cortex and the action of typical and atypical antipsychotic drugs. Eur Arch Psychiatry Clin Neurosci 249(Suppl 4):90–98
Kapur S, Remington G (1996) Serotonin–dopamine interaction and its relevance to schizophrenia. Am J Psychiatry 153(4):466–476
Kennett GA, Wood MD, Bright F, Cilia J, Piper DC, Gager T, Thomas D, Baxter GS, Forbes IT, Ham P, Blackburn TP (1996) In vitro and in vivo profile of SB 206553, a potent 5-HT2C/5-HT2B receptor antagonist with anxiolytic-like properties. Br J Pharmacol 117(3):427–434
Kuroki T, Meltzer HY, Ichikawa J (2003) 5-HT 2A receptor stimulation by DOI, a 5-HT 2A/2C receptor agonist, potentiates amphetamine-induced dopamine release in rat medial prefrontal cortex and nucleus accumbens. Brain Res 972(1–2):216–221
Laruelle M (2000) Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab 20(3):423–451
Le Masurier M, Houston G, Cowen P, Grasby P, Sharp T, Hume S (2004) Tyrosine-free amino acid mixture attenuates amphetamine-induced displacement of [11C]raclopride in striatum in vivo: a rat PET study. Synapse 51(2):151–157
Lucas G, Spampinato U (2000) Role of striatal serotonin2A and serotonin2C receptor subtypes in the control of in vivo dopamine outflow in the rat striatum. J Neurochem 74:693–701
McKenna DJ, Repke DB, Peroutka SJ (1990) Differential interactions of indolealkylamines with 5-hydroxytryptamine receptor subtypes. Neuropharmacology 29:193–198
Meltzer HY, Matsubara S, Lee JC (1989) Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotinin2 pK i values. J Pharmacol Exp Ther 251:238–246
Meltzer HY, Li Z, Kaneda Y, Ichikawa J (2003) Serotonin receptors: their key role in drugs to treat schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 27(7):1159–1172
Meltzer HY, Arvanitis L, Bauer D, Rein W, Meta-Trial Study Group (2004) Placebo-controlled evaluation of four novel compounds for the treatment of schizophrenia and schizoaffective disorder. Am J Psychiatry 161(6):975–984
Montgomery AJ, Asselin MC, Farde L, Grasby PM (2007) Measurement of methylphenidate-induced change in extrastriatal dopamine concentration using [11C]FLB 457 PET. J Cereb Blood Flow Metab 27(2):369–377
Moresco RM, Pietra L, Henin M, Panzacchi A, Locatelli M, Bonaldi L, Carpinelli A, Gobbo C, Bellodi L, Perani D, Fazio F (2007) Fluvoxamine treatment and D2 receptors: a pet study on OCD drug-naive patients. Neuropsychopharmacology 32(1):197–205
Myers R, Hume S (2002) Small animal PET. Eur Neuropsychopharmacol 12:545–555
Nash JF (1990) Ketanserin pre-treatment attenuates MDMA-induced dopamine release in the striatum as measured by in vivo microdialysis. Life Sci 47(26):2401–2408
Navailles S, Moison D, Ryczko D, Spampinato U (2006) Region-dependent regulation of mesoaccumbens dopamine neurons in vivo by the constitutive activity of central serotonin2C receptors. J Neurochem 99(4):1311–1319
Ng NK, Lee HS, Wong PT (1999) Regulation of striatal dopamine release through 5-HT1 and 5-HT2 receptors. J Neurosci Res 55(5):600–607
Olijslagers JE, Werkman TR, McCreary AC, Siarey R, Kruse CG, Wadman WJ (2004) 5-HT2 receptors differentially modulate dopamine-mediated auto-inhibition in A9 and A10 midbrain areas of the rat. Neuropharmacology 46(4):504–510
Porras G, Di Matteo V, Fracasso C, Lucas G, De Deurwaerdère P, Caccia S, Esposito E, Spampinato U (2002) 5-HT2A and 5-HT2C/2B receptor subtypes modulate dopamine release induced in vivo by amphetamine and morphine in both the rat nucleus accumbens and striatum. Neuropsychopharmacology 26:311–324
Potkin SG, Shipley J, Bera RB, Carreon D, Fallon J, Alva G, Keator D (2001) Clinical and PET effects of M100907, a selective 5-HT-2A receptor antagonist. Schizophr Res 49(Suppl. 1):242
Prisco S, Pagannone S, Esposito E (1994) Serotonin-dopamine interaction in the rat ventral tegmental area: an electrophysiological study in vivo. J Pharmacol Exp Ther 271(1):83–90
Rinaldi-Carmona M, Congy C, Santucci V, Simiand J, Gautret B, Neliat G, Labeeuw B, Le Fur G, Soubrie P, Breliere JC (1992) Biochemical and pharmacological properties of SR 46349B, a new potent and selective 5-hydroxytryptamine2 receptor antagonist. J Pharmacol Exp Ther 262(2):759–768
Robb RA, Hanson DP (1991) A software system for interactive and quantitative visualization of multidimensional biomedical images. Australas Phys Eng Sci Med 14:930
Roth BL, Ciaranello RD, Meltzer HY (1992) Binding of typical and atypical antipsychotic agents to transiently expressed 5-HT1C receptors. J Pharmacol Exp Ther 260:1361–1365
Rothman RB, Baumann MH (2006) Balance between dopamine and serotonin release modulates behavioral effects of amphetamine-type drugs. Ann N Y Acad Sci 1074:245–260
Schmidt CJ, Fadayel GM (1996) Regional effects of MK-801 on dopamine release: effects of competitive NMDA or 5-HT2A receptor blockade. J Pharmacol Exp Ther 277:1541–1549
Schmidt CJ, Fadayel GM, Sullivan CK, Taylor VL (1992) 5-HT2 receptors exert a state-dependent regulation of dopaminergic function: studies with MDL 100,907 and the amphetamine analogue, 3,4-methylenedioxymethamphetamine. Eur J Pharmacol 223:65–74
Shah F, Hirani E, Hume SP, Osman S, Pike VW (1998) [N-methyl-C-11]SR 46349B—examination as a radioligand for brain 5-HT2A receptors in rat in vivo. J Nucl Med 39(5):237P–237P 1044 Suppl. S
Sorensen SM, Kehne JH, Fadayel GM, Humphreys TM, Ketteler HJ, Sullivan CK, Taylor VL, Schmidt CJ (1993) Characterization of the 5-HT2 receptor antagonist MDL 100907 as a putative atypical antipsychotic: behavioral, electrophysiological and neurochemical studies. J Pharmacol Exp Ther 266(2):684–691
Tan PZ, Baldwin RM, Van Dyck CH, Al-Tikriti M, Roth B, Khan N, Charney DS, Innis RB (1999) Characterization of radioactive metabolites of 5-HT2A receptor PET ligand [18F]altanserin in human and rodent. Nucl Med Biol 26(6):601–608
Tassin JP (2008) Uncoupling between noradrenergic and serotonergic neurons as a molecular basis of stable changes in behavior induced by repeated drugs of abuse. Biochem Pharmacol 75(1):85–97
Tsukada H, Nishiyama S, Kakiuchi T, Ohba H, Sato K, Harada N (1999) Is synaptic dopamine concentration the exclusive factor which alters the in vivo binding of [11C]raclopride?: PET studies combined with microdialysis in conscious monkeys. Brain Res 841(1–2):160–169
Villemagne VL, Wong DF, Yokoi F, Stephane M, Rice KC, Matecka D, Clough DJ, Dannals RF, Rothman RB (1999) GBR12909 attenuates amphetamine-induced striatal dopamine release as measured by [(11)C]raclopride continuous infusion PET scans. Synapse 33(4):268–273
Vollenweider FX, Vontobel P, Hell D, Leenders KL (1999) 5-HT modulation of dopamine release in basal ganglia in psilocybin-induced psychosis in man—a PET study with [11C]raclopride. Neuropsychopharmacology 20(5):424–433
Weiner DM, Burstein ES, Nash N, Croston GE, Currier EA, Vanover KE et al (2001) 5-Hydroxytryptamine2a receptor inverse agonists as antipsychotics. J Pharmacol Exp Ther 299(1):268–276
Acknowledgments
The early stages of the work presented here were initiated by Dr Susan P. Hume, and we are very grateful for her important contribution to this research. These investigations were also partly funded by GlaxoSmithKline. A portion of this work has previously been presented in preliminary form (Ahmad et al. 2005; Egerton and Grasby 2007).
Disclosure/conflict of interest
Dr Alice Egerton is funded by GlaxoSmith Kline through Imperial College and reports no competing interests. Rabia Ahmad and Dr Ella Hirani report no competing interests. Prof. Paul M Grasby has served as an occasional consultant to GlaxoSmithKline, Merck, and Pfizer and reports no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Egerton, A., Ahmad, R., Hirani, E. et al. Modulation of striatal dopamine release by 5-HT2A and 5-HT2C receptor antagonists: [11C]raclopride PET studies in the rat. Psychopharmacology 200, 487–496 (2008). https://doi.org/10.1007/s00213-008-1226-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-008-1226-4