
Abstract
Rationale
Tramadol is in an unscheduled atypical analgesic with low rates of diversion and abuse and mixed pharmacologic actions, including modest opioid agonist activity.
Objectives
The purpose of the current study was to characterize the opioid withdrawal suppression efficacy of oral tramadol.
Materials and methods
Residential, opioid-dependent adults (n = 10) were maintained on morphine (15 mg subcutaneously, quad in diem) for approximately 6 weeks. Spontaneous opioid withdrawal was produced by substituting placebo for scheduled morphine doses 17.5 h before experimental sessions that occurred twice weekly. The acute effects of placebo, tramadol (50, 100, 200, and 400 mg orally), naloxone (0.1 and 0.2 mg intramuscularly [IM]), and morphine (15 and 30 mg IM) were tested under double-blind, double-dummy, randomized conditions. Outcomes included observer- and subject-rated measures, physiologic indices, and psychomotor/cognitive task performance.
Results
Naloxone and morphine produced prototypic opioid antagonist and agonist effects, respectively. Tramadol 50 and 100 mg produced effects most similar to placebo. Tramadol 200 and 400 mg initially produced significant dose-related increases in ratings of “bad effects” and “feel sick,” followed by evidence of opioid withdrawal suppression. Tramadol did not produce significant increases on measures of positive drug effects nor any clinically significant physiologic changes.
Conclusions
Tramadol 200 and 400 mg show evidence of opioid withdrawal suppression without significant observer- and subject-rated opioid agonist effects. The profile of action did not suggest a high risk for tramadol abuse in opioid dependent individuals. Tramadol may be a useful medication for treating opioid withdrawal.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tramadol (Ultram®) is an analgesic medication used to treat moderate to severe pain. It provides analgesia through a dual mechanism of action: mu-opioid receptor agonism and monoamine reuptake inhibition (Collart et al. 1993; Desmeules et al. 1996; Enggaard et al. 2006; Raffa et al. 1992). The racemic parent compound, (±)-trans-2-(dimethylaminomethyl)-1-(m-methoxyphenyl)-cyclohexanol hydrochloride (tramadol), acts primarily to block the reuptake of norepinephrine and serotonin and has little opioid activity, while its hepatic metabolite, (+)-O-desmethyltramadol (M1), has mild to moderate affinity and intrinsic agonist activity at the mu-opioid receptor (Collart et al. 1993; Desmeules et al. 1996; Enggaard et al. 2006; Gillen et al. 2000; Hennies et al. 1988; Lai et al. 1996; Raffa et al. 1992). Oral and parenteral tramadol are estimated to have approximately one tenth and one third the analgesic potency of oral and parenteral morphine, respectively (Gutstein and Akil 2001; Lehmann et al. 1990).
Tramadol was initially approved in Germany in 1977 for pain treatment. In 1994, it was approved in the USA as an unscheduled analgesic after years of a positive epidemiologic experience in Germany with tramadol (lack of diversion and abuse), and several preclinical (Friderichs et al. 1978; Murano et al. 1978; Yanagita 1978) and clinical laboratory studies (Barth et al. 1987; Cami et al. 1994; Jasinski et al. 1993; Preston and Jasinski 1989; Preston et al. 1991) suggested that tramadol had low abuse liability. More recently, subjective opioid agonist-like effects of oral tramadol have been reported among drug users (Epstein et al. 2006; Zacny 2005) along with classic opioid withdrawal symptoms after tramadol cessation or dose reductions (Senay et al. 2003), indicating that tramadol may exert a clinically relevant amount of opioid activity. Despite this opioid activity, extensive postmarketing surveillance in the USA has demonstrated low rates of diversion and abuse for tramadol (Cicero et al. 1999, 2005; Inciardi et al. 2006; Woody et al. 2003).
The favorable abuse liability profile of tramadol along with its opioid activity may make it a useful medication for opioid dependence treatment particularly in places, such as the USA, where there are substantial legal and regulatory restrictions on using scheduled drugs for treatment of this disorder. Three retrospective reports have suggested that tramadol may be helpful in relieving opioid withdrawal; two reported no difference in treatment retention compared to buprenorphine (Tamaskar et al. 2003; Threlkeld et al. 2006), but these results were confounded by concomitant clonidine use, and one reported better treatment retention and less opioid withdrawal compared to clonidine (Sobey et al. 2003). A fourth study, which was prospective, found tramadol similar in efficacy to low-dose methadone for the treatment of opioid withdrawal (Salehi et al. 2005). Our laboratory recently completed a clinical pharmacology study that evaluated whether tramadol could suppress spontaneous opioid withdrawal among subjects maintained on oral hydromorphone (10 mg quad in diem [QID]) (Carroll et al. 2006). Experimental sessions took place 24 h after the last hydromorphone dose when mild spontaneous opioid withdrawal was present and assessed the acute effects of: oral tramadol (50, 100, 200, and 400 mg), hydromorphone (opioid agonist control), naloxone (opioid antagonist control), and placebo. There were decreases in opioid withdrawal for tramadol and hydromorphone conditions compared to placebo, but these were not statistically significant. The small sample size (n = 6) and mild severity of spontaneous opioid withdrawal produced by oral hydromorphone maintenance may have limited the ability to detect significant withdrawal suppression with the opioid agonist control condition and tramadol.
The purpose of the current study was to evaluate the opioid agonist effects of oral tramadol utilizing an opioid withdrawal suppression experimental design. The study included both positive and negative opioid control conditions and employed a larger sample size compared to the Carroll et al. 2006 study. The present study also used morphine as the maintenance medication (in lieu of hydromorphone). This design was based upon the original 24-h opioid substitution procedure developed in the 1950s by the US Public Health Service Addiction Research Center in Lexington, KY. This experimental procedure was a valid and reliable technique for determining if a test drug was morphine like, as morphine-like substances suppressed morphine withdrawal while nonmorphine-like substances did not (for review, see Jasinski 1977). In the present study, withdrawal suppression efficacy and subjective, physiologic, and psychomotor/cognitive effects were evaluated to characterize tramadol’s opioid effects, the threshold dose that produces evidence of opioid withdrawal suppression, and the safety profile in the context of spontaneous morphine withdrawal.
Materials and methods
Participants
Participants were ten adult heroin-dependent volunteers (eight men/two women, five Caucasian/five African-American) that used heroin by the intravenous (n = 8) and intranasal route (n = 2) a mean of 27.9 out of the 30 days before study enrollment (range = 20–30 days) and spent a mean of $21/day on heroin (range = $10–60). The mean number of years since first use of heroin was 17.6 years (range = 5–35 years). Other drugs used in the 30 days before study participation included cocaine by seven subjects (mean = 12.4 days in the last 30 days), benzodiazepines by four subjects (mean = 3.0 days in the last 30 days), marijuana by two subjects (mean = 1.5 days in the last 30 days), and alcohol to intoxication by two subjects (mean = 20 days in the last 30 days). Volunteers had a mean age of 36.8 years (range = 26–54 years) and a mean of 11.4 years of education (range = 10–14 years). All tested positive for opiates on urine toxicology and were diagnosed with Opioid Dependence using the Structured Clinical Interview for Diagnostic and Statistical Manual of Mental Disorders—Fourth Edition (Spitzer et al. 1992). Volunteers underwent routine medical screening, which included a history and physical examination, electrocardiogram, serum chemistry and hematology, and urinalysis testing. Exclusion criteria included current physical dependence on alcohol or a sedative/hypnotic drug, major mental illness (e.g., schizophrenia), significant medical problem (e.g., history of seizure or hypertension), and pregnancy. The Institutional Review Board approved the study, and all participants gave written informed consent and were paid for their participation. Participants were informed that this study was testing whether tramadol may be helpful for the treatment of opioid dependence and that they would be maintained on morphine daily; however, they were informed that their morphine doses may change without their knowledge.
Study setting
Participants resided at the residential Behavioral Pharmacology Research Unit for approximately 6 weeks. On a random schedule throughout the stay, urine and breath samples were collected and tested for illicit drugs and alcohol, respectively. Urine testing was completed with an on-site enzyme-multiplied immunoassay technique toxicology system (Behring Diagnostics, San Jose, CA), and breath samples were analyzed with an Alco-Sensor IV breathalyzer (AlcoPro, Knoxville, TN); no tests were positive. Participants were maintained on a caffeine-free diet and were allowed to smoke ad libitum up to 45 min before sessions. Magnesium hydroxide, ducosate, acetaminophen, fiber supplement, and antacid were available to subjects as needed but were never given after midnight preceding study session days.
Study design and procedure
This was a randomized, placebo-controlled, within-subjects crossover study. Participants were administered subcutaneous (SC) morphine 15 mg QID (06:00, 10:00, 16:00, and 22:00 hours) to maintain a mild to moderate degree of opioid physical dependence. After at least 7 days of morphine administration, participants began a series of ten laboratory sessions. Opiate withdrawal was produced by placebo substitution for morphine (see below) before each session to evaluate the withdrawal suppression efficacy of tramadol. The first session was always a double-blind, placebo-training session to ensure that participants understood the test measures and were experiencing opiate withdrawal. These training sessions were excluded from data analyses. In the following nine experimental sessions, subjects received both oral capsules and an intramuscular (IM) injection; however, only one active drug condition was tested in each session. The nine double-blind drug conditions were: oral tramadol (50, 100, 200, and 400 mg), IM morphine (7.5 and 15 mg), IM naloxone (0.1 and 0.2 mg), and placebo (saline injection and lactose capsules). Morphine and naloxone served as opioid agonist and antagonist control conditions, respectively. An additional 11th session was scheduled in case there were methodological problems (e.g., missing data or computer error) with one of the previous nine sessions and needed to be repeated (five subjects had a 11th session that was used in data analyses).
Morphine doses scheduled for 22:00 hours on the day before and 06:00 hours on the day of the session were substituted with placebo in a double-blind fashion to elicit spontaneous opioid withdrawal. Thus, at the time of session commencement (09:30 hours), 17.5 h had passed since the last active morphine dose was administered. Research staff verified the presence of opioid withdrawal using a modified Himmelsbach scale (range 0–14; Kolb and Himmelsbach 1938), which has been previously shown to be sensitive to detecting acute opioid withdrawal effects (Stoller et al. 2001). The mean Himmelsbach score was 5.3 (average based on ten subjects × nine experimental sessions per subject = 90 predrug administration assessments); this score is consistent with a mild to moderate amount of opiate withdrawal.
Drugs
Tramadol HCl (Ortho-McNeil Pharmaceuticals, Raritan, NJ), naloxone HCl (Endo Laboratories, Chadds Ford, PA), and morphine sulfate (Baxter Healthcare, Deerfield, IL) were obtained from commercial sources. Tramadol was administered under an investigator-obtained Investigational New Drug Application (IND) from the Food and Drug Administration (IND no. 69,537). All drugs were aseptically prepared under a laminar flow hood by filtering the solution through a 0.22-μm Millex-GS Millipore filter (Millipore Products Division, Bedford, MA) into a sterile, pyrogen-free vial (American Pharmaceutical Partners, Los Angeles, CA). Naloxone doses were prepared from a 0.4 mg/ml-solution, and morphine doses were prepared from a 15 mg/ml-solution; both were diluted with bacteriostatic 0.9% saline for injection (Hospira, Lake Forest, IL) using sterile pyrogen-free plastic syringes (Becton Dickinson, Franklin Lakes, NJ) to achieve the desired doses in a 1-ml volume. Tramadol tablet(s) (50 mg each) were broken and overencapsulated along with lactose monohydrate powder, N.F. (Ruger Chemical, Irvington, NJ) in size 0 capsules (Capsugel, Greenwood, SC). Each tramadol dose (50, 100, 200, and 400 mg) contained the same number of capsules (four) to keep the study the blind. Capsules without tramadol contained lactose monohydrate powder, N.F. (Ruger Chemical).
Experimental sessions
Experimental test sessions occurred twice weekly with at least 72 h between session days (e.g., Monday/Thursday or Tuesday/Friday). Sessions took place in a quiet room separate from the residential unit, and participants were monitored continuously throughout the test session by a research assistant and/or nursing staff. The room contained two chairs, an Apple computer, circular lights apparatus, and physiologic monitoring equipment. Subjective and observer measures along with cognitive performance measures were presented on the computer screen, and responses were entered using a keypad. Physiologic measures were recorded directly to the computer.
Sessions lasted 4 h and ran from approximately 09:30 to 13:30 hours; at 10:00 hours, one of the nine experimental drug conditions was administered (scheduled morphine doses at 10:00 hours on session days were omitted). Thirty minutes of data were collected before drug administration; data from 15 min before drug administration served as baseline data for analyses. Data collection continued for 210 min after drug administration. Measures described below were collected every 15 min throughout each session except when noted otherwise.
Participant and observer measures
Participants completed visual analog scales (VAS) and a drug class identification questionnaire. The VAS consisted of six items relating to possible drug effects: Any Drug Effects, High, Good Effects, Bad Effects, Like Drug, and Feel Sick. Participants clicked the computer mouse along a line labeled “None” on one end and “Extremely” on the other end for each item, and scores were determined from 0 (none) to 100 (extremely). The drug class questionnaire asked the participant to select one of ten drug classes to characterize what drug they believe they received. Choices were: “Blank or placebo,” “opioid,” “opioid antagonist,” “antipsychotics/neuroleptics,” “barbiturate,” “antidepressant,” “PCP or hallucinogen,” “benzodiazepine,” “cocaine or stimulant,” and “other.” These drug classes, along with examples for each class, initially were described to participants.
Both participants and an observer (a research assistant answering questions based on how the participant appeared at that moment) completed an opioid adjective rating questionnaire, which consisted of 37 items rated on a scale from 0 (not at all) to 4 (extremely; Stoller et al. 2001). These items consisted of two scales; a 16-item agonist scale that is associated with morphine-like effects (score range 0–64) and a 21-item antagonist scale that is associated with opioid withdrawal effects (range 0–84). In addition, observers completed an assessment of seven signs of opioid withdrawal scored from 0 to 2 for each item: lacrimation, rhinorrhea, yawning, perspiration, piloerection, bowel sounds, and restlessness (modified Himmelsbach scale based on Kolb and Himmelsbach 1938). Scores for each item were summed to give a total modified Himmelsbach score (range 0–14).
Physiological measures
Heart rate, systolic and diastolic blood pressure, oxygen saturation, and skin temperature were collected using a Criticare Model 507E Noninvasive Patient Monitor (Criticare Systems, Waukesha, WI). These data were collected every minute and then averaged across 15-min intervals. Respiratory rate was evaluated once every 15 min. Pupil diameter was measured by photographs using a Polaroid camera with a 2× magnification. Baseline pupil diameter was the median diameter of three photos taken 15 min before drug administration. After drug administration, pupil diameter was measured every 15 min with a single photograph.
Psychomotor/cognitive performance measures
Participants completed a circular lights task (Griffiths et al. 1983) and three cognitive performance computerized tasks: digit symbol substitution test (DSST; McLeod et al. 1982) and trails A and B based on the trail-making task (Reitan 1958). The dependent measure for circular lights was the number of correct buttons pressed during a 60-s trial. For the DSST, dependent measures were the number of attempted trials during a 90-s trial and accuracy (proportion of attempted trials correctly replicated). In addition, participants’ cognitive awareness of their performance was assessed before (preperformance estimate) and after (postperformance estimate) completing the DSST by asking participants immediately before and after each DSST how well they thought they would perform on the task compared to normal (preperformance estimate) and how well they thought they had performed compared to normal (postperformance estimate), respectively. Participants made these ratings on a VAS labeled “much worse” at the left extreme and “much better” at the right extreme, with normal labeled in the middle. Scores range from −50 (much worse) to 50 (much better). For trails A and B, the dependent measures were the number of sequence errors and the total line length (Strain et al. 2000).
Data analysis
Drug identification questionnaire responses are presented descriptively. To characterize the time course and magnitude of tramadol effects for other measures, three sets of analyses were completed using data from all subjects (n = 10) for each of the nine drug conditions: (1) time course analyses, (2) time to peak maximum and minimum drug effect (maximum only for VAS drug effects) analyses, and (3) peak maximum and minimum drug effect (maximum only for VAS drug effects) analyses. All measures were analyzed with peak drug effect analyses. Time course analyses were completed for physiologic and subjective and observer measures, and time to peak drug effect analyses were completed for subjective and observer measures. For all analyses, baseline values for each measure were subtracted from each postdrug administration time point (i.e., 15, 30, 45, 60, 75 … 195, 210 min) to correct for any potential baseline differences among drug conditions, and all active drug conditions were compared to placebo using Tukey’s honestly significant difference (HSD). The mean square error term needed to perform these tests was calculated using a two-factor analysis of variance (drug condition and time) for the time course analyses and a one-factor analysis of variance (drug condition) for the time to peak and peak analyses. Comparisons for which p values from the Tukey HSD were less than 0.05 are reported as statistically significant.
Results
Visual Analog Scale items
Figure 1 illustrates VAS time course results. Results for tramadol 50 and 100 mg are not shown because these doses typically resulted in no significant differences compared to placebo. In addition, only the higher doses of morphine and naloxone are shown because these are the opioid agonist and antagonist control condition doses, respectively, that produced consistent differences from placebo across time. In the first 120 min of sessions, tramadol (200 and 400 mg) produced significantly higher ratings compared to placebo on Any Drug Effects, Bad Effects, and Feel Sick, but the majority of these tramadol effects were lower in magnitude than effects produced by naloxone 0.2 mg. Inspection of the data revealed that the onset of oral tramadol effects was later (e.g., 45–60 min for Any Drug Effects and 60 min for Feel Sick) than the onset of effects for both IM morphine 15 mg (30 min for Any Drug Effects) and IM naloxone 0.2 mg (30 min for Any Drug Effects and 15 min for Feel Sick), as determined by the time to the first significant increase (relative to placebo). The duration of tramadol effects on these three measures was also shorter; for example, for Any Drug Effects, tramadol’s effects lasted approximately 1–1.25 h, compared to almost 2 h for naloxone 0.2 mg and 1.75 h for morphine 15 mg. The time course for ratings of High, Good Effects, and Like Drug demonstrated that morphine 15 mg produced immediate drug effects that were significantly different from placebo and that lasted approximately 2–3 h, while neither naloxone nor tramadol produced any significant changes on these measures.

Time course of mean Visual Analog Scale scores. SEM bars not shown to preserve clarity of figure. A darkened symbol indicates a significant difference from placebo at that time point
Peak maximum values for VAS items are displayed in Fig. 2. Tramadol was not significantly different from placebo on any VAS item; however, dose-related increases for ratings of Bad Effects and Feel Sick were seen, such that tramadol doses of 200 and 400 mg appeared similar to naloxone. For High, Good Effects, and Like Drug, tramadol peak ratings were low in intensity (all means less than 12) in contrast to morphine 15 mg, which produced mean peak ratings greater than 20 on these measures.

Mean peak visual analog scale drug effect scores. Bars indicate ±1 SEM. A darkened symbol indicates a significant difference from placebo. a, Significant difference from naloxone 0.2 mg. b, Significant difference from morphine 15 mg. Morphine and naloxone were administered IM; tramadol was administered PO
Drug class identifications
Table 1 displays the percentage of drug class identification responses for each drug condition. Tramadol 50 and 100 mg doses were most frequently identified as placebo, while higher tramadol doses were increasingly identified as not placebo (>50%) for each dose. Opioid agonist and antagonist identifications were the most common active drug classes identified for tramadol 200 and 400 mg. There was a wide range in patterns of subjects’ drug identifications during tramadol 200- and 400-mg sessions. For example, some subjects identified tramadol 200 mg as having drug effects most similar to an opioid antagonist (n = 3), antidepressant (n = 1), or placebo (n = 3), while others identified a combination of drug effects (opioid agonist and antagonist [n = 1] and opioid agonist and other [n = 2]). Identifications of tramadol 200 mg did not reliably predict the pattern of identifications for tramadol 400 mg. That is, of those subjects who identified tramadol 200 mg as primarily either an opioid agonist or antagonist, only 50% identified tramadol 400 mg more frequently in that same drug class. In addition, tramadol 200 and 400 mg antagonist identifications typically occurred earlier than agonist identifications. Subjects first identified these doses as an antagonist starting at a mean time of 34 min (range = 15–45 min) and as an agonist at 81 min (range = 15–135 min).
Opioid adjective rating questionnaire and modified Himmelsbach scale
Figure 3 illustrates the time course of drug effects on the opioid adjective rating questionnaire and modified Himmelsbach scale. On the observer-rated agonist scale (top left panel), only morphine 15 mg produced significantly higher scores compared to placebo, although these were intermittent effects. On the participant-rated agonist scale (top right panel), morphine 15 mg produced higher scores compared to placebo that were sustained from 45 to 150 min after drug administration. Naloxone and tramadol did not produce sustained changes on this scale. On the observer-rated antagonist adjective scale (middle left panel), morphine and tramadol produced evidence of withdrawal suppression that continued through the last time point; however, morphine immediately reduced antagonist adjective scale scores while tramadol 200 and 400 mg reduced scores beginning at 120 and 90 min, respectively.

Time course of mean opioid adjective agonist and antagonist scale scores and modified Himmelsbach scale score. SEM bars not shown to preserve clarity of figure. A darkened symbol indicates a significant difference from placebo at that time point
Naloxone 0.2 mg produced significant increases on participant-rated antagonist scale scores (middle right panel) over time that peaked within 1 h of drug administration and then dissipated, similar to its effects on the observer-rated antagonist scale and consistent with naloxone-precipitated opioid withdrawal. Tramadol 200 and 400 also decreased participant-rated antagonist scale scores, but only tramadol 400 mg was significantly different from placebo. Consistent with the pattern of results on the antagonist adjective scales, total scores on the modified Himmelsbach scale (bottom panel) demonstrated significant decreases compared to placebo for tramadol 200 and 400 mg and morphine 15 mg.
Results from peak maximum increase and decrease change from baseline analyses are presented for each drug condition in Table 2. Morphine 15 mg and naloxone 0.2 mg produced typical opioid agonist and antagonist effects, respectively. For instance, compared to placebo, morphine 15 mg produced significantly greater maximum decreases on the observer- and participant-rated antagonist adjective scale, and naloxone 0.2 mg produced significantly greater maximum increases on the observer-rated antagonist adjective scale. In contrast, tramadol was not significantly different from placebo on these measures.
Physiologic measures
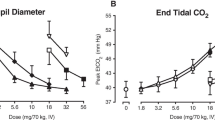
There were no clinically significant vital sign abnormalities during any session. Time course data for systolic and diastolic blood pressure, heart rate, and pupil diameter are illustrated in Fig. 4. Compared to placebo, morphine 15 mg significantly decreased systolic and diastolic blood pressure along with pupil diameter. Naloxone 0.2 mg increased heart rate at three time points relative to placebo. Tramadol produced few significant differences compared to placebo. When significant tramadol effects were present, they were intermittent. For example, compared to placebo, tramadol 400 mg increased pupil diameter at 45, 75, and 90 min after drug administration but then decreased pupil diameter at 210 min.

Time course of mean systolic and diastolic blood pressure, pulse, and pupil diameter. SEM bars not shown to preserve clarity of figure. A darkened symbol indicates a significant difference from placebo at that time point
Peak maximum change from baseline analyses showed that naloxone 0.2 mg increased peak heart rate by 2.8 bpm, which was significantly greater than placebo (−1.6 bpm) and tramadol 200 and 400 mg (−1.4 and −1.0 bpm, respectively). There were no other significant drug effects compared to placebo for peak maximum or minimum analyses. There were no significant results for skin temperature, and oxygen saturation changes were small in magnitude across all drug conditions (range = −1.1–1.0%).
Psychomotor performance measures
Peak change data for psychomotor performance measures are shown in Table 2. Tramadol 400 mg produced significantly greater peak increases and decreases in DSST preperformance estimates compared to placebo, indicating that participants believed they were going to do better on the DSST after receiving this tramadol dose. However, DSST accuracy was not significantly better compared to placebo. Tramadol effects on the remaining psychomotor measures, in general, appeared most similar to placebo or morphine and most different from naloxone. For example, on trails B, peak increases in errors and line length were smaller in magnitude for all tramadol doses compared to naloxone 0.1 mg. Morphine 15 mg produced a smaller peak increase in sequence errors on trails B and smaller peak decreases on circular lights and on the DSST (number of trials attempted and accuracy) compared to placebo.
Time-to-peak effects
The time-to-peak analyses revealed a different time–action function for opposing pharmacodynamic effects. That is, the time to tramadol’s peak maximum Good Effects occurred later than the time to tramadol’s peak maximum Bad Effects. For the tramadol 400 mg condition, peak Good Effects occurred at 76.5 min, while peak Bad Effects occurred at 37.5 min. Overall, analyses of time-to-peak effects demonstrated no significant differences between tramadol and morphine or tramadol and naloxone on VAS items, the opioid adjective rating questionnaires, and the modified Himmelsbach scale.
Discussion
This study evaluated the withdrawal suppression efficacy and subjective, physiologic, and psychomotor/cognitive effects of tramadol among opioid-dependent volunteers maintained on morphine (15 mg SC QID) and experiencing spontaneous opioid withdrawal after a double-blind substitution of morphine dosing with placebo. The acute effects of single oral doses of tramadol were compared to placebo and IM morphine and naloxone. Both morphine and naloxone produced dose-related increases in typical opioid agonist and antagonist effects, respectively, with the highest dose under each control condition consistently resulting in significant differences compared to placebo. Tramadol drug effects varied between subjects (as evidenced by drug class identification responses and wide standard error bars on VAS measures in Fig. 2) and were both time and dose dependent. Tramadol 50 and 100 mg produced effects most similar to placebo across all measures, while the 200- and 400-mg conditions produced evidence of opioid withdrawal suppression without significant subjective opioid agonist-like effects.
The onset of tramadol’s withdrawal suppression was later than that seen with morphine (Fig. 3), and the time to peak suppression was delayed approximately 30–45 min compared to morphine, although this difference was not statistically significant. In part, this delay in withdrawal suppression may reflect the differing routes of drug administration used in this study (oral tramadol vs IM morphine). However, oral tramadol also produces delayed drug action compared to other orally administered opioids like oxycodone (Epstein et al. 2006). In that study, acute doses of oral tramadol produced peak “feel the drug” and “liking” ratings along with miosis at least 1 h later compared to oral oxycodone. This suggests the delay in tramadol-induced withdrawal suppression seen in the current study may not be due only to differences in route of administration but may be related to its conversion to the active metabolite, M1. This conversion is hepatic and is mediated by the P450 2D6 enzyme system. Notably, it has been shown that adults who are poor metabolizers of tramadol, with respect to the polymorphic isoenzyme cytochrome P450 2D6 (CYP2D6), do not develop miosis after tramadol ingestion in contrast to those who have intact CYP2D6 (Fliegert et al. 2005). In addition, M1 has a T max of 2 h (Rouini et al. 2006), which is consistent with the delay in onset of significant withdrawal suppression in the current study and time to maximum withdrawal suppression.
The withdrawal suppression procedure employed in this study was adapted from the 24-h substitution procedure developed in the mid-1950s at the US Public Health Service Addictions Research Center in Lexington, KY (Jasinski 1977). That procedure maintained subjects on morphine (typically 15–60 mg SC QID) and substituted three consecutive doses of test compound (in lieu of morphine maintenance doses) to determine whether the test compound doses could suppress the emergence of opioid withdrawal (Fraser et al. 1960; Fraser and Rosenberg 1964; Jasinski et al. 1975, 1971). In contrast, the current study substituted two placebo doses (in lieu of morphine maintenance doses) before administering a single dose of the study drug. This allowed for the evaluation of the withdrawal suppression efficacy of single doses of the test drug in the context of spontaneous opioid withdrawal. The confirmed presence of mild opioid withdrawal before drug administration and the use of the opioid agonist and antagonist control conditions in this study suggest that this procedure may be useful in future laboratory studies aimed at testing the efficacy of single doses of compounds for the treatment of opioid withdrawal.
Despite the efficacy of tramadol in suppressing withdrawal, it did not elicit significant endorsement of feeling high, drug liking, or good effects that would suggest robust opioid agonist effects (Figs. 1 and 2). These results are in contrast to the significant increases produced by morphine (15 mg) on these measures and suggest that acute doses of tramadol have low abuse liability when administered in the context of opioid withdrawal. Even in the context of nonwithdrawal states (Epstein et al. 2006), oral tramadol has delayed opioid agonist effects that are suggestive of a more favorable abuse liability profile compared to prototypic opioids that have more rapid onsets of opioid effects (Balster and Bigelow 2003).
The relative lack of subjective opioid agonist-like effects (i.e., drug liking) produced by tramadol in the context of its withdrawal suppression efficacy is interesting. Tramadol has a complex pharmacology. It is possible that tramadol is not suppressing opioid withdrawal solely through its opioid agonist actions. It is possible that the acute effects of tramadol on the monoamine system, which are often aversive, might be countering or dampening opioid agonist subjective effects. With regard to the possibility that withdrawal suppression through nonopioid mechanisms might occur, it is well known that nonopioid drugs such as clonidine, a presynaptic alpha-2 adrenoreceptor agonist, can reduce opioid withdrawal symptoms by decreasing noradrenergic outflow that is associated with opioid withdrawal (Collins et al. 2005; Singewald and Philippu 1998; Umbricht et al. 2003). Interestingly, tramadol administration in rats activates the presynaptic alpha-2 adrenoreceptor resulting in suppression of noradrenergic activity (Berrocoso et al. 2006). Theoretically, this suppression could decrease opioid withdrawal symptoms, but tramadol did not produce significant decreases in blood pressure in this study, which is characteristic of alpha-2 adrenoreceptor agonists (Walsh et al. 2003).
Tramadol (200 and 400 mg) produced initial dose-related increases in ratings of Bad Effects and Feel Sick, which peaked and dissipated by 2 h postdrug administration, before the onset of withdrawal suppression (Fig. 1). These initial aversive effects may have countered or dampened subsequent opioid agonist subjective effects and may be secondary to tramadol’s dose-related inhibition of the serotonin and norepinephrine transporter (Barann et al. 2006; Raffa et al. 1992, 1993), which commonly produces side effects such as nausea and gastrointestinal distress (Schatzberg et al. 1997). Although tramadol was generally well tolerated, two subjects vomited after receiving tramadol 200 and 400 mg, and eight subjects reported increased ratings of “stomach turning” and “sick to stomach” after ingestion of these tramadol doses. Previous studies have reported similar side effects with increasing acute doses of tramadol (Bodalia et al. 2003; Gutstein and Akil 2001; Preston et al. 1991). Thus, these initial unpleasant effects may have influenced the subjective experience of the drug even once these effects dissipated. While these initial negative effects may protect against the abuse of tramadol, tolerance may develop to these side effects as they do with other monoamine reuptake inhibitors (e.g., serotonin-selective reuptake inhibitors) after chronic daily dosing.
The initial unpleasant effects of tramadol also could be evidence of it acting as a partial agonist (i.e., further displacing morphine from the mu-opioid receptor), but this seems unlikely for two reasons. First, tramadol and M1’s affinity for the mu-opioid receptor is more than 1,000× and 10× lower, respectively, than that of morphine (Frink et al. 1996; Hennies et al. 1988; Lai et al. 1996). Second, clinical laboratory studies maintaining opioid-dependent adults on 30 and 60 mg of methadone have failed to demonstrate that acute doses of tramadol can precipitate opioid withdrawal (Cami et al. 1994; Carroll et al. 2006).
The negative effects of tramadol may make it unappealing to potential patients, but it is possible that these effects could be avoided if tramadol was administered in smaller doses such as 50–100 mg. This smaller dose could be administered more frequently (e.g., every 4–6 h) throughout the day so that at least 200 mg, the threshold dose in the current study that produced evidence of opioid withdrawal suppression, was ingested. This is a dosing strategy that was used in several retrospective studies that reported modest efficacy of tramadol in the treatment of opioid withdrawal (Sobey et al. 2003; Tamaskar et al. 2003; Threlkeld et al. 2006).
Tramadol produced a favorable safety profile; there were no clinically significant vital sign abnormalities, and psychomotor/cognitive performance did not worsen after tramadol ingestion. Only tramadol 400 mg produced significant changes in pupil size. Initially, there was mydriasis significantly greater than that seen with placebo (Fig. 4), consistent with norepinephrine reuptake blockade. Later (at 210 min), there was evidence of miosis. The delay in tramadol’s miotic effects is likely secondary to the delay in metabolism to the M1 metabolite as has been described previously (Epstein et al. 2006; Knaggs et al. 2004; Zacny 2005).
There are limitations to this study. The duration of withdrawal suppression produced by tramadol was not determined because suppression continued through the last time point (210 min) without evidence of dissipation. Thus, peak suppression of tramadol may have occurred later. Without these data, it is difficult to determine the relative potency of tramadol vs morphine in suppressing withdrawal. In addition, the current study evaluated withdrawal suppression efficacy among adults with a low-moderate amount of opioid physical dependence (60 mg of morphine daily). Future work will need to evaluate the withdrawal suppression efficacy of tramadol with higher levels of opioid physical dependence, over longer periods of time and under conditions of chronic dosing.
In summary, the present study demonstrated that acute oral tramadol doses of 200 and 400 mg can suppress both subjective and observer ratings of opioid withdrawal among adults with a mild to moderate level of opioid physical dependence. This suppression is preceded by transient and dose-related increases in unpleasant side effects, which may protect against the abuse of tramadol and may account for why withdrawal suppression effects were not accompanied by typical opioid agonist subjective effects. These results highlight tramadol’s activity via both opioid and nonopioid mechanisms of actions. Given positive results from previous retrospective reports of tramadol in the treatment of opioid withdrawal and these results here suggesting that at least 200 mg of tramadol is necessary to produce modest opioid suppression, there are sufficient data to evaluate prospectively the efficacy of tramadol as a medication for the treatment of opioid withdrawal.
References
Balster RL, Bigelow GE (2003) Guidelines and methodological reviews concerning drug abuse liability assessment. Drug Alcohol Depend 70:S13–S40
Barann M, Urban B, Stamer U, Dorner Z, Bonisch H, Bruss M (2006) Effects of tramadol and O-demethyl-tramadol on human 5-HT reuptake carriers and human 5-HT3A receptors: a possible mechanism for tramadol-induced early emesis. Eur J Pharmacol 531:54–58
Barth H, Durra S, Giertz H, Goroll D, Flohe L (1987) Long-term administration of the centrally acting analgesic tramadol did not induce dependence or tolerance. Pain 4:S231
Berrocoso E, Mico JA, Ugedo L (2006) In vivo effect of tramadol on locus coeruleus neurons is mediated by alpha2-adrenoceptors and modulated by serotonin. Neuropharmacology 51:146–153
Bodalia B, McDonald CJ, Smith KJ, O’Brien C, Cousens L (2003) A comparison of the pharmacokinetics, clinical efficacy, and tolerability of once-daily tramadol tablets with normal release tramadol capsules. J Pain Symptom Manage 25:142–149
Cami J, Lamas X, Farre M (1994) Acute effects of tramadol in methadone-maintained volunteers. Drugs 47(Suppl 1):39–43
Carroll CP, Walsh SL, Bigelow GE, Strain EC, Preston KL (2006) Assessment of agonist and antagonist effects of tramadol in opioid-dependent humans. Exp Clin Psychopharmacol 14:109–120
Cicero TJ, Adams EH, Geller A, Inciardi JA, Munoz A, Schnoll SH, Senay EC, Woody GE (1999) A postmarketing surveillance program to monitor Ultram (tramadol hydrochloride) abuse in the United States. Drug Alcohol Depend 57:7–22
Cicero TJ, Inciardi JA, Adams EH, Geller A, Senay EC, Woody GE, Munoz A (2005) Rates of abuse of tramadol remain unchanged with the introduction of new branded and generic products: results of an abuse monitoring system, 1994–2004. Pharmacoepidemiol Drug Saf 14:851–859
Collart L, Luthy C, Favario-Constantin C, Dayer P (1993) Duality of the analgesic effect of tramadol in humans. Schweiz Med Wochenschr 123:2241–2243
Collins ED, Kleber HD, Whittington RA, Heitler NE (2005) Anesthesia-assisted vs buprenorphine- or clonidine-assisted heroin detoxification and naltrexone induction: a randomized trial. JAMA 294:903–913
Desmeules JA, Piguet V, Collart L, Dayer P (1996) Contribution of monoaminergic modulation to the analgesic effect of tramadol. Br J Clin Pharmacol 41:7–12
Enggaard TP, Poulsen L, Arendt-Nielsen L, Brosen K, Ossig J, Sindrup SH (2006) The analgesic effect of tramadol after intravenous injection in healthy volunteers in relation to CYP2D6. Anesth Analg 102:146–150
Epstein DH, Preston KL, Jasinski DR (2006) Abuse liability, behavioral pharmacology, and physical-dependence potential of opioids in humans and laboratory animals: lessons from tramadol. Biol Psychol 73:90–99
Fliegert F, Kurth B, Gohler K (2005) The effects of tramadol on static and dynamic pupillometry in healthy subjects—the relationship between pharmacodynamics, pharmacokinetics and CYP2D6 metaboliser status. Eur J Clin Pharmacol 61:257–266
Fraser HF, Isbell H, Vanhorn GD (1960) Human pharmacology and addiction liability of norcodeine. J Pharmacol Exp Ther 129:172–177
Fraser HF, Rosenberg DE (1964) Studies on the human addiction liability of 2¢-hydroxy-5-9-dimethyl-2-(3,3-dimethylallyl)-6,7-benzomorphan (Win 20,228): a weak narcotic antagonist. J Pharmacol Exp Ther 143:149–156
Friderichs E, Felgenhauer F, Jongschaap P, Osterloh G (1978) Pharmacological studies on analgesia, dependence on and tolerance of tramadol, a potent analgetic drug (author’s transl). Arzneimittelforschung 28:122–134
Frink MC, Hennies HH, Englberger W, Haurand M, Wilffert B (1996) Influence of tramadol on neurotransmitter systems of the rat brain. Arzneimittelforschung 46:1029–1036
Gillen C, Haurand M, Kobelt DJ, Wnendt S (2000) Affinity, potency and efficacy of tramadol and its metabolites at the cloned human mu-opioid receptor. Naunyn Schmiedeberg’s Arch Pharmacol 362:116–121
Griffiths RR, Bigelow GE, Liebson I (1983) Differential effects of diazepam and pentobarbital on mood and behavior. Arch Gen Psychiatry 40:865–873
Gutstein H, Akil H (2001) Opioid analgesics. In: Hardman J, Limbird L, Gilman A (eds) Goodman and Gilman’s the pharmacological basis of therapeutics. McGraw-Hill, New York, pp 569–619
Hennies HH, Friderichs E, Schneider J (1988) Receptor binding, analgesic and antitussive potency of tramadol and other selected opioids. Arzneimittelforschung 38:877–880
Inciardi JA, Cicero TJ, Munoz A, Adams EH, Geller A, Senay EC, Woody GE (2006) The Diversion of Ultram, Ultracet, and generic tramadol HCL. J Addict Dis 25:53–58
Jasinski DR (1977) Assessment of the abuse potentiality of morphinelike drugs (methods used in man). In: Martin WR (ed) Drug addiction I. Springer, Berlin, pp 197–258
Jasinski DR, Griffith JD, Carr CB (1975) Etorphine in man. I. Subjective effects and suppression of morphine abstinence. Clin Pharmacol Ther 17:267–272
Jasinski DR, Martin WR, Hoeldtke R (1971) Studies of the dependence-producing properties of GPA-1657, profadol, and propiram in man. Clin Pharmacol Ther 12:613–649
Jasinski DR, Preston KL, Sullivan JT, Testa M (1993) Abuse potential of oral tramadol. NIDA Res Monogr 132:103
Knaggs RD, Crighton IM, Cobby TF, Fletcher AJ, Hobbs GJ (2004) The pupillary effects of intravenous morphine, codeine, and tramadol in volunteers. Anesth Analg 99:108–112
Kolb L, Himmelsbach CK (1938) Clinical studies of drug addiction. III. A critical review of the withdrawal treatments with method of evaluating abstinence syndromes. Am J Psychiatry 94:759–797
Lai J, Ma SW, Porreca F, Raffa RB (1996) Tramadol, M1 metabolite and enantiomer affinities for cloned human opioid receptors expressed in transfected HN9.10 neuroblastoma cells. Eur J Pharmacol 316:369–372
Lehmann KA, Kratzenberg U, Schroeder-Bark B, Horrichs-Haermeyer G (1990) Postoperative patient-controlled analgesia with tramadol: analgesic efficacy and minimum effective concentrations. Clin J Pain 6:212–220
McLeod D, Griffiths RR, Yingling J (1982) An automated version of the digit symbol substitution test (DSST). Behav Res Methods Instrum Comput 14:463–466
Murano T, Yamamoto H, Endo N, Kudo Y, Okada N, Masuda Y, Yano I (1978) Studies on dependence on tramadol in rats. Arzneimittelforschung 28:152–158
Preston KL, Jasinski DR (1989) Effects of tramadol in humans: assessment of its abuse potential. NIDA Res Monogr 95:392
Preston KL, Jasinski DR, Testa M (1991) Abuse potential and pharmacological comparison of tramadol and morphine. Drug Alcohol Depend 27:7–17
Raffa RB, Friderichs E, Reimann W, Shank RP, Codd EE, Vaught JL (1992) Opioid and nonopioid components independently contribute to the mechanism of action of tramadol, an ‘atypical’ opioid analgesic. J Pharmacol Exp Ther 260:275–285
Raffa RB, Friderichs E, Reimann W, Shank RP, Codd EE, Vaught JL, Jacoby HI, Selve N (1993) Complementary and synergistic antinociceptive interaction between the enantiomers of tramadol. J Pharmacol Exp Ther 267:331–340
Reitan R (1958) Validity of the trail making test as an indicator of organic brain damage. Percept Mot Skills 8:271–276
Rouini MR, Ardakani YH, Soltani F, Aboul-Enein HY, Foroumadi A (2006) Development and validation of a rapid HPLC method for simultaneous determination of tramadol, and its two main metabolites in human plasma. J Chromatogr B 830:207–211
Salehi M, Amanatkar M, Barekatain M (2005) Tramadol versus methadone for the management of acute opioid withdrawal: an add-on study. J Res Med Sci 11:185–189
Schatzberg A, Cole J, DeBattista C (1997) Manual of clinical psychopharmacology, 3rd edn. American Psychiatric, Washington, DC
Senay EC, Adams EH, Geller A, Inciardi JA, Munoz A, Schnoll SH, Woody GE, Cicero TJ (2003) Physical dependence on Ultram (tramadol hydrochloride): both opioid-like and atypical withdrawal symptoms occur. Drug Alcohol Depend 69:233–241
Singewald N, Philippu A (1998) Release of neurotransmitters in the locus coeruleus. Prog Neurobiol 56:237–267
Sobey PW, Parran TV Jr, Grey SF, Adelman CL, Yu J (2003) The use of tramadol for acute heroin withdrawal: a comparison to clonidine. J Addict Dis 22:13–25
Spitzer RL, Williams JB, Gibbon M, First MB (1992) The structured clinical interview for DSM-III-R (SCID). I. History, rationale, and description. Arch Gen Psychiatry 49:624–629
Stoller KB, Bigelow GE, Walsh SL, Strain EC (2001) Effects of buprenorphine/naloxone in opioid-dependent humans. Psychopharmacology (Berl) 154:230–242
Strain EC, Stoller K, Walsh SL, Bigelow GE (2000) Effects of buprenorphine versus buprenorphine/naloxone tablets in non-dependent opioid abusers. Psychopharmacology (Berl) 148:374–383
Tamaskar R, Parran TV Jr, Heggi A, Brateanu A, Rabb M, Yu J (2003) Tramadol versus buprenorphine for the treatment of opiate withdrawal: a retrospective cohort control study. J Addict Dis 22:5–12
Threlkeld M, Parran TV, Adelman CA, Grey SF, Yu J (2006) Tramadol versus buprenorphine for the management of acute heroin withdrawal: a retrospective matched cohort controlled study. Am J Addict 15:186–191
Umbricht A, Hoover DR, Tucker MJ, Leslie JM, Chaisson RE, Preston KL (2003) Opioid detoxification with buprenorphine, clonidine, or methadone in hospitalized heroin-dependent patients with HIV infection. Drug Alcohol Depend 69:263–272
Walsh SL, Strain EC, Bigelow GE (2003) Evaluation of the effects of lofexidine and clonidine on naloxone-precipitated withdrawal in opioid-dependent humans. Addiction 98:427–439
Woody GE, Senay EC, Geller A, Adams EH, Inciardi JA, Schnoll S, Munoz A, Cicero TJ (2003) An independent assessment of MEDWatch reporting for abuse/dependence and withdrawal from Ultram (tramadol hydrochloride). Drug Alcohol Depend 72:163–168
Yanagita T (1978) Drug dependence potential of 1-(m-methoxyphenyl)-2-dimethylaminomethyl)-cyclohexan-1-ol hydrochloride (tramadol) tested in monkeys. Arzneimittelforschung 28:158–163
Zacny JP (2005) Profiling the subjective, psychomotor, and physiological effects of tramadol in recreational drug users. Drug Alcohol Depend 80:273–278
Acknowledgments
The authors thank Afsheen Siddiqi, Elliot Joseph, Mary Misenhimer, John Yingling, Linda Felch, and the residential nursing staff for assistance in volunteer recruitment, data collection, and analysis. This research was supported by the National Institute on Drug Abuse R01 DA018125, K02 DA00332, and T32 DA07209.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lofwall, M.R., Walsh, S.L., Bigelow, G.E. et al. Modest opioid withdrawal suppression efficacy of oral tramadol in humans. Psychopharmacology 194, 381–393 (2007). https://doi.org/10.1007/s00213-007-0847-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-007-0847-3