
Abstract
Rationale
Paliperidone (9-OH-risperidone) is the main metabolite of the atypical antipsychotic risperidone. While both drugs are potent dopamine (D)2 antagonists, they have quantitative differential affinities for serotonin (5-HT) and norepinephrine (NE) receptor binding sites.
Objectives
The present study aimed to determine if paliperidone exerts distinct effects on 5-HT and NE neuronal activity from those of risperidone.
Materials and methods
Risperidone and paliperidone were administered to Sprague–Dawley rats. Neuronal activity of 5-HT and NE neurons was assessed using in vivo electrophysiology.
Results
Acute administration of risperidone but not paliperidone inhibited the firing of 5-HT neurons, as previously reported. This inhibition was partially antagonized by the NE reuptake inhibitor desipramine, by the 5-HT1A receptor antagonist WAY 100635, and completely reversed when both drugs were given consecutively. Risperidone inhibited the firing of 5-HT neurons after 2 and 14 days of administration, with or without escitalopram. Paliperidone did not alter the firing rate of NE neurons by itself, but it reversed the suppression of NE neurons induced by escitalopram, as it was previously reported for risperidone.
Conclusion
These results indicate that although risperidone and paliperidone share a qualitatively similar receptor binding profile in vitro, they differentially alter the firing of 5-HT and NE neurons in vivo. The capacity of paliperidone to reverse the selective serotonin reuptake inhibitor (SSRI)-induced inhibition of NE neuronal firing, without interfering with the effect of SSRIs of 5-HT neuronal activity, suggests that paliperidone may be a very effective adjunct in SSRI-resistant depression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atypical antipsychotic drugs act as dopamine (D)2 and serotonin (5-HT)2A receptor antagonists. Their affinity for 5-HT2A receptors is, however, higher than for D2 receptors. In contrast, the classical antipsychotic haloperidol is a potent D2 receptor antagonist, and its affinity to 5-HT2A receptors is insignificant (Schotte et al. 1996). Addition of atypical antipsychotics to the regimen of selective serotonin reuptake inhibitor (SSRI)-resistant depressed patients is an effective therapeutic option (Papakostas et al. 2005; Rapaport et al. 2006; Simon and Nemeroff 2005; Tohen et al. 2003). Haloperidol is, however, devoid of intrinsic effectiveness in depression. Because both classical and atypical antipsychotics are D2 antagonists, it was suggested that this beneficial effect of atypical antipsychotics in SSRI-resistant depression is mediated via their 5-HT2A receptor antagonistic property (Szabo and Blier 2002).
It was previously shown that 5-HT2A receptors play an important role in the interaction between 5-HT and NE systems in the brain (Szabo and Blier 2001a). The 5-HT2A receptors are expressed on γ-aminobutyric (GABA) cells innervating NE neurons in the locus coeruleus (LC). The elevation in synaptic availability of 5-HT by SSRIs activates GABA neurons through excitatory 5-HT2A receptors, which then results in the inhibition of NE neuronal activity (Szabo and Blier 2001b). Atypical antipsychotic drugs, because of their ability to block 5-HT2A receptors, reverse the SSRI-induced inhibition of the firing rate and burst activity of NE neurons, as it was demonstrated for the combination of the atypical antipsychotics olanzapine and risperidone with the SSRIs fluoxetine and escitalopram, respectively (Dremencov et al. 2007; Seager et al. 2005). In support of this assertion, the selective 5-HT2A antagonist M100907 but not the 5-HT2C antagonist SB242084 nor haloperidol antagonized the effect of escitalopram on NE neuronal firing activity (Dremencov et al. 2007).
Risperidone is a potent 5-HT2A, 5-HT2C, 5-HT1D, D2, histamine (H1), α1-, and α2-adrenergic receptor antagonist (Richelson and Souder 2000). Acute administration of risperidone elevates extracellular 5-HT levels in the rat dorsal raphe nucleus (DRN) and prefrontal cortex and decreases the firing activity of 5-HT neurons (Hertel et al. 1997a; Hertel et al. 2001). The ability of risperidone to elevate extracellular 5-HT was explained by its 5-HT1D antagonistic property (Hertel et al. 1999). The risperidone-induced suppression of the firing rate of 5-HT neurons was explained by the elevation of extracellular 5-HT in the DRN, which leads to the activation of 5-HT1A autoreceptors (Hertel et al. 2001). Given the α2-adrenoceptor antagonistic property of risperidone, this drug has the potential to elevate the firing activity of NE neurons. Repeated administration of risperidone slightly increased NE neuronal firing activity in one study (Nasif et al. 2000). However, another study showed that sustained administration of risperidone did not alter the firing activity of NE neurons by itself but did so when given in rats pretreated with a 5-HT synthesis inhibitor (+)p-chlorophenylalanine (Dremencov et al. 2007).
Paliperidone (9-OH-risperidone) is the major metabolite of risperidone (Ereshefsky and Lacombe 1993; Riedel et al. 2005). It was recently approved by the US Food and Drug Administration as a new atypical antipsychotic drug. Paliperidone and risperidone have similar affinity for 5-HT1D, 5-HT2B, 5-HT7, D2, and D3 receptors (Leysen et al. 1994; Richelson and Souder 2000; Schotte et al. 1995). According to one study, paliperidone has a weaker affinity for 5-HT2A and α1/α2-adrenergic receptors than risperidone (Richelson and Souder 2000). Other studies demonstrate that risperidone has similar in vitro binding potential to these receptors. However, these studies show that the binding of risperidone in vitro to 5-HT2A and α1/α2-adrenergic receptors is higher than that of paliperidone (Leysen et al. 1994; Schotte et al. 1995). The aim of the current study was to compare the effects of risperidone and paliperidone on 5-HT and NE neuronal firing activity in vivo to determine whether its quantitatively different in vitro affinity profile for the three abovementioned receptors was physiologically significant.
Materials and methods
Animals
The experiments were carried out in male Sprague–Dawley rats (Charles River, St. Constant, QC) weighing between 300 and 350 g and kept under standard laboratory conditions (12:12 light–dark cycle with access to food and water ad libitum). All animal procedures were approved by the Ottawa Health Research Institute Animal Care Committee and were carried out in accordance with the guidelines of the Canadian Council on Animal Care.
Drugs
Escitalopram (Lundbeck, Copenhagen, DK) was dissolved in distilled water and administered via osmotic minipumps (Alza, Palo Alto, CA). The pumps were implanted subcutaneously under isoflurane (Abbot, Montreal, QC) anesthesia. Risperidone and paliperidone (Janssen, Titusville, NJ) were dissolved in 10% tartaric acid (Sigma, St. Louis, MO) and then in distilled water (1:100). Desipramine and WAY 100635 (Sigma) were dissolved in distilled water.
Treatments
Escitalopram was administered via osmotic minipumps for 2 and 14 days at a daily dosage of 10 mg kg−1 day−1. Control animals were implanted with minipumps containing distilled water. Acute administration of risperidone and paliperidone were performed using two and five cumulative intravenous (i.v.) injections of 0.2 mg/kg, respectively. Repeated administration of risperidone and paliperidone were performed using subcutaneous injections of 1 mg kg−1 day−1 for 2 and 14 days, alone or in combination with escitalopram. The doses of escitalopram and risperidone were chosen on the basis of previous experiments (Dremencov et al. 2007).
Electrophysiological experiments
Rats were anaesthetized with chloral hydrate (Sigma; 400 mg/kg, intraperitoneal) and mounted in a stereotaxic apparatus (David Kopf Instuments, Tujunga, CA). Supplemental doses were given to prevent any nociceptive reaction to pinching of the hind paw. Body temperature was maintained at 37°C throughout the experiments utilizing a thermostat-controlled heating pad. Extracellular unitary recordings were conducted with single-barreled glass electrodes filled with a 2-M NaCl solution. Their impedance range was between 4 and 6 MΩ. Serotonin neurons were recorded with micropipettes lowered at 1.0 to 1.5 mm anterior to the interaural line. Spontaneously active 5-HT neurons of the DRN were identified using the following criteria: regular firing rate (0.5–2.5 Hz) and positive action potential of long duration. NE neurons of the LC were recorded with micropipettes lowered at 0.8 to 1.1 mm posterior to the interaural and 0.5 to 0.7 mm lateral to the midline. Spontaneously active NE neurons were identified using the following criteria: regular firing rate (0.5–5.0 Hz) and positive action potential of long duration (0.8–1.2 ms) exhibiting a characteristic burst discharge in response to nociceptive pinch of the contralateral hind paw. First locating the mesencephalic fifth motor nucleus neurons, which respond to lower jaw depression, and then moving the electrode medially to record LC NE neurons provided additional indication for the validity of the site of recordings. Both NE and 5-HT neurons were recorded for at least 1 min after stabilization to establish their basal firing rate.
Characterization of pharmacological basis for the effect of risperidone on the firing activity of 5-HT neurons
Rats were anesthetized, mounted in a stereotaxic apparatus, and a gauge-26 catheter was inserted in a lateral tail vein. The electrodes were lowered into the DRN. After a 5-HT neuron was identified and its firing activity was deemed stable, it was recorded for at least 1 min. Risperidone was given intravenously at the dose of 0.2 mg/kg dissolved in approximately 0.1 ml of saline. When the firing rate of the neuron reached a plateau after this injection, it was recorded for at least 1 min. An additional dose of risperidone (0.2 mg/kg) was subsequently injected. The NE reuptake inhibitor desipramine (5 mg/kg, i.v.) or the selective 5-HT1A receptor antagonist WAY 100635 (0.05, 0.10, 0.15, and 0.20 mg/kg, i.v.) were then given in an attempt to reverse risperidone-induced inhibition of the firing activity of the neuron. In the animals that received desipramine, WAY 100635 (0.05 mg/kg, i.v.) was injected after the desipramine administration, and in the animals administered WAY 100635, desipramine (5 mg/kg) was given after WAY 100635 administration.
Analysis of burst firing
The firing patterns of NE neurons were analyzed by interspike interval burst analysis, using the Spike 2 program (Cambridge Electronic Design, Cambridge, UK). The onset of a burst was defined as the concurrence of two spikes with interspike interval shorter than 0.08 s. The termination of burst was defined as an interspike interval of 0.16 s or longer (Dawe et al. 2001).
Statistical analysis
All results were expressed as means (±SEM) of single neuron values. Statistical comparisons of the differences in neuronal firing resulted from the 2 or 14-day administration of escitalopram, risperidone, or paliperidone were carried out using two-way analysis of variance (ANOVA). Statistical comparisons of the differences in 5-HT neuronal firing resulted from the acute consecutive i.v. administration of risperidone, desipramine, and WAY 100635 were carried out using the ANOVA for repeated measurements. Statistically significant differences were determined using the p < 0.05 criterion.
Results
Effects of acute administrations of risperidone and paliperidone on 5-HT neuronal firing activity
The firing rate of 5-HT neurons in control rats was 0.92 + 0.10 Hz (n = 4 rats). The rate and the pattern of the firing were typical for 5-HT neurons, as previously described (Aghajanian and Vandermaelen 1982; Hajos et al. 1995). The acute (i.v.) administration of risperidone of 0.2 and 0.4 mg/kg of risperidone dose-dependently decreased the firing activity of 5-HT neurons (Figs. 1 and 2), as expected (Hertel et al. 1997b). Paliperidone, however, failed to inhibit 5-HT neurons at the cumulative dose of 0.4 mg/kg nor at higher doses (Fig. 3). The subsequent administration of 0.10, 0.15, and 0.20 mg/kg of the 5-HT1A receptor antagonist WAY 100635 partially and dose-dependently antagonized the risperidone-induced inhibition of 5-HT neurons (Figs. 1a and 2a). However, the complete reversal of risperidone-induced inhibition of 5-HT neurons was observed only when the NE reuptake inhibitor desipramine (5 mg/kg) was subsequently given (n = 5 rats). When desipramine (5 mg/kg) was first given after risperidone (Figs. 1b and 2b), it partially antagonized risperidone-induced inhibition of the 5-HT neuronal firing rate. The dose of 0.05 mg/kg of WAY 100635, given subsequently to desipramine, completely reversed risperidone-induced inhibition of the 5-HT neuronal firing rate (n = 5 rats).

a Representative single-unit recordings from the dorsal raphe nucleus serotonin neuron during the consecutive acute injections of risperidone (two doses of 0.2 mg/kg each, i.v.) and then the selective 5-HT1A receptor antagonist WAY 100635 (four doses of 0.05 mg/kg each) and the norepinephrine reuptake inhibitor desipramine (5 mg/kg, i.v.). b Desipramine was given before WAY 100635

Effect of acute consecutive administrations of risperidone, WAY 100635, and desipramine on the firing activity of serotonin neurons. a The animals were consecutively administered (i.v.) atypical antipsychotics risperidone (two doses of 0.2 mg/kg each), selective 5-HT1A receptor antagonist WAY 100635 (four doses of 0.05 mg/kg each), and norepinephrine reuptake inhibitor desipramine (5 mg/kg). b Other animals were consecutively administered risperidone (two doses of 0.2 mg/kg each), desipramine (5 mg/kg), and WAY 100635 (0.05 mg/kg). Significant effect of the time was observed (F[df 7, 28] = 6.24, p < 0.05 and F[df 4, 16] = 9.71, p < 0.01, respectively). Asterisk, p < 0.05, in comparison with baseline or with 0.2 mg/kg of risperidone; Number sign, p < 0.05, in comparison with 0.4 mg/kg of risperidone

Lack of the effect of acute administration of paliperidone on firing activity of serotonin neurons. The animals (n = 5) were consecutively given five i.v. injections of paliperidone of 0.2 mg/kg each
Effect of repeated administration of risperidone and paliperidone on 5-HT neuronal firing activity in vehicle- and escitalopram-treated rats
A 2-day regimen of 10 mg kg−1 day−1 of escitalopram (n = 5 rats), 1 mg kg−1 day−1 of risperidone (n = 5 rats), and their combination (n = 4 rats) significantly decreased the firing rate of 5-HT neurons (Fig. 5). The 2-day regimen of 1 mg kg−1 day−1 of paliperidone did not alter the firing activity of 5-HT neurons in vehicle- or escitalopram-administered rats (n = 5 rats in each group). The firing rate of 5-HT neurons was normalized after 14 days of escitalopram administration (n = 5 rats), as expected (El Mansari et al. 2005). However, the 14-day administration of risperidone significantly decreased the firing rate of 5-HT neurons (n = 5 rats), as well as its 2-day regimen. Moreover, risperidone coadministered with escitalopram for 14 days prevented the normalization of the 5-HT neuronal firing rate (n = 5 rats). In contrast, the 14-day regimen of paliperidone did not alter the firing rate of 5-HT neurons, neither by itself nor in combination with escitalopram (Figs. 4 and 5, n = 5 rats in each group).

Representative electrode descents from the dorsal raphe nucleus showing the spontaneous 5-HT neuronal firing activity in a control rat (a), and in a rat administered risperidone (b) or paliperidone (c) for 14 days. Each individual histogram represents the number of action potential per 10-s period. The number above each neuronal recording represents the distance in millimeters of the neuron from the brain surface

Effect of the risperidone or paliperidone regimen on the 5-HT neuronal firing rate in the rats administered vehicle or escitalopram. The animals were implanted with minipumps containing the vehicle (water) or escitalopram (10 mg/kg/day) for 2 (a) or 14 days (b) and received no cotreatment (control) or were coadministered (s.c.) risperidone or paliperidone (1 mg kg−1 day−1 each). After 2 days, there was a significant effect of the treatment (escitalopram or vehicle, F[df 1, 243] = 26.16, p < 0.001), cotreatment (risperidone, paliperidone or no cotreatment, F[df 2, 243] = 3.99, p < 0.05), and treatment × cotreatment interaction (F[df 2, 243] = 3.93, p < 0.05). After 14 days, there was a significant effect of cotreatment (F[df 2, 221] = 6.61, p < 0.01). The number of neurons recorded in each group is provided within the histograms. Triple asterisk, p < 0.001, double asterisk, p < 0.01, and asterisk, p < 0.05, in comparison with control animals
Effect of paliperidone on NE neuronal firing activity in vehicle- and escitalopram-treated rats
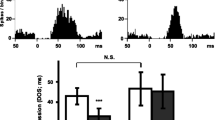
The firing rate of NE neurons in control rats was 1.48 + 0.23 Hz (n = 4 rats). The rate and the pattern of the firing were typical for NE neurons, as previously described (Aghajanian and Vandermaelen 1982; Dremencov et al. 2007; Seager et al. 2005). A 2-day regimen of 10 mg kg−1 day−1 of escitalopram significantly (n = 5) decreased the firing rate (Fig. 6a) and the burst activity (Fig. 7a) of NE neurons. Paliperidone coadministered with escitalopram restored the suppression of the NE neuronal firing rate (n = 7 rats). However, it did not alter the NE neuronal firing rate by itself (n = 5 rats). Burst activity of NE neurons was not attenuated by paliperidone when it was given by itself and in combination with escitalopram. A 14-day regimen of 10 mg kg−1 day−1 of escitalopram (n = 5 rats) significantly decreased the firing rate (Fig. 6b) and burst activity (Fig. 7b) of NE neurons. Paliperidone coadministered with escitalopram restored the suppression of the NE neuronal firing rate (n = 5 rats) and the percentage of neurons exhibiting burst firing. However, it did not alter the NE neuronal firing activity by itself (n = 6 rats).

Effect of the paliperidone regimen on the NE neuronal firing rate in the rats administered vehicle or escitalopram. The animals were implanted with minipumps containing vehicle (water) or escitalopram (10 mg kg−1 day−1) for 2 (a) or 14 days (b) and received no cotreatment (control) or were coadministered (s.c.) paliperidone (1 mg kg−1 day−1). After 2 days, there was a significant effect of the treatment (escitalopram or vehicle, F[df 1, 122] = 14.76, p < 0.001) and cotreatment (paliperidone or no cotreatment, F[df 1, 122] = 8.64, p < 0.01) After 14 days, there was a significant effect of the treatment F[df 1, 165] = 5.45, p < 0.05), cotreatment (paliperidone or no cotreatment, F[df 1, 165] = 17.18, p < 0.001), and treatment × cotreatment interaction (F[df 1, 165] = 3.98, p < 0.05). The number of neurons recorded in each group is provided within the histograms. Triple asterisk, p < 0.001, double asterisk, p < 0.01, in comparison with control animals; triple number sign, p < 0.001, double number sign, p < 0.01, in comparison with animals administered escitalopram alone

Effect of the paliperidone regimen on the burst activity of norepinephrine neurons in the rats administered vehicle or escitalopram. The animals were implanted with minipumps containing vehicle (water) or escitalopram (10 mg kg−1 day−1) for 2 (a) or 14 days (b) and received no cotreatment (control) or were coadministered (s.c.) paliperidone (1 mg kg−1 day−1) for 2 days. After 2 days, there was a significant effect of the treatment (escitalopram or vehicle) on the burst occurrence (F[df 1, 122] = 9.52, p < 0.005), on the percent of spikes occurring in bursts (F[df 1, 122] = 14.67, p < 0.001) and on the percent of neurons exhibits burst firing (F[df 1, 25] = 6.62, p < 0.05). After 14 days, there was a significant effect of the treatment on the percent of spikes occurring in bursts (F[df 1, 165] = 6.62, p < 0.05) and on the percent of neurons exhibits burst firing (F[df 1, 23] = 4.84, p < 0.05) and a significant effect of cotreatment (paliperidone or no cotreatment) on the percent of neurons exhibiting burst firing (F[df 1, 23] = 4.62, p < 0.05). Triple asterisk, p < 0.001, double asterisk, p < 0.01, and asterisk, p < 0.05, in comparison with control animals; number sign, p < 0.05, in comparison with animals administered escitalopram alone
Discussion
The results of the present study showed that subacute administration of risperidone but not of paliperidone suppressed the firing activity of 5-HT neurons. The risperidone-induced inhibition of 5-HT neuronal firing activity was partially antagonized by the 5-HT1A receptor antagonist WAY 100635 and by the NE reuptake inhibitor desipramine. A complete reversal was observed only when both drugs were given. Paliperidone did not alter 5-HT or NE neuronal firing by itself but prevented the escitalopram-induced suppression of firing of NE neurons when the two drugs were coadministered for 2 or 14 days. This ability of paliperidone to reverse the escitalopram-induced suppression of NE neuronal firing is similar to that of risperidone (Dremencov et al. 2007). However, unlike risperidone, paliperidone given for 2 days did not elevate the firing activity of NE neurons in escitalopram-administered rats above the value observed in control animals, and it did not reverse the escitalopram-induced suppression of burst firing of NE neurons after 2 days of coadministration with escitalopram.
The acute administration of risperidone (Figs. 1 and 2) but not of paliperidone (Fig. 3) suppressed the firing activity of 5-HT neurons, as was previously reported (Hertel et al. 1997b). The risperidone-induced suppression of the firing rate of 5-HT neurons was previously explained entirely by the elevation of extracellular 5-HT levels in the DRN resulting in the activation of 5-HT1A autoreceptors (Hertel et al. 1997b). However, the results of the current study indicated that the selective 5-HT1A receptor antagonist WAY 100635, given also at 0.1 mg/kg, did not entirely reverse the risperidone-induced inhibition of 5-HT neuronal firing activity, although the same dose completely reversed the escitalopram-induced inhibition of the 5-HT neuronal firing activity (El Mansari et al. 2005). Only the higher cumulative doses of WAY 100635 (0.15 and 0.20 mg/kg) partially reversed the firing activity of 5-HT neurons. Thus, other receptor(s), such as α1-adrenoceptors, may be involved in risperidone-induced suppression of the 5-HT neuronal firing rate.
It was observed in the current study that the NE reuptake inhibitor desipramine partially antagonized risperidone-induced inhibition of the 5-HT neuronal firing activity and completely reversed it when it was injected after WAY 100635 administration. Desipramine was previously reported to reverse the α1-adrenoceptor antagonist prazosin-mediated inhibition of the 5-HT neuronal firing activity (Gartside et al. 1997). Furthermore, desipramine reversed the inhibitory effect of olanzapine and clozapine, whereas only WAY 100635 reversed the inhibitory effect of atypical antipsychotics ziprasidone and aripiprazole (Sprouse et al. 1999; Stark et al. 2007). It can therefore be concluded that risperidone inhibits the firing activity of 5-HT neurons via both the activation of 5-HT1A autoreceptors and blockage of α1-adrenoceptors, olanzapine and clozapine via the antagonism of α1-adrenoceptors and aripiprazole and ziprasidone via the activation of 5-HT1A autoreceptors. Paliperidone is thus unique among atypical antipsychotic drugs because it does not inhibit the firing of 5-HT neurons (Figs. 3, 4, and 5).
The firing rate of 5-HT neurons, suppressed by escitalopram after 2 days of administration, recovered to the levels of control animals after the 14-day regimen (Fig. 5a), as expected (El Mansari et al. 2005). This normalization of the 5-HT neuronal firing rate after long-term SSRI administration was first explained by the desensitization of the cell body 5-HT autoreceptors (Blier and de Montigny 1983). The 14-day regimen of risperidone significantly suppressed the firing activity of 5-HT neurons similar to the 2-day regimen. Moreover, the 14-day risperidone coadministration with escitalopram prevented the restoration of the 5-HT neuronal firing rate (Fig. 5). Thus, risperidone possibly prevents the desensitization of 5-HT1A autoreceptors or α1-adrenergic heteroreceptors in the rat DRN. Further studies should be performed to clarify the mechanism of the long-term effect of risperidone on 5-HT and NE receptors in the rat brain.
The current study showed (Fig. 5) that risperidone coadministered with escitalopram for 14 days prevented the recovery of the 5-HT neuronal firing activity observed when escitalopram was given alone for a same time course (El Mansari et al. 2005). Paradoxically, risperidone added to the SSRI regimen increases their clinical efficiency (Rapaport et al. 2006; Shelton and Stahl 2004). It might therefore be suggested that risperidone coadministered with SSRIs increases 5-HT neurotransmission independently of the firing of 5-HT neurons. Indeed, risperidone elevates 5-HT levels in the prefrontal cortex via the 5-HT1D receptor-mediated pathway (Hertel et al. 1999).
Paliperidone given alone did not change the firing activity of NE neurons after the 2- or 14-day regimens (Fig. 6). Nevertheless, it reversed the escitalopram-induced suppression of the NE neuronal firing activity, as it was reported for risperidone (Dremencov et al. 2007). However, differently from risperidone, paliperidone did not elevate the firing rate of NE neurons in the LC of escitalopram-administered rats above the value observed in control animals after 2 days of coadministration, as well as not reversing the escitalopram-induced suppression of burst firing of NE neurons (Fig. 7). The ability of risperidone to reverse escitalopram-induced inhibition of burst firing of NE neurons and to elevate the firing rate of NE neurons in the LC of escitalopram-administered animals might be mediated via its 5-HT2A and α2-adrenoceptor antagonistic properties, respectively (Dremencov et al. 2007). Thus, the difference between risperidone and paliperidone in their effect on the NE neuronal firing activity in vivo may be explained by the weaker binding of paliperidone for 5-HT2A and α2-adrenergic receptors observed in vitro (Leysen et al. 1994; Schotte et al. 1995).
Because paliperidone is a major metabolite of risperidone (Ereshefsky and Lacombe 1993; Riedel et al. 2005), animals receiving risperidone are also exposed to paliperidone. However, most of risperidone is metabolized to paliperidone. Nevertheless, a small plasma concentration of risperidone was enough to attenuate the firing of 5-HT neurons.
In summary, paliperidone and risperidone differentially affect the neuronal firing activity of 5-HT and NE neurons in vivo. The capacity of paliperidone to reverse the SSRI-induced inhibition of the NE neuronal firing rate, without the decreasing of the 5-HT neuronal activity like risperidone, suggests that paliperidone may be a very effective adjunct in SSRI-resistant depression.
References
Aghajanian GK, Vandermaelen CP (1982) Intracellular identification of central noradrenergic and serotonergic neurons by a new double labeling procedure. J Neurosci 2:1786–1792
Blier P, de Montigny C (1983) Effects of quipazine on pre- and postsynaptic serotonin receptors: single cell studies in the rat CNS. Neuropharmacology 22:495–499
Dawe GS, Huff KD, Vandergriff JL, Sharp T, O’Neill MJ, Rasmussen K (2001) Olanzapine activates the rat locus coeruleus: in vivo electrophysiology and c-fos immunoreactivity. Biol Psychiatry 50:510–520
Dremencov E, El Mansari M, Blier P (2007) Noradrenergic augmentation of escitalopram response by risperidone: electrophysiologic studies in the rat brain. Biol Psychiatry 61:671–678
El Mansari M, Sanchez C, Chouvet G, Renaud B, Haddjeri N (2005) Effects of acute and long-term administration of escitalopram and citalopram on serotonin neurotransmission: an in vivo electrophysiological study in rat brain. Neuropsychopharmacology 30:1269–1277
Ereshefsky L, Lacombe S (1993) Pharmacological profile of risperidone. Can J Psychiatry 38(Suppl 3):S80–S88
Gartside SE, Umbers V, Sharp T (1997) Inhibition of 5-HT cell firing in the DRN by non-selective 5-HT reuptake inhibitors: studies on the role of 5-HT1A autoreceptors and noradrenergic mechanisms. Psychopharmacology (Berl) 130:261–268
Hajos M, Gartside SE, Sharp T (1995) Inhibition of median and dorsal raphe neurones following administration of the selective serotonin reuptake inhibitor paroxetine. Naunyn-Schmiedeberg’s Arch Pharmacol 351:624–629
Hertel P, Nomikos GG, Schilstrom B, Arborelius L, Svensson TH (1997a) Risperidone dose-dependently increases extracellular concentrations of serotonin in the rat frontal cortex: role of alpha 2-adrenoceptor antagonism. Neuropsychopharmacology 17:44–55
Hertel P, Nomikos GG, Svensson TH (1997b) Risperidone inhibits 5-hydroxytryptaminergic neuronal activity in the dorsal raphe nucleus by local release of 5-hydroxytryptamine. Br J Pharmacol 122:1639–1646
Hertel P, Nomikos GG, Svensson TH (1999) The antipsychotic drug risperidone interacts with auto- and hetero-receptors regulating serotonin output in the rat frontal cortex. Neuropharmacology 38:1175–1184
Hertel P, Lindblom N, Nomikos GG, Svensson TH (2001) Receptor-mediated regulation of serotonin output in the rat dorsal raphe nucleus: effects of risperidone. Psychopharmacology (Berl) 153:307–314
Leysen JE, Janssen PM, Megens AA, Schotte A (1994) Risperidone: a novel antipsychotic with balanced serotonin–dopamine antagonism, receptor occupancy profile, and pharmacologic activity. J Clin Psychiatry 55(Suppl):5–12
Nasif FJ, Cuadra GR, Ramirez OA (2000) Effects of chronic risperidone on central noradrenergic transmission. Eur J Pharmacol 394:67–73
Papakostas GI, Petersen TJ, Kinrys G, Burns AM, Worthington JJ, Alpert JE, Fava M, Nierenberg AA (2005) Aripiprazole augmentation of selective serotonin reuptake inhibitors for treatment-resistant major depressive disorder. J Clin Psychiatry 66:1326–1330
Rapaport MH, Gharabawi GM, Canuso CM, Mahmoud RA, Keller MB, Bossie CA, Turkoz I, Lasser RA, Loescher A, Bouhours P, Dunbar F, Nemeroff CB (2006) Effects of risperidone augmentation in patients with treatment-resistant depression: results of open-label treatment followed by double-blind continuation. Neuropsychopharmacology 31:2505–2513
Richelson E, Souder T (2000) Binding of antipsychotic drugs to human brain receptors focus on newer generation compounds. Life Sci 68:29–39
Riedel M, Schwarz MJ, Strassnig M, Spellmann I, Muller-Arends A, Weber K, Zach J, Muller N, Moller HJ (2005) Risperidone plasma levels, clinical response and side-effects. Eur Arch Psychiatry Clin Neurosci 255:261–268
Schotte A, Bonaventure P, Janssen PF, Leysen JE (1995) In vitro receptor binding and in vivo receptor occupancy in rat and guinea pig brain: risperidone compared with antipsychotics hitherto used. Jpn J Pharmacol 69:399–412
Schotte A, Janssen PF, Gommeren W, Luyten WH, Van Gompel P, Lesage AS, De Loore K, Leysen JE (1996) Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology (Berl) 124:57–73
Seager MA, Barth VN, Phebus LA, Rasmussen K (2005) Chronic coadministration of olanzapine and fluoxetine activates locus coeruleus neurons in rats: implications for bipolar disorder. Psychopharmacology (Berl) 181:126–133
Shelton RC, Stahl SM (2004) Risperidone and paroxetine given singly and in combination for bipolar depression. J Clin Psychiatry 65:1715–1719
Simon JS, Nemeroff CB (2005) Aripiprazole augmentation of antidepressants for the treatment of partially responding and nonresponding patients with major depressive disorder. J Clin Psychiatry 66:1216–1220
Sprouse JS, Reynolds LS, Braselton JP, Rollema H, Zorn SH (1999) Comparison of the novel antipsychotic ziprasidone with clozapine and olanzapine: inhibition of dorsal raphe cell firing and the role of 5-HT1A receptor activation. Neuropsychopharmacology 21:622–631
Stark AD, Jordan S, Allers KA, Bertekap RL, Chen R, Mistry Kannan T, Molski TF, Yocca FD, Sharp T, Kikuchi T, Burris KD (2007) Interaction of the novel antipsychotic aripiprazole with 5-HT1A and 5-HT 2A receptors: functional receptor-binding and in vivo electrophysiological studies. Psychopharmacology (Berl) 190:373–382
Szabo ST, Blier P (2001a) Response of the norepinephrine system to antidepressant drugs. CNS Spectr 6:679–684
Szabo ST, Blier P (2001b) Serotonin (1A) receptor ligands act on norepinephrine neuron firing through excitatory amino acid and GABA(A) receptors: a microiontophoretic study in the rat locus coeruleus. Synapse 42:203–212
Szabo ST, Blier P (2002) Effects of serotonin (5-hydroxytryptamine, 5-HT) reuptake inhibition plus 5-HT(2A) receptor antagonism on the firing activity of norepinephrine neurons. J Pharmacol Exp Ther 302:983–991
Tohen M, Vieta E, Calabrese J, Ketter TA, Sachs G, Bowden C, Mitchell PB, Centorrino F, Risser R, Baker RW, Evans AR, Beymer K, Dube S, Tollefson GD, Breier A (2003) Efficacy of olanzapine and olanzapine–fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 60:1079–1088
Acknowledgments
This research was supported by Janssen Pharmaceuticals. PB received a Canada Research Chair in Psychopharmacology and an Endowed Chair from the University of Ottawa Institute of Mental Health Research. ED received a postdoctoral fellowship from Bar-Ilan University (Israel). We thank Dr. Owen Kelly for his help with statistical analyses and Lundbeck A/S for supplying escitalopram.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dremencov, E., El Mansari, M. & Blier, P. Distinct electrophysiological effects of paliperidone and risperidone on the firing activity of rat serotonin and norepinephrine neurons. Psychopharmacology 194, 63–72 (2007). https://doi.org/10.1007/s00213-007-0818-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-007-0818-8