
Abstract
Rationale
Animal research suggests that anticipation of reward can elicit dopamine release in the nucleus accumbens (NAcc). Human functional magnetic resonance imaging (FMRI) research further suggests that reward anticipation can increase local blood oxygen level dependent (BOLD) signal in the NAcc. However, the physiological relationship between dopamine release and BOLD signal increases in the NAcc has not yet been established.
Objectives
This review considers pharmacological MRI (phMRI) evidence for a directional relationship between NAcc dopamine release and BOLD signal, as well as implications for human psychopathological symptoms.
Results
Accumulating phMRI evidence supports a simple model in which NAcc dopamine release activates postsynaptic D1 receptors, which changes postsynaptic membrane potential, eventually increasing local BOLD signal. This continuing influence can change on a second-to-second basis.
Conclusions
Dopamine release in the NAcc appears to increase local BOLD signal via agonism of postsynaptic D1 receptors. Such a physiological mechanism implies that FMRI may be used to track symptoms related to NAcc dopaminergic dysregulation in psychiatric disorders including schizophrenia and attention deficit/hyperactivity disorder.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
It was a brilliant mistake. Having just arrived at Donald Hebb’s laboratory for a postdoctoral fellowship, social psychologist James Olds had little experience implanting rats. But with the help of surgically skilled graduate student Peter Milner, he hoped to arouse a rat by electrically stimulating an area deep in the midbrain, known at the time as the “ascending reticular activating system.” Despite showing no evidence of the predicted alerting response to stimulation, one particular rat kept returning to the corner of the table where it had been stimulated before. Olds ingeniously devised an apparatus that allowed the rat to press a bar to stimulate its own brain. To the researchers’ amazement, the rat pressed the bar over and over—to the exclusion of all other activities. Olds and Milner reported that: “...the control exercised over the animal’s behavior is extreme, possibly exceeding that exercised by any other reward previously used in animal experimentation” (Olds and Milner 1954).
Subsequent X-rays revealed that the investigators had misplaced their electrode: instead of the midbrain, they had implanted a region higher up and further forward, near the ventral striatum (Milner 1989). In fact, later research verified that self-stimulation behavior could be elicited from specific sites lying along a path extending from the midbrain over the lateral hypothalamus to the ventral striatum (including the nucleus accumbens, NAcc) and up into the lower and middle reaches of the frontal cortex in all mammalian species studied (see Fig. 1; Olds and Fobes 1981). Subsequent technical advances by Swedish chemists coincidentally revealed that the midbrain regions housed the cell bodies of dopamine neurons, which sent ascending projections to many of the striatal and forebrain regions that supported self-stimulation (Falck and Hillarp 1959). Cumulative findings now indicate that rats will work tirelessly to self-administer both electrical stimulation and compounds that stimulate dopamine receptors (i.e., dopamine agonists) to these regions. Conversely, drugs that block dopamine receptors (i.e., dopamine antagonists) halt electrical and chemical self-stimulation (McBride et al. 1999). Thus, dopamine release (possibly triggered by descending projections to the midbrain) ultimately appeared to play a key role in modulating self-stimulation behavior (Shizgal 1997). Based on this evidence, dopamine release in some mesolimbic regions was hypothesized to signal reward (Wise and Rompre 1989).

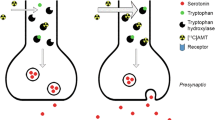
NAcc dopamine release and postsynaptic effects. Ventral tegmental area dopamine neurons project to subcortical regions including the NAcc as well as cortical regions including the orbital and mesial prefrontal cortex and medial temporal cortex. When dopamine is released in the NAcc, it diffuses as much as 12,000 nm from the synapse (which has an estimated width of only 15 nm and a diameter of 300 nm; Garris et al. 1994). Dopamine release thus stimulates both presynaptic autoreceptors (i.e., D2 type only) and postsynaptic receptors (i.e., both D1 and D2 types). This pre- versus postsynaptic distinction in receptor distribution implies that occupancy of presynaptic D2 receptors can regulate and decrease dopamine release, whereas occupancy of postsynaptic D1 or D2 receptors should locally change postsynaptic activity but not alter release. After a single neural impulse and subsequent dopamine release, changes in the potential of postsynaptic neural membranes begin at 200 ms and can last up to 1,000 ms (Gonon 1997). After release, dopamine is sucked back into the presynaptic neuron by reuptake mechanisms where it is either repackaged or broken down by enzymes. Most of the dopamine is cleared within 2,000 ms of initial release, although this process also depends to some extent on how much dopamine has already been released (Wightman et al. 1988). The timing of this entire process has been verified using physiological recordings featuring subsecond temporal resolution (Garris et al. 1994). Given an average firing rate of five impulses per second, extracellular dopamine levels should have the capacity to change on a second-to-second timescale, either increasing after burst firing or decreasing after inhibition of firing. These changes should then translate into second-to-second fluctuations in postsynaptic receptor occupancy, which should alter the postsynaptic membrane potential
More recent findings, however, suggest that dopamine plays a critical role not so much in the experience elicited by stimulation as in the anticipation of that experience. Using tasks developed by Pavlov, electrophysiologists observed that midbrain dopamine neurons fire not only in response to unexpected rewards, but also in response to cues that a monkey has learned will predict reward (Schultz et al. 1997). Using similar tasks in rats, researchers then verified that in addition to unpredicted rewards, reward-predicting cues cause midbrain projections to release dopamine in regions of the ventral striatum such as the NAcc (Roitman et al. 2004).
These discoveries were made possible by technological advances that enabled investigators to visualize subsecond changes in dopamine release in small subcortical regions (like the NAcc) of awake animals. Microdialysis had previously enabled investigators to analyze dopamine released into the extracellular fluid in rats, but the temporal resolution was limited to 120–180 s (Westerink 1995). However, a newer technique called cyclic voltammetry currently allows investigators to infer extrasynaptic dopamine release at a temporal resolution of less than 0.1 s (Wightman and Robinson 2002). In an analogous advance in human neuroimaging, variants of positron emission tomography (PET) utilize displaceable dopamine receptor ligands to allow researchers to visualize dopamine release, but only at a temporal resolution approximating 3,600 s. On the other hand, whereas functional magnetic resonance imaging (FMRI) does not index changes in dopamine release, it does allow researchers to visualize changes in oxygenation in subcortical brain regions at a temporal resolution of less than 1 s.Footnote 1 A multitude of findings utilizing these new techniques is beginning to generate a surprisingly coherent picture of human mesolimbic function.

Linking inferential level I. Association of a NAcc activation with b anticipation (ant; first white stripe) of gaining $5.00 (black and white circles) versus $0.00 (gray circles; p < .001), and c correlation of +$5.00 cue-induced positive arousal with +$5.00 cue-induced percent signal change in the NAcc volume of interest (n = 24)
The first study to combine injections of dopamine-releasing compounds with FMRI did so in humans. In a study of cocaine addicts, Breiter et al. (1997) found that cocaine injection (0.6 mg/kg) increased blood oxygen level dependent (BOLD) signal in many regions, including mesolimbic regions such as the ventral tegmental area, NAcc, and mesial prefrontal cortex (MPFC; Breiter et al. 1997; but see also Kufahl et al. 2005). Subsequent investigators observed a more circumscribed pattern of mesolimbic increases in BOLD signal in naïve subjects injected with amphetamine (0.15 mg/kg), again including the NAcc and MPFC (Vollm et al. 2004). Whereas stimulant-induced increases in BOLD signal evolved over the course of minutes, event-related FMRI studies indicated that mesolimbic BOLD signal could also reliably increase on a second-to-second basis. Specifically, nonpharmacological rewarding stimuli ranging from pleasant tastes to monetary gain can increase BOLD signal in mesolimbic regions, and cues signalling these rewards also increase BOLD signal in the NAcc (Knutson and Cooper 2005; O’Doherty 2004). For instance, cues that signal potential monetary gain preferentially and proportionally increase BOLD signal in the NAcc, whereas cues that signal potential monetary losses do not (Knutson et al. 2001).
Together, these findings suggest that injections of dopamine-releasing agents can increase BOLD signal in the NAcc and, further, that reward cues can also increase BOLD signal in the NAcc. Thus, reward cues might increase NAcc BOLD signal by eliciting endogenous dopamine release. However, it is also possible that dopamine-releasing agents activate the NAcc through dopamine-independent mechanisms. Thus, a missing link in this line of reasoning involves the precise relationship of NAcc dopamine release to increased BOLD signal.
After the initial FMRI study of humans undergoing cocaine injections, researchers began to systematically investigate the effects of pharmacological manipulations on BOLD signal using animal models. In a typical study, experimental animals are injected with a dopamine-releasing agent (e.g., amphetamine) while undergoing FMRI. Other dopaminergic manipulations can then be included. These exciting new pharmacological MRI (phMRI) techniques have the potential to address critical mechanistic questions about how NAcc dopamine release might translate into increased BOLD signal, with broader implications for human brain imaging, and possible extensions to psychiatric disorders.
phMRI findings
Can dopamine release increase NAcc BOLD signal, and if so, how? A number of mechanisms are possible. First, agonism of both postsynaptic and presynaptic neural dopamine receptors might increase BOLD signal. Second, agonism of postsynaptic dopamine receptors alone might increase BOLD signal. Third, dopamine-releasing compounds may globally dilate cerebral veins over the entire brain, which could also locally increase NAcc BOLD signal as a side effect. Fourth, dopamine may directly agonize dopamine receptors on local veins rather than on postsynaptic neurons. Fifth, dopamine may locally agonize dopamine receptors on glia, altering the energy exchange between neurons and veins. Below, we review phMRI evidence for a link between NAcc dopamine and BOLD signal and consider the plausibility of each of these potential mechanisms.
NAcc dopamine release increases BOLD signal
Consistent with the findings of the initial FMRI study of cocaine in humans, phMRI experiments indicated that dopamine release correlated with increased BOLD signal in the NAcc of anesthetized rats. Specifically, researchers found that injection of dopamine-releasing agents (e.g., d-amphetamine) as well as agents that block dopamine reuptake (e.g., 2beta-carbomethoxy-3beta-(4-fluorophenyl) tropane, or CFT) at doses that typically induce locomotion increased BOLD signal in several mesolimbic regions (including the NAcc). Simultaneous microdialysis verified that changes in NAcc extracellular dopamine and BOLD signal shared a similar temporal profile, including a steep rise at injection, the time to peak (5–20 min), and return to baseline (70–90 min; Chen et al. 1997). Lesioning dopamine neurons on one side of the rat’s striatum (verified with PET) abolished this effect on the lesioned side, but left the effect intact on the unlesioned side (Chen et al. 1997). Grafting fetal dopamine cells into the lesioned side of the striatum restored both amphetamine-induced dopamine release (verified with microdialysis) and BOLD signal increases (Chen et al. 1999). Cocaine injections also dose-dependently increased BOLD signal and blood volume (a measure which correlates with BOLD signal) in mesolimbic regions of rats (including the NAcc; Mandeville et al. 2001; Marota et al. 2000). These results generalize to primates as well, as amphetamine injections increased blood volume in mesolimbic regions (particularly in the NAcc) in monkeys. Dopamine lesioning reduced amphetamine’s effects, which depended upon the integrity of dopamine synapses (verified with CFT PET; Jenkins et al. 2004). Together, these findings suggest that NAcc dopamine release increases BOLD signal (and correlated blood volume). Further studies investigated which specific pharmacological mechanisms mediate this effect.
D1 receptor blockade decreases effect
The study indicating that cocaine injection could increase NAcc blood volume also revealed that antagonism of D1 receptors blocked this effect (Marota et al. 2000). D1 receptor antagonism also blocks increases in NAcc BOLD signal induced by amphetamine injection (Dixon et al. 2005). Microdialysis confirmed that dopamine was still released in the NAcc under D1 receptor blockade, although no associated increase in BOLD signal occurred (Choi et al. 2006). Thus, a critical event for increasing NAcc BOLD signal is not dopamine release per se, but rather the resulting increase in D1 receptor agonism. Together, these findings are consistent with the notion that dopamine release increases NAcc BOLD signal through a postsynaptic mechanism, as D1 receptors reside exclusively on postsynaptic membranes and veins (Edvinsson et al. 1985), but not on the presynaptic membrane of NAcc dopamine neurons.
D2 receptor blockade increases effect
Unlike postsynaptic D1 receptors, D2 receptors occupy both postsynaptic and presynaptic membranes in the NAcc. Presynaptic D2 receptors can act as autoreceptors, reducing dopamine release, and have higher affinity for dopamine than do postsynaptic receptors (Cooper et al. 2003). Thus, low doses of D2 agonists can decrease dopamine release in the NAcc by agonizing autoreceptors, whereas D2 antagonists can increase dopamine release by blocking autoreceptors. Accordingly, phMRI research has documented that administration of low doses of D2 agonists slightly decreases NAcc blood volume and can blunt amphetamine-induced increases in NAcc blood volume (Chen et al. 2005). In contrast, whereas administration of D2 antagonists has no intrinsic effect on NAcc blood volume, D2 antagonists can potentiate amphetamine-induced increases in NAcc blood volume (Schwarz et al. 2004a). Microdialysis indicates that D2 antagonists’ ability to enhance amphetamine effects is proportional to increases in NAcc dopamine release. Together, these findings suggest that NAcc dopamine release does not increase BOLD signal via a presynaptic mechanism, which should be mediated by D2 autoreceptors.
NAcc dopamine effects on BOLD are local
Both the regional specificity of the effects described above and the ability of dopamine lesions to unilaterally obliterate them argue against an account in which these effects reflect local byproducts of global changes in brain vascular tone. Further evidence against global modulation comes from the fact that changes in peripheral carbon dioxide, heart rate, and blood pressure are not correlated in time or magnitude with amphetamine injection induced increases in NAcc BOLD signal (Chen et al. 1997, 1999). Finally, injection of rats with cocaine methiodide (a cocaine variant with identical peripheral effects but no central effects) does not increase NAcc BOLD signal, supporting a critical role for dopamine release in the brain (Luo et al. 2003).
Ruling out other mechanisms
The phMRI research summarized above provides convergent evidence that increased postsynaptic D1 agonism constitutes a critical event that increases NAcc BOLD signal. However, many other factors might also influence BOLD signal in this region. Researchers have investigated and ruled out a number of alternative mechanisms that might account for such a coupling. Most phMRI studies have been conducted in anesthetized animals. As different anesthetics can alter the membrane properties of neurons, findings might not generalize to awake animals. Additionally, peripheral effects of stimulants (e.g., increasing heart rate and respiration) might generate correlated NAcc BOLD signal through peripheral nervous feedback. Thus, researchers trained awake rats to undergo phMRI while receiving intracranial injections of cocaine. They continued to observe mesolimbic increases in BOLD signal in response to intracranial injections of cocaine in regions including the NAcc, despite an absence of significant peripheral changes in heart rate and respiration (Febo et al. 2004). These findings suggest that stimulant-induced increases in NAcc BOLD signal generalize to the waking state and are not merely a byproduct of peripheral feedback.
Injection of stimulants might also influence other neurochemicals in the NAcc with known vasodilatory properties. For instance, nitric oxide is released during synaptic activity and can also increase vasodilation (Lindauer et al. 1999). However, global nitric oxide inhibition does not blunt amphatemine injection-induced increases in NAcc blood volume (Choi et al. 2006). Stimulants like amphetamine and cocaine can also release and block the reuptake of other neurotransmitters including norepinephrine, serotonin, and acetylcholine. Altered concentrations of these other neurotransmitters might account for increases in NAcc blood volume that occur after stimulant injection. Thus, it is important to note that phMRI researchers have repeatedly shown that chemically selective lesions of dopamine neurons (e.g., via 6-hydroxydopamine injections) abolish the ability of stimulant injections to increase NAcc BOLD signal (Chen et al. 1997, 1999; Nguyen et al. 2000), although they may continue to induce release of other neurotransmitters. Early studies also demonstrated that the increased blood flow caused by amphetamine injection was not mediated by either lactate or K+ and H+ ions (Astrup et al. 1978), ruling out other potential coupling agents. Finally, dopamine neurons also corelease glutamate (an excitatory neurotransmitter) in the prefrontal cortex of awake animals (Lavin et al. 2005) and in the striatum in slice preparation (Chuhma et al. 2004). Additionally, the NAcc receives glutamatergic projections from the prefrontal cortex and midline thalamus (Gerfen and Wilson 1996), and it is possible that these projections can elicit synaptic dopamine release independent from changes in dopamine neuron firing. Thus, glutamate and dopamine may have a synergistic effect on postsynaptic activity in the NAcc, but the effects of glutamatergic manipulations on BOLD signal have yet to be examined. Future phMRI experiments might systematically manipulate glutamate in the NAcc to determine whether this affects the BOLD signal.
Even if postsynaptic D1 receptors mediate the coupling between dopamine release and BOLD signal increase in the NAcc, this still leaves open the question of whether the critical receptors are localized on postsynaptic neurons, glia, or directly on veins. Dopamine receptors comprise two variants: D1-like (which include D5 as well as D1 receptors) and D2-like (which include D3 and D4 as well as D2 receptors). Whereas D1-like receptors are localized on neural membranes and veins, D2-like receptors are localized on neural membranes and glia (Choi et al. 2006; see Fig. 1). Thus, if a D1-related mechanism mediates the link between dopamine release and increased NAcc BOLD signal, this may occur by acting on postsynaptic neurons, or directly on veins, but probably not by acting directly on glia. The findings of one intriguing study imply that as the timing of peak dopamine release induced by cocaine injections temporally precedes the peak increase of blood volume in the striatum (i.e., by 5–10 min, as confirmed by microdialysis), BOLD signal increases may predominantly reflect postsynaptic D1 activation rather than direct agonism of D1 receptors on veins (Schwarz et al. 2004b).
Whereas many methodological details remain to be resolved (Steward et al. 2005), phMRI research has made critical advances in clarifying a relationship between dopamine release and BOLD signal. In the NAcc in particular, accumulating evidence suggests that this link depends on agonism of D1 receptors on postsynaptic neural membranes, with D2 receptors playing a more indirect modulatory role.
A process model
Whereas phMRI potentially affords temporal resolution on the order of seconds, stimulant injections cause long-lasting changes in neural activity that can persist for minutes or more. Nonetheless, the existing phMRI evidence may inform a more temporally sensitive model of how second-to-second changes in endogenous dopamine release influence NAcc BOLD signal. Such a model could help shed light on how NAcc BOLD signal might correlate with specific psychiatric symptoms related to dopaminergic dysfunction.
Linking postsynaptic D1 agonism and BOLD signal in the NAcc warrants careful consideration of the timing of each process. Dopamine neurons have been described as firing in fast “phasic” bursts (i.e., lasting 100–300 ms; greater than ten impulses per second) or at more continuous “tonic” rates (i.e., 1–10 impulses/s; Grace 1991). Theorists have associated these fast and continuous firing rates with various behavioral states. For instance, fast bursts have been associated with changes in reward prediction, whereas continuous firing rates have been associated with general levels of motivation or activity. However, typical distributions of dopamine firing rates are unimodal rather than bimodal (median, approximately four impulses per second, skewed towards faster firing rates), suggesting that dopamine neurons also fire at intermediate rates (Schultz 2002). Additionally, in the NAcc, dopamine typically diffuses some distance from the synapse before occupying postsynaptic receptors, depending upon the amount released and the speed of reuptake (Cragg and Rice 2004; Garris et al. 1994). Together, these facts are consistent with observations that even fast bursts of dopamine firing can produce increases in extracellular dopamine in the NAcc that persist from 2–3 s (see Fig. 1), and support the inference that subsequent agonism of postsynaptic D1 receptors fluctuates at a second-to-second timescale.
The BOLD signal typically measured with event-related FMRI also has well-established temporal properties. In the NAcc, BOLD signal related to reward cues typically rises at 2 s, peaks from 4–6 s, and may take anywhere from 10–12 s to fall back to baseline (Aguirre et al. 1998; Knutson et al. 2003). An immediate undershoot may also occur, but this phenomenon has currently only been visualized at high magnet field strengths in visual cortex (not the NAcc). Physiological correlates of the BOLD signal have been reviewed in detail elsewhere (Logothetis and Wandell 2004), but changes in BOLD signal correlate more closely with changes in postsynaptic local field potentials than with changes in presynaptic firing rates. Thus, assuming a constant rise time, the fast onset of the BOLD signal may afford visualization of second-to-second changes in postsynaptic activity. The slow offset of the BOLD signal could potentially obscure subsequent changes, but this problem can be avoided by presenting stimuli repeatedly and at varying intervals, allowing deconvolution of the BOLD signal. With a properly designed event-related study, adequate signal-to-noise ratio, and the absence of artifacts, second-to-second changes in NAcc BOLD signal can be visualized (see Fig. 2).
Combined with phMRI evidence that NAcc dopamine release may increase BOLD signal via a D1-dependent postsynaptic mechanism, consideration of these temporal characteristics suggests a simple process model in which: (1) released dopamine diffuses away from the synapse, activating postsynaptic D1 and D2 receptors (0–2 s after firing), which (2) changes postsynaptic membrane polarity and engages metabolic processes (0–2 s after firing), which (3) immediately utilizes oxygen from nearby capillaries, possibly generating an immediate small decrease in BOLD signal (0–2 s after firing), followed by a delayed large increase in BOLD signal in draining veins (4–6 s after firing—the signal typically visualized with FMRI), as (4) dopamine transporters continuously suck unbound dopamine and metabolites back into the presynaptic neuron, and agonism of presynaptic D2 receptors simultaneously decreases subsequent dopamine release (ongoing; Montague et al. 2004). Generally, this model implies that in the NAcc, postsynaptic D1 agonism and BOLD signal can synchronously fluctuate on a second-to-second timescale.
Implications
This simple process model implies that changes in NAcc BOLD signal reflect changes in postsynaptic D1 agonism. Further, this association might occur not only at longer timescales associated with pharmacological manipulations, but also at shorter timescales associated with endogenous bursts of dopamine release (Grace 1991; Schultz 2002). Critical research establishing generalization of phMRI findings to shorter timescales has yet to be conducted. For instance, future experiments might combine dopamine depletion or antagonism with stimuli known to elicit fast increases in NAcc BOLD signal (e.g., sexually arousing odors in animals or monetary cues in humans; Ferris et al. 2004; Knutson et al. 2001; Leyton et al. 2004). If fast increases in NAcc BOLD signal depend upon postsynaptic D1 agonism (and not, for instance, glutamate release), they should be blunted by dopamine depletion or antagonism. Functional correlates related to changes in NAcc BOLD signal should also change at these shorter timescales. Some of these predicted correlates are pharmacological, whereas others are psychological. All hold potential relevance to specific psychiatric symptoms.
One pharmacological implication of the model is straightforward: Holding other inputs constant, increases in postsynaptic D1 receptor agonism should increase NAcc BOLD signal, whereas decreases in postsynaptic D1 receptor agonism should decrease NAcc BOLD signal. Although D1 receptor agonism has been linked to excitatory postsynaptic effects (West et al. 2003), the excitatory or inhibitory effect of agonism should not matter as much as the change in postsynaptic agonism (Logothetis and Wandell 2004). Increased postsynaptic D1 agonism should alter membrane potential, increasing energy utilization and the BOLD signal. On the other hand, decreased postsynaptic D1 agonism should reduce changes in membrane potential, decreasing energy utilization and the BOLD signal (Attwell and Iadecola 2002). Similar findings have been documented in visual cortex in which cessation of neurotransmission leads to decreases in BOLD signal (Shmuel et al. 2006). Of course, the link between dopamine release and postsynaptic BOLD signal might be further modulated by individual differences in the resting level of dopamine, availability and affinity of dopamine receptors, and efficacy of reuptake (Cools and Robbins 2004).
Another set of pharmacological predictions involves combining pharmacological agents likely to influence continuously available “tonic” dopamine (i.e., which changes over the course of minutes or longer) with brief cognitive probes designed to elicit faster “phasic” release of endogenous dopamine in the NAcc (i.e., which changes on the order of seconds). First, D1 antagonists should blunt increases in NAcc BOLD signal. Some evidences support this prediction, as typical but not atypical neuroleptics blunted NAcc BOLD signal in schizophrenics during the anticipation of monetary gain (Juckel et al. 2006a). Second, low doses of D2 antagonists should enhance momentary increases in NAcc BOLD signal. This prediction has not yet been tested in humans. Third, dopamine reuptake inhibitors (e.g., amphetamine, cocaine) should prolong fast increases in NAcc BOLD signal, as reuptake inhibition should prolong dopamine synaptic availability. Some research also supports this prediction, as oral administration of amphetamine blunted but also prolonged the timecourse of second-to-second increases in NAcc BOLD signal during anticipation of monetary gain, compared to double-blind placebo administration (Knutson et al. 2004). Blunting is consistent with the notion that prolonged dopamine availability caused by reuptake inhibition agonized D2 autoreceptors. Comparative research has verified amphetamine’s ability to prolong and blunt NAcc dopamine release (with voltammetry; Schmitz et al. 2001). The effects of acute administration of D1 and D2 agonists on increases in NAcc BOLD signal are more difficult to predict, as agonists and endogenous dopamine may compete for the same receptors yet may differ in their rate of occupancy or postsynaptic diffusion. Additionally, receptors can often rapidly adjust to prolonged increases or decreases in continuous dopamine levels via alterations in sensitivity or through receptor internalization. Generally, the process model implies that psychiatric drugs that influence dopamine may have pronounced effects on momentary changes in NAcc BOLD signal. The effect of drugs that affect other biogenic amines (e.g., serotonin or norepinephrine) remains to be systematically explored.
Psychological predictions can also be derived from this simple model of dopamine’s influence on NAcc BOLD signal. Elsewhere, we have argued that increases in NAcc BOLD signal might index increased positive arousal or an affective state marked by both positive valence and arousal (e.g., excitement, enthusiasm), which tends to occur during reward anticipation (Knutson et al. 2001). The inverse of such a state is a negative, unaroused state (e.g., boredom, tiredness; Watson and Tellegen 1985). If the capacity for positive arousal depends upon momentary increases in NAcc dopamine, then such a capacity may be diminished in psychiatric disorders in which dopamine release is dysregulated. For instance, radioligand PET research indicates that unmedicated schizophrenics show excessive and chronic dopamine release in the NAcc (Abi-Dargham et al. 1998). Indeed, an event-related FMRI study of unmedicated schizophrenics indicated not only that they show blunted increases in NAcc BOLD signal during anticipation of monetary reward relative to healthy controls, but also that the degree of blunting corresponded with the degree to which the schizophrenics reported suffering from negative symptoms or symptoms involving low positive arousal (e.g., anhedonia; Juckel et al. 2006b). On the other hand, radioligand PET studies suggest that adolescents with attention deficit/hyperactivity disorder (AD/HD) may show reduced release of dopamine in the NAcc (Rosa-Neto et al. 2005). Interestingly, an event-related FMRI study of unmedicated adolescents with AD/HD indicated that they too showed reduced increases in NAcc BOLD signal during anticipation of monetary reward relative to healthy controls, and that the degree of diminution correlated with hyperactive symptoms (Scheres et al. 2006).
Whereas apparently inconsistent, the same absence of positive arousal that leads to a lack of motivation in the case of schizophrenics may also encourage hyperactive attempts to seek environmental stimulation in the case of adolescents with AD/HD. One implication is that the absolute level of NAcc dopamine is not as important for enabling positive arousal as is the ability of NAcc dopamine to respond rapidly to environmental contingencies. Of course, such speculations must withstand empirical testing. But together, emerging findings generally raise the possibility that NAcc BOLD signal may show more irregularities in people who have disorders involving symptoms that respond to dopaminergic medications (e.g., schizophrenia, AD/HD) than in disorders involving symptoms that respond to other types of medications (e.g., serotonergic medications applied to anxiety disorders and unipolar depression).
Although this review focuses anatomically on the NAcc, many other regions are innervated by ventral tegmental area dopamine neurons, including orbital and mesial prefrontal cortex and medial temporal lobe. These regions subserve distinct functions, show different dopamine kinetics, and are not physiologically homogenous (for instance, the prefrontal cortex lacks dopamine autoreceptors), so one cannot generalize from the NAcc to these other regions. Based on the inferential links we have traced, NAcc dopamine release should increase NAcc BOLD signal, which may correlate with a state of positive arousal. However, reverse inferences along these lines pose more potential hazards than do forward inferences, as other variables besides increased NAcc BOLD signal could correlate with increases in self-reported positive arousal, and other variables besides increased NAcc dopamine release could lead to increased NAcc BOLD signal (see Fig. 3). Nonetheless, the existing findings are consistent with a model in which dopamine release increases NAcc BOLD signal, which indexes positive arousal. These links can be tested with interdisciplinary scientific methods. Often, physiological questions lead to more physiological details, and further away from behavior. Fortunately, these physiological details lead back out, to testable behavioral predictions with implications for mental health.

Linking inferential level II. Venn diagram indicating that increased NAcc D1 agonism increases NAcc BOLD signal, which may index increased positive arousal. However, due to increased multiple causation at more molar levels of description, backward inference to the molecular level is not as robust as forward inference
A half-century ago, Olds and Milner fortuitously “short-circuited” a neural pathway critical for survival. Now, neuroimaging methods with enhanced spatiotemporal resolution offer the promise of modeling what goes into that pathway, what comes out, and what happens in between. Thus, these new methods may enable researchers to use neural activity not only to better understand what came before, but also to better predict what is yet to come.
Notes
Measuring NAcc activity. Because dopamine release fluctuates rapidly in the NAcc (see Fig. 1), both spatial and temporal resolutions present critical considerations for researchers. Presently available methods differ in spatial and temporal resolution (current estimates are listed below for each). However, future innovations may well increase the resolution of any of these methods. Because they are invasive, microdialysis and cyclic voltammetry are primarily used in rodents, whereas PET and FMRI are less invasive and so commonly used in humans. a Microdialysis (0.3 mm, 120 s): A probe containing concentric tubes is positioned in the brain region of interest. Synthetic extracellular fluid is passed from the outer tube to the inner tube. Extracellular molecules (e.g., dopamine) are also sucked into the inner tube, which can then be analyzed for chemical content with high performance liquid or gas chromatography. b Cyclic voltammetry (0.01 mm, 0.100 s): A probe containing two adjacent electrical wires is positioned in the brain region of interest. A variable current is passed from one wire to the other, and electron flow through intermediary material is measured. Certain molecules increase current flow at specific voltages. Given the absence of other similarly reactive molecules (e.g., norepinephrine), and controlling for background current, the presence of dopamine can be inferred. c Radioligand positron emission tomography (8 mm, 3,600 s): A compound containing radioactively tagged molecules that mimic dopamine is injected into the subject. The tagged molecules travel to the brain and attach to dopamine receptors. The tags decay, emitting two positrons in opposite directions, and a camera detects the emissions and computes their point of origin. As the tags decay, subjects receive an experimental treatment that releases dopamine (e.g., receive an amphetamine injection) or a control treatment that does not (e.g., receive a placebo injection). Once released, dopamine displaces tagged molecules from receptors, and a difference in positron emission in that specific locale between experimental and control conditions can later be computed. d Functional magnetic resonance imaging: (4 mm, 1 s): A large magnet aligns protons in subjects’ brains. A smaller coil then emits magnetic pulses that flip the protons’ orientation and subsequently assesses the speed and spin with which protons return to their original orientation. Subjects engage in a task that elicits more or less synaptic activity. In brain regions with increased synaptic activity, oxygen consumption increases, followed by a disproportionate increase in local venous oxygen with an approximate lag of 4–6 s. “Ballooning” of these veins with oxygenated blood creates a small venous pocket of demagnetization, which has been called “blood oxygen level dependent” (BOLD) signal. Changes in regional BOLD signal can be compared across experimental versus control task conditions. Whereas microdialysis and radioligand PET have greater chemical specificity for detecting dopamine, cyclic voltammetry and FMRI have greater temporal resolution for detecting rapid changes in NAcc activity. As each method has different strengths, each can make unique contributions to researchers’ understanding of changes in NAcc dopamine.
References
Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl JP, Bowers M, van Dyck CH, Charney DS, Innis RB, Laruelle M (1998) Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry 155:761–767
Aguirre GK, Zarahn E, D’Esposito M (1998) The variability of human, BOLD hemodynamic responses. NeuroImage 8:360–369
Astrup J, Heuser D, Lassen NA, Nilsson B, Norberg K, Siesjo BK (1978) Evidence against H+ and K+ as main factors for the control of cerebral blood flow: a microelectrode study. Ciba Foundation Symposium, pp 313–337
Attwell D, Iadecola C (2002) The neural basis of functional brain imaging signals. Trends Neurosci 25:621–625
Breiter HC, Gollub RL, Weisskoff RM, Kennedy DN, Makris N, Berke JD, Goodman JM, Kantor HL, Gastfriend DR, Riorden JP, Mathew RT, Rosen BR, Hyman SE (1997) Acute effects of cocaine on human brain activity and emotion. Neuron 19:591–611
Chen YI, Galpern WR, Brownell AL, Matthews RT, Bogdanov M, Isacson O, Keltner JR, Beal MF, Rosen BR, Jenkins BG (1997) Detection of dopaminergic neurotransmitter activity using phMRI: correlation with PET, microdialysis, and behavioral data. Magn Reson Med 38:389–398
Chen YI, Brownell AL, Galpern W, Isacson O, Bogdanov M, Beal MF, Livni E, Rosen BR, Jenkins BG (1999) Detection of dopaminergic cell loss and neural transplantation using pharmacological MRI, PET and behavioral assessment. NeuroReport 10:2881–2886
Chen YC, Choi JK, Anderson SL, Rosen BR, Jenkins BG (2005) Mapping dopamine D2/D3 receptor function using pharmacological magnetic resonance imaging. Psychopharmacology 180:704–715
Choi JK, Chen YI, Hamel E, Jenkins BG (2006) Brain hemodynamic changes mediated by dopamine receptors: Role of the cerebral microvasculature in dopamine-mediated neurovascular coupling. NeuroImage 30:700–712
Chuhma N, Zhang H, Masson J, Zhuang X, Sulzer D, Hen R, Rayport S (2004) Dopamine neurons mediate a fast excitatory signal via their glutamatergic synapses. J Neurosci 24:972–981
Cools R, Robbins TW (2004) Chemistry of the adaptive mind. Philos Trans R Soc Lond 362:2871–2888
Cooper JR, Bloom FE, Roth RH (2003) The biochemical basis of neuropharmacology, 8th edn. Oxford University Press, Oxford
Cragg SJ, Rice ME (2004) DAncing past the DAT at a DA synapse. Trends Neurosci 27:270–277
Dixon AL, Prior M, Morris PM, Shah YB, Joseph MH, Young AM (2005) Dopamine antagonist modulation of amphetamine response as detected using pharmacological MRI. Neuropharmacology 48:236–245
Edvinsson L, McCulloch J, Sharkey J (1985) Vasomotor responses of cerebral arterioles in situ to putative dopamine receptor agonists. Br J Pharmacol 85:403–410
Falck B, Hillarp NA (1959) On the cellular localization of catechol amines in the brain. Acta Anat 38:277–279
Febo M, Segarra AC, Tenney JR, Brevard ME, Duong TQ, Ferris CF (2004) Imaging cocaine-induced changes in the mesocorticolimbic dopaminergic system of conscious rats. J Neurosci Methods 139:167–176
Ferris CF, Snowdon CT, King JA, Sullivan JMJ, Ziegler Te, Olson DP, Schultz-Darken NJ, Tannenbaum PL, Ludwig R, Wu Z, Einspanier A, Vaughan JT, Duong TQ (2004) Activation of neural pathways associated with sexual arousal in non-human primates. J Magn Reson Imaging 19:168–174
Garris PA, Ciolkowski EL, Pastore P, Wightman RM (1994) Efflux of dopamine from the synaptic cleft in the nucleus accumbens of the rat brain. J Neurosci 14:6084–6093
Gerfen CR, Wilson CJ (1996) The basal ganglia. In: Bjorklund A, Hokfelt T (eds) Handbook of chemical neuroanatomy. Elsevier Science, London, UK, pp 371–468
Gonon F (1997) Prolonged and extrasynaptic excitatory action of dopamine mediated by D1 receptors in the rat striatum in vivo. J Neurosci 17:5972–5978
Grace AA (1991) Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience 41:1–24
Jenkins BG, Sanchez-Pernaute R, Brownell AL, Chen YC, Isacson O (2004) Mapping dopamine function in primates using pharmacological magnetic resonance imaging. J Neurosci 24:9553–9560
Juckel G, Schlagenhauf F, Koslowski M, Filonov D, Wustenberg T, Villringer A, Knutson B, Kienast T, Gallinat J, Wrase J, Heinz A (2006a) Dysfunction of ventral striatal reward prediction in schizophrenic patients treated with typical but not atypical neuroleptics. Psychopharmacology 187:222–228
Juckel G, Schlagenhauf F, Koslowski M, Wustenberg T, Villringer A, Knutson B, Wrase J, Heinz A (2006b) Dysfunction of ventral striatal reward prediction in schizophrenia. NeuroImage 29:409–416
Knutson B, Cooper JC (2005) Functional magnetic resonance imaging of reward prediction. Curr Opin Neurol 18:411–417
Knutson B, Adams CM, Fong GW, Hommer D (2001) Anticipation of increasing monetary reward selectively recruits nucleus accumbens. J Neurosci 21:RC159
Knutson B, Fong GW, Bennett SM, Adams CM, Hommer D (2003) A region of mesial prefrontal cortex tracks monetarily rewarding outcomes: characterization with rapid event-related FMRI. NeuroImage 18:263–272
Knutson B, Bjork JM, Fong GW, Hommer D, Mattay VS, Weinberger DR (2004) Amphetamine modulates human incentive processing. Neuron 43:261–269
Kufahl PR, Li Z, Risinger RC, Rainey CJ, Wu G, Bloom AS, Li S-J (2005) Neural responses to acute cocaine administration in the human brain detected by fMRI. NeuroImage 28:904–914
Lavin A, Nogueira L, Lapish CC, Wightman RM, Phillips PE, Seamans JK (2005) Mesocortical dopamine neurons operate in distinct temporal domains using multimodal signaling. J Neurosci 25:5013–5023
Leyton M, Dagher A, Boileau I, Casey K, Baker GB, Diksic M, Gunn R, Young SN, Benkelfat C (2004) Decreasing amphetamine-induced dopamine release by acute phenylalanine/tyrosine depletion: A PET/[11C]Raclopride study in healthy men. Neuropsychopharmacology 29:427–432
Lindauer U, Megow D, Matsuda H, Dirnagl U (1999) Nitric oxide: a modulator, but not a mediator of neurovascular coupling in rat somatosensory cortex. Am J Physiol 277:H799–H811
Logothetis NK, Wandell BA (2004) Interpreting the BOLD signal. Annu Rev Physiol 66:735–769
Luo F, Wu G, Li Z, Li S-J (2003) Characterization of effects of mean arterial blood pressure induced by cocaine and cocaine methiodide on BOLD signals in rat brain. Magn Reson Med 49:264–270
Mandeville JB, Jenkins BG, Kosofsky BE, Moskowitz MA, Rosen BR, Marota JJ (2001) Regional sensitivity of BOLD and CBV changes during stimulation of rat brain. Magn Reson Med 45:443–447
Marota JJ, Mandeville JB, Weisskoff RM, Moskowitz MA, Rosen BR, Kosofsky BE (2000) Cocaine activation discriminates dopaminergic projections by temporal response: an FMRI study in rat. NeuroImage 11:13–23
McBride WJ, Murphy JM, Ikemoto S (1999) Localization of brain reinforcement mechanisms: intracranial self-administration and intracranial place-conditioning studies. Behav Brain Res 101:129–152
Milner PM (1989) The discovery of self-stimulation and other stories. Neurosci Biobehav Rev 13:61–67
Montague PR, McClure SM, Baldwin PR, Phillips PEM, Budygin EA, Stuber GD, Kilpatrick MR, Wightman RM (2004) Dynamic gain control of dopamine delivery in freely moving animals. J Neurosci 24:1754–1759
Nguyen TV, Brownell AL, Chen YC, Livni E, Coyle JT, Rosen BR, Cavagna F, Jenkins BG (2000) Detection of the effects of dopamine receptor supersensitivity using pharmacological MRI and correlations with PET. Synapse 36:57–65
O’Doherty JP (2004) Reward representations and reward-related learning in the human brain: insights from neuroimaging. Curr Opin Neurobiol 14:769–776
Olds ME, Fobes JL (1981) The central basis of motivation: intracranial self-stimulation studies. Annu Rev Psychol 32:523–574
Olds J, Milner P (1954) Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol 47:419–427
Roitman MF, Stuber GD, Phillips PEM, Wightman RM, Carelli RM (2004) Dopamine operates as a subsecond modulator of food seeking. J Neurosci 24:1265–1271
Rosa-Neto P, Lou HC, Cumming P, Pryds O, Karrebaek H, Lunding J, Gjedde A (2005) Methylphenidate-evoked changes in striatal dopamine correlate with inattention and impulsivity in adolescents with attention deficit hyperactivity disorder. NeuroImage 25:868–876
Scheres A, Milham MP, Knutson B, Castellanos FX (2006) Ventral striatal hyporesponsiveness during reward anticipation in attention deficit/hyperactivity disorder. Biol Psychiatry (in press). DOI 10.1016/j.biopsych.2006.04.042
Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D (2001) Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci 21:5916–5924
Schultz W (2002) Getting formal with dopamine and reward. Neuron 36:241–263
Schultz W, Dayan P, Montague PR (1997) A neural substrate of prediction and reward. Science 275:1593–1599
Schwarz A, Gozzi A, Reese T, Bertain S, Crestan V, Hagan J, Heidbreder C, Bifone A (2004a) Selective dopamine D(3) receptor antagonist SB-277011-A potentiates phMRI response to acute amphetamine challenge in the rat brain. Synapse 54:1–10
Schwarz AJ, Zocchi A, Reese T, Gozzi M, Varnier G, Curcuruto O, Sartori I, Girlanda E, Biscaro B, Crestan V, Bertani S, Heidbreder C, Bifone A (2004b) Concurrent pharmacological MRI and in situ microdialysis of cocaine reveal a complex relationship between the central hemodynamic response and local dopamine concentration. NeuroImage 23:296–304
Shizgal P (1997) Neural basis of utility estimation. Curr Opin Neurobiol 7:198–208
Shmuel A, Augath M, Oeltermann A, Logothetis NK (2006) Negative functional MRI response correlates with decreases in neuronal activity in monkey visual area V1. Nat Neurosci 9:569–577
Steward CA, Marsden CD, Prior MJW, Morris PG, Shah YB (2005) Methodological considerations in rat brain BOLD contrast pharmacological MRI. Psychopharmacology 180:687–704
Vollm BA, de Araujo IE, Cowen PJ, Rolls ET, Kringelbach ML, Smith KA, Jezzard P, Heal RJ, Matthews PM (2004) Methamphetamine activates reward circuitry in drug naive human subjects. Neuropsychopharmacology 29:1715–1722
Watson D, Tellegen A (1985) Toward a consensual structure of mood. Psychol Bull 98:219–235
West AR, Floresco SB, Charara A, Rosenkranz JA, Grace AA (2003) Electrophysiological interactions between striatal glutamatergic and dopaminergic systems. Ann NY Acad Sci 1003:53–74
Westerink BHC (1995) Brain microdialysis and its application for the study of animal behaviour. Behav Brain Res 70:103–124
Wightman RM, Robinson DL (2002) Transient changes in mesolimbic dopamine and their association with ‘reward’. J Neurochem 82:721–735
Wightman RM, Amatore C, Engstrom RC, Hale PD, Kristensen EW, Kuhr WG, May LJ (1988) Real-time characterization of dopamine overflow and uptake in the rat striatum. Neuroscience 25:513–523
Wise RA, Rompre PP (1989) Brain, dopamine, and reward. Annu Rev Psychol 40:191–225
Acknowledgment
We thank G. Elliott Wimmer, Peter Shizgal, and three anonymous reviewers for helpful comments on prior drafts of the manuscript. During manuscript preparation, BK was supported by NIDA grant DA020615-01.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Knutson, B., Gibbs, S.E.B. Linking nucleus accumbens dopamine and blood oxygenation. Psychopharmacology 191, 813–822 (2007). https://doi.org/10.1007/s00213-006-0686-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-006-0686-7