
Abstract
Cisplatin is a well-studied and widely used chemotherapeutic agent and is effective in the treatment of the advanced human non-small cell lung cancer (NSCLC). Curcumin is a yellow pigment derived from the rhizome of Curcuma longa and has been proved to have antioxidant and antitumor properties. XRCC1 is an important scaffold protein involved in base excision repair and plays an important role in the development of lung cancer. In this study, we characterize the role of curcumin in the cytotoxicity, p38 MAPK activation, and XRCC1 expression affected by cisplatin in NSCLC cells. We show that curcumin enhanced the cytotoxicity induced by cisplatin in two NSCLC cells, A549 and H1703. Treatment with cisplatin alone increased XRCC1 mRNA and protein expression through p38 MAPK activation. Moreover, SB2023580 (p38 inhibitor) decreased the XRCC1 mRNA and protein stability upon cisplatin treatment. Knockdown of XRCC1 in NSCLC cells by transfection of XRCC1 siRNA or inactivation of p38 MAPK resulted in enhancing the cytotoxicity and cell growth inhibition induced by cisplatin. Curcumin inhibited the expression of XRCC1 in cisplatin-exposed NSCLC cells. Furthermore, transfection with constitutive active MKK6 or HA-p38 MAPK vectors rescued the XRCC1 protein level and also the cell survival suppressed by cisplatin and curcumin combination in A549 and H1703 cells. These findings suggested that the downregulation of XRCC1 expression by curcumin can enhance the chemosensitivity of cisplatin in NSCLC cells.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lung cancer is the leading cause of cancer death worldwide and classified as non-small-cell lung cancer (NSCLC) and small-cell lung cancer (Silvestri and Rivera 2005). NSCLC accounts for 85 % of lung cancer cases, and the majority of people diagnosed with NSCLC are unsuitable for surgery due to the NSCLC patients with metastasis (Massarelli and Herbst 2006). The first-line therapeutic option for advanced NSCLC patients includes chemotherapy with a platinum-containing compound such as cisplatin in combination with second- or third-generation cytotoxic agents (Pfister et al. 2004). Cisplatin is a cytotoxic platinum compound that triggers DNA crosslinking-induced cell death (Kelland 2007), and cellular response to cisplatin exposure includes activation of MAPKs signal transduction pathway (Persons et al. 1999). Despite advances in early detection and chemotherapy, its prognosis is usually poor (Schiller et al. 2002). This poor treatment outcome is due to the development of multiple mechanisms to overcome cisplatin-induced DNA damage and apoptosis and lead to cisplatin resistance (Rosell et al. 2002; Rudin et al. 2003; Siddik 2003; Galluzzi et al. 2012). One of the major systems activated is enhanced capability of DNA repair pathways in cancer cells to remove the cisplatin-induced DNA damage (Usanova et al. 2010).
X-ray repair cross complementing group 1 (XRCC1) is a key mediator of DNA single-strand breaks (SSBs) repair, and deficiency in XRCC1 results in embryonic lethality in mice (Tebbs et al. 1999; Tebbs et al. 2003). XRCC1 is involved in the repair process of cisplatin-induced DNA damage in HepG2 cells (Zhang et al. 2010). Downregulation of XRCC1 expression in human breast cancer cell lines resulted in decreased SSBs repair capacity and hypersensitivity to methyl methanesulfonate (Brem and Hall 2005). Moreover, previous studies have suggested an association between XRCC1 and cisplatin resistance (Kudo et al. 2012; Siddiqui-Jain et al. 2012); however, the contribution of XRCC1 to cisplatin resistance in NSCLC and underlying mechanisms is not fully understood.
Curcumin, a well-known chemopreventive agent, has been reported as a candidate for development of potential therapeutic strategies for treatment of cancer (Shishodia et al. 2005). It exhibits antioxidant, anti-inflammatory, antibacterial, antiviral, and hepatoprotective roles (Kumar et al. 2016). Curcumin can sensitize human ovarian cancer cells to cisplatin or oxaliplatin (Montopoli et al. 2009). Moreover, curcumin has the ability to suppress several cellular signaling molecules, including the MAPK family signaling pathways (Chen and Tan 1998; Han et al. 2002). For example, curcumin induces cell death in cisplatin-resistant human ovarian cancer cells by modulating p38 MAPK and AKT activity (Weir et al. 2007). However, it is unclear whether curcumin could enhance the sensitivity of cisplatin through modulation of p38 MAPK and XRCC1 expression in NSCLC.
In the present study, we used two different NSCLC cell types, including bronchioloalveolar cell carcinoma (A549) and squamous cell carcinoma (H1703) to investigate the molecular mechanisms of curcumin in combination with cisplatin for generation of cytotoxic effects. Moreover, a recent study indicated that irinotecan, a topoisomerase-1 inhibitor, has a synergistic effect with cisplatin in cisplatin-resistant gastric cancer cells through suppression of XRCC1 (Xu et al. 2014). Therefore, we also wanted to know whether the curcumin could affect the sensitivity of cisplatin for NSCLC cells through modulating expression of XRCC1. These results may provide a rational design for future drug regimens incorporating cisplatin and curcumin for the treatment of NSCLC.
Materials and methods
Chemical agents and cell culture
Curcumin, cisplatin, cycloheximide, and actinomycin D were obtained from Sigma (St Louis, MO). SB2023580 was purchased from Calbiochem-Novabiochem (San Diego, CA, USA). Curcumin, actinomycin D, and SB2023580 were dissolved in dimethyl sulfoxide (DMSO). Cycloheximide was dissolved in Milli-Q-purified water (Millipore, Billerica, MA, USA). Human lung carcinoma cells A549 and H1703 were obtained from the American Type Culture Collection (Manassas, VA), and the cells were cultured at 37 °C in a humidified atmosphere containing 5 % CO2 in RPMI-1640 complete medium supplemented with sodium bicarbonate (2.2 %, w/v), l-glutamine (0.03 %, w/v), penicillin (100 units/ml), streptomycin (100 μg/ml), and fetal calf serum (10 %).
Western blot analysis
After different treatments, equal amounts of proteins from each set of experiments were subjected to Western blot analysis. The specific phospho-p38 MAPK (Thr180/Tyr182) and phospho-MKK3 (Ser189)/MKK6 (Ser207) antibodies were purchased from Cell Signaling (Beverly, MA). Rabbit polyclonal antibodies against XRCC1 (H-300) (sc-11429), p38(C-20) (sc-535), MEK3 (N-20) (sc-959), HA (F-7) (sc-7392), and actin (I-19) (sc-1616) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Transfection with MKK6E, HA-p38 MAPK vectors, small interfering RNA
Plasmids transfection of MKK6E (a constitutively active form of MKK6) and HA-p38 MAPK was achieved as previously described (Cheng et al. 2006; Chuang et al. 2008). Exponentially growing human lung cancer cells (106) were plated for 18 h, and then MKK6E expression vectors were transfected into A549 or H1703 cells using Lipofectamine (Invitrogen). The XRCC1 siRNA (sc-36859) or scramble control siRNA were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Cells were transfected with siRNA duplexes (200 nM) for 24–48 h using Lipofectamine 2000 (Invitrogen) for 24 h.
Quantitative real-time polymerase chain reaction
Polymerase chain reactions (PCRs) were performed using ABI Prism 7900HT according to the manufacturer’s instructions. Amplification of specific PCR products was performed using the SYBR Green PCR Master Mix (Applied Biosystems). For each sample, the data were normalized to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The designed primers in this study were XRCC1 forward primer, 5′-GGGACCGGGTCAAAATTGTT-3′; XRCC1 reverse primer, 5′-ACCGTACAAAACTCAAGCCAAAG-3′; GAPDH forward primer, 5′-CATGAGAAGTATGACAACAGCCT-3′; GAPDH reverse primer, 5′-AGTCCTTCCACGATACCAAAGT-3′. Analysis was performed using the comparative Ct value method. For each sample, the data were normalized to the housekeeping gene GAPDH.
MTS assay
Cells were cultured at 5000 per well in 96-well tissue culture plates. To assess cell viability, drugs were added after plating. At the end of the culture period, 20 μL of MTS solution (CellTiter 96 Aqueous One Solution Cell Proliferation Assay; Promega, Madison, WI, USA) was added, the cells were incubated for a further 2 h, and the absorbance was measured at 490 nm using an ELISA plate reader (Biorad Technologies, Hercules, CA). The cell viability was calculated as follows: cell viability (%) = (OD of drug-treated sample/OD of untreated sample) × 100. The values of cell viability were calculated from the mean amount of the three independent experiments.
Combination index analysis
The cytotoxicity induced by the combined treatment with cisplatin and/or curcumin was compared with the cytotoxicity induced by each drug using the combination index (CI), where CI < 0.9, CI = 0.9–1.1, and CI > 1.1 indicate synergistic, additive, and antagonistic effects, respectively. The combination index analysis was performed using CalcuSyn software (Biosoft, Oxford, UK). The mean of CI values at a fraction affected (FA) of 0.90, 0.75, 0.50 were averaged for each experiment, and the values were used to calculate the mean between the three independent experiments.
Trypan blue dye exclusion assay
The cells were treated with cisplatin and/or curcumin for 1–3 days. After treatment, the cells were harvested, and the proportion of viable cells was determined by hemocytometer, counting the number of cells non-stained with trypan blue. Trypan blue dye can be excluded from living cells but is able to penetrate dead cells. The dead cells were calculated as follows: trypan blue (+) cells ratio (%) = (stained cell number / total cell number) × 100.
Colony-forming ability assay
Immediately after drug treatment, the cells were washed with phosphate-buffered saline and trypsinized to determine the cell numbers. The cells were plated at a density of 500–1000 cells on a 60 mm-diameter Petri dish in triplicate for each treatment and cultured for 12–14 days. The cell colonies were stained with 1 % crystal violet solution in 30 % ethanol. Cytotoxicity was determined by the number of colonies in the treated cells divided by the number of colonies in the untreated control.
Statistical analyses
For each protocol, three or four independent experiments were performed. Results were expressed as the mean ± SEM. Statistical calculations were performed using SigmaPlot 2000 software (Systat Software, San Jose, CA). Differences in measured variables between the experimental and control groups were assessed by unpaired t-test. P < 0.05 was considered statistically significant.
Results
The combination of cisplatin and curcumin showed a synergistic effect on the reduction of cell viability.
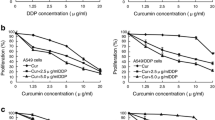
Previous study indicated that the combination of curcumin and cisplatin enhanced growth suppression in head and neck squamous cell carcinoma (Duarte et al. 2010); therefore, we attempted to determine whether curcumin could enhance the cytotoxic effects of cisplatin in NSCLC cells. According to a previous study, NSCLC cells treatment with chemotherapeutic drug mitomycin C (MMC) plus curcumin at a ratio of 1:2 resulted in increasing the sensitivity of NSCLC cells to MMC (Weng et al. 2012); therefore, cisplatin and curcumin were combined at the 1:2 or 1:4, and the effect of combined treatment on cell viability was examined by MTS assay. Combined treatment with curcumin and cisplatin for 24 h resulted in a greater loss of cell viability in A549 and H1703 cells than treatment with either curcumin or cisplatin alone (Fig. 1a). The CI values for cisplatin and curcumin were <1, indicating that the combination treatment had a synergistic effect (Fig. 1b). In addition, A549 and H1703 cells were exposed to cisplatin and/or curcumin for 1–3 days and cell proliferation was determined by MTS and trypan blue dye exclusion assay (Fig. 1c, d). Cisplatin and curcumin co-treatment had a greater cell growth inhibition effect than either treatment alone (Fig. 1c, d). Colony-forming assays were conducted to investigate whether curcumin affected cisplatin-mediated long-term clonogenic cell survival in NSCLC cells. In Fig. 1e, curcumin significantly suppressed the cell colony-forming ability of cisplatin-exposed A549 and H1703 cells. The results showed that combined cisplatin and curcumin had a synergistic cytotoxic effect on human NSCLC cells.

Curcumin co-treatment with cisplatin synergistically enhanced cytotoxicity. a Cisplatin and curcumin were combined at a ratio of 1:2 or 1:4 and the MTS assay was used to analyze cell viability. The mean of cell viability was calculated amount of three independent experiments. b The mean CI values at each fraction affected (FA) of 0.50, 0.75, and 0.90 for cisplatin and curcumin combined treatment were calculated form three independent experiments. c Cells were treated with cisplatin (1 μM) and/or curcumin (10 or 20 μM) for 1–3 days after which living cells were determined by MTS assay. **p < 0.01 using the Student’s t test for comparison between cells treated with a drug alone or with a cisplatin/curcumin combination. d Cells were treated with cisplatin (20 μM) and/or curcumin (40 μM) for 1–3 days after which living cells were determined by trypan blue dye exclusion assay. **p < 0.01 using Student’s t test for comparison between cells treated with cisplatin alone or with a cisplatin and curcumin combination. e Cisplatin (1 μM) and/or curcumin (10 μM) were added to cells for 24 h, and cytotoxicity was determined by colony-forming ability assay. **p < 0.01, using Student’s t test for comparison between the cells treated with cisplatin alone or cisplatin/curcumin combination
Cisplatin increased phospho-p38, phospho-MKK3/6, XRCC1 protein, and mRNA expression in A549 and H1703 cells.
Next, to know whether cisplatin treatment could affect XRCC1 expression. we assessed A549 or H1703 cells treated with cisplatin (20 μM) for 4–24 h or cisplatin (2.5, 5, 10, 20 μM) for 24 h, and the real-time PCR was used for determination of the XRCC1 mRNA level. The protein levels of XRCC1 were determined by Western blot analysis. In Fig. 2a, b, cisplatin induced XRCC1 mRNA and protein expression in a time and dose-dependent manner and also increased phospho-p38 and phospho-MKK3/6 protein levels. In addition, to determine whether p38 activation was involved in up-regulation of XRCC1 by cisplatin, A549 or H1703 cells were pretreated with p38 inhibitors (SB2023580) to block cisplatin-induced p38 activation. In Fig. 3a, b, the addition of SB2023580 decreased cellular and cisplatin-induced p38 activation. Moreover, SB2023580 pretreatment decreased XRCC1 mRNA and protein levels in cisplatin-exposed cells. Furthermore, knockdown the p38 MAPK expression by specific p38 MAPK siRNA in A549 or H1703 cells decreased the XRCC1 expression in response to cisplatin as compared with the control cells (Fig. 3c, d). In contrast, transfection with MKK6E (a constitutively active form of MKK6) increased cellular p38 MAPK phosphorylation and XRCC1 mRNA and protein expression compared with transfection with the pcDNA3 control vector (Fig. 3e, f). Therefore, we concluded that cisplatin increased XRCC1 expression in a p38 MAPK activation manner.

Cisplatin increased XRCC1 expression in a dose- and time-dependent manner. a A549 or H1703 cells (106) were cultured in complete medium for 18 h and then exposed to cisplatin (20 μM) for 4–24 h or various concentrations of cisplatin (2.5–20 μM) for 24 h in complete medium. The total RNA was isolated and subjected to real-time PCR for XRCC1 mRNA expression. The results (mean ± SEM) were from three independent experiments. b After treatment, the cell extracts were examined by Western blot for determination of XRCC1, phospho-MKK3/6, phospho-p38, actin, MKK3, and p38 MAPK protein levels

Cisplatin increased XRCC1 expression in a p38 MAPK activation manner. a Upper panel various concentrations of SB2023580 were added to the cells for 24 h. Lower panel SB2023580 (10 μM) was added to A549 or H1703 cells for 1 h before cisplatin treatment for 24 h. The total RNA was isolated and subjected to real-time PCR for XRCC1 mRNA expression. The results (mean ± SEM) were from three independent experiments. **p < 0.01 using Student’s t test for comparison between the cells treated with cisplatin–DMSO or an cisplatin–SB2023580 combination. b After treatment as above, the cell extracts were examined by Western blot. c and d A549 or H1703 cells (5 × 105) were cultured in a 60-mm Petri dish for 18 h and then transfected with si-p38 RNA. After incubation for 1 day, the cells were treated with 10 μM cisplatin for 24 h. **p < 0.01, respectively, using Student’s t test to compare cells treated with cisplatin in si-p38 RNA vs. si-control-transfected cells. e and f MKK6E expression vectors were transferred into cells using lipofection and allowed to express for 2 days. **p < 0.01, respectively, using Student’s t test to compare cells transfected with MKK6E expression vectors vs. pcDNA3-transfected cells. After treatment as the above, total RNA was isolated and subjected to real-time PCR for XRCC1 mRNA expression (c and e). The cell extracts were examined by Western blot for determination of XRCC1, phospho-p38, p38, and actin protein levels (d and f)
Inactivation of p38 activity enhanced XRCC1 mRNA and protein instability upon cisplatin treatment.
Next, we examined whether p38 signal was involved in post-transcriptional regulation of XRCC1 transcripts under cisplatin treatment. To evaluate the mRNA stability of XRCC1 in cisplatin-exposed A549 or H1703 cells, we treated the cells with actinomycin D to block de novo RNA synthesis, and then measured the levels of existing XRCC1 mRNA using real-time PCR at 8, 12, and 16 h after treatment. In Fig. 4a, after actinomycin D co-treatment, SB2023580 pretreatment could enhance XRCC1 mRNA instability in cisplatin-exposed A549 and H1703 cells. Then, cycloheximide (an inhibitor of de novo protein synthesis) was added to cisplatin treatment for 8, 12, and 16 h, and the remaining XRCC1 protein was analyzed by Western blot. In Fig. 4b, XRCC1 protein levels were progressively reduced with time in the presence of cycloheximide. However, cisplatin treatment significantly prevented XRCC1 degradation after cycloheximide treatment compared with untreated cells. In Fig. 4b, less XRCC1 protein remained with SB2023580 co-treatment, compared with cisplatin alone. Therefore, we concluded that the inactivation of p38 kinase activity decreased cisplatin-induced XRCC1 expression via promoting mRNA and protein instability in A549 and H1703 cells.

Cisplatin increased XRCC1 mRNA and protein stability in NSCLC cells in a p38 MAPK activation manner. a A549 or H1703 cells (5 × 105) were pretreated with SB2023580 for 1 h; the cells were exposed to cisplatin (10 μM) or DMSO for 16 h in the presence or absence of actinomycin D (2 μg/mL) for 8, 12, or 16 h; total RNA was isolated and subjected to real-time PCR for XRCC1 mRNA expression. b A549 or H1703 cells were pretreated with SB2023580 for 1 h, then the cells were exposed to cisplatin (20 μM) for 16 h followed by co-treatment with cycloheximide (CHX; 0.1 mg/mL) for 8, 12, or 16 h. Whole-cell extracts were collected for Western blot analysis
Knockdown of XRCC1 or blocking p38 activation enhanced cisplatin-induced cytotoxicity and growth inhibition in NSCLC cells.
We next examined the effect of siRNA-mediated XRCC1 knockdown on cisplatin-induced cytotoxicity and cell growth inhibition in NSCLC cells. At 24 h post-transfection of si-XRCC1, real-time PCR and Western blot analysis showed a decrease in XRCC1 mRNA and protein in cisplatin-treated A549 and H1703 cells (Fig. 5a). However, knockdown XRCC1 expression did not affect the cisplatin-induced p38 MAPK activity (Fig. 5a). Furthermore, suppression of XRCC1 protein expression by si-XRCC1 RNA resulted in increased sensitivity to cisplatin compared to si-control transfected cells (Fig. 5b). We also conducted a cell growth inhibition assay to evaluate the synergistic effects of XRCC1 knockdown with cisplatin treatment. More inhibition of cell growth was induced by the combination of XRCC1 siRNA and cisplatin than by cisplatin alone in A549 or H1703 cells (Fig. 5c). Therefore, downregulation of XRCC1 expression could enhance cisplatin-induced cytotoxicity and growth inhibition in NSCLC cells.

Knockdown of XRCC1 expression by si-RNA transfection enhanced the cytotoxicity induced by cisplatin. a The cells were transfected with siRNA duplexes (200 nM) specific to XRCC1 or scrambled (control) in complete medium for 24 h prior to treatment with cisplatin in complete medium for 24 h; total RNA was isolated and subjected to real-time PCR for XRCC1 mRNA expression (upper panel). Whole-cell extracts were collected for Western blot analysis using specific antibodies against XRCC1 (lower panel). b After the above-mentioned treatment, cytotoxicity was determined by the MTS assay. c After the cells were transfected with si-XRCC1 or si-scrambled RNA, the cells were treated with cisplatin (5, 10 μM) for 1–3 days, after which living cells were determined by MTS assay. The results (mean ± SEM) were from three independent experiments. **p < 0.01 using Student’s t test for comparison between the cells treated with cisplatin in si-XRCC1 RNA or si-scrambled RNA-transfected cells. d and e A549 or H1703 cells were pretreated with SB2023580 (1, 5, 10 μM) for 1 h and then co-treated with cisplatin (5 μM) for 24 h. Cytotoxicity was determined by MTS assay (d) and trypan blue dye exclusion assay (e)
Next, the role of p38 MAPK in the cytotoxic effect of cisplatin was examined; the p38 inhibitor SB2023580 was added to block p38 activation, respectively. Co-treatment with SB2023580 further decreased cell viability significantly in cisplatin-exposed A549 or H1703 cells, compared with cisplatin treatment alone (Fig. 5d, e). Taken together, the inactivation of the MKK3/6-p38 MAPK signal enhanced cisplatin-induced cytotoxicity in NSCLC cells.
Curcumin decreased cisplatin-induced XRCC1 protein and mRNA expression in human lung cancer cells.
Previously, Xu et al. reported irinotecan enhanced effect of cisplatin on resistant gastric cancer cells via promoting the degradation of XRCC1 (Xu et al. 2014); therefore, we hypothesized that curcumin may affect XRCC1 expression in cisplatin treated NSCLC cells. To test this hypothesis, A549 and H1703 cells were exposed to various concentrations of cisplatin and curcumin for 24 h. In Fig. 6a, b, curcumin decreased XRCC1 mRNA and protein levels in A549 and H1703 cells. As a result, XRCC1 mRNA and protein levels were induced by cisplatin treatment in a dose-dependent manner but inhibited by curcumin treatment (Fig. 6a, b). In addition, curcumin suppressed the protein levels of phospho-p38, phospho-MKK3/6 in cisplatin-exposed NSCLC cells (Fig. 6b).

Curcumin decreased XRCC1 protein and mRNA levels in cisplatin-exposed NSCLC cells. a and b A549 or H1703 cells (106) were cultured in complete medium for 18 h and then were exposed to cisplatin (5, 10, 20 μM) and curcumin (10 μM) for 24 h. After treatment as the above, total RNA was isolated and subjected to real-time PCR for XRCC1 mRNA expression (a). The cell extracts were examined by Western blot for determination of XRCC1, phospho-p38, actin, and p38 protein levels (b). The means ± standard deviation (SD) from four independent experiments. **p < 0.01, respectively, using Student’s t test for comparison between the cells treated with cisplatin/curcumin alone or combined. c and d MKK6E (5 μg) or pcDNA3 (5 μg) expression plasmids were transfected into cells using lipofectamine. After expression for 24 h, the cells were treated with cisplatin and curcumin for an additional 24 h, and total RNA was isolated and subjected to real-time PCR for XRCC1 mRNA expression. The means ± standard deviation (SD) from four independent experiments. **p < 0.01, respectively, using Student’s t-test to compare cells treated with cisplatin and curcumin in MKK6E vs. pcDNA3-transfected cells (c). The whole-cell extracts were collected for Western blot analysis (d). e and f HA-p38 (5 μg) or pcDNA3 (5 μg) expression plasmids were transfected into cells using lipofectamine. After expression for 24 h, the cells were treated with cisplatin (5 μM) and curcumin (20 μM) for an additional 24 h, and total RNA was isolated and subjected to real-time PCR for XRCC1 mRNA expression. The means ± SD from four independent experiments. **p < 0.01, respectively, using Student’s t test to compare cells treated with cisplatin and curcumin in HA-p38 vs. pcDNA3-transfected cells (e). The whole-cell extracts were collected for Western blot analysis (f). g and h After MKK6E (5 μg) or HA-p38 expression plasmids transfection, cells were treated with cisplatin and curcumin for 24 h. Cytotoxicity was determined by assessment with the MTS assay. **p < 0.01, *p < 0.05 by Student’s t test to compare cells treated with cisplatin and curcumin in MKK6E/HA-p38 vs. pcDNA3-transfected A549 or H1703 cells
Transfection with MKK6E or HA-p38 MAPK vectors enhanced the XRCC1 protein level and the cell survival suppressed by curcumin and cisplatin.
We investigated whether curcumin-mediated XRCC1 downregulation was correlated with MKK3/6-p38 downregulation in cisplatin and curcumin-exposed NSCLC cells. Overexpression of MKK6E (Fig. 6c, d) or HA-p38 MAPK vector (Fig. 6e, f) could rescue the cellular XRCC1 mRNA and protein levels that were suppressed by curcumin and cisplatin. Also, both MKK6E and HA-p38 MAPK vector transfection could rescue A549 and H1703 cell viability after being decreased by curcumin and cisplatin (Fig. 6g, h). Therefore, downregulation of p38 MAPK-mediated XRCC1 expression by curcumin enhanced the cisplatin-induced cytotoxicity in A549 and H1703 cells.
Discussion
The safety of curcumin has been widely demonstrated and has a variety of potential pharmaceutical applications. It has shown to prevent tumor initiation, proliferation, and metastasis process in many human cancers (Reuter et al. 2008). It has been reported that curcumin and 5-fluorouracil synergistic inhibition of the growth of human colon cancer cell line HT-29 was associated with the decreased expression of cyclooxygenase-2 protein (Du et al. 2006). In our previous study, enhancement of the cytotoxicity to cisplatin by administration of curcumin is mediated by downregulation of the expression levels of ERCC1 and thymidine phosphorylase in NSCLC cells (Tsai et al. 2011). The present data showed that curcumin decreased cisplatin-induced XRCC1 expression to enhance cytotoxicity in human lung cancer cells. Moreover, we found that curcumin could enhance cisplatin sensitivity in human fibroblasts HFW cells (Supplementary Fig. S1). Therefore, curcumin could be used as potential candidate for lung cancer therapy.
In the previous study, curcumin-inhibited activation of p38 may be achieved through activation of MAPK phosphatase-1 (MKP-1) which acts as a negative regulator of p38 MAPK in HT22 hippocampal cells (Pae et al. 2009). Our present data showed that XRCC1 expression by cisplatin was correlated with p38 activity. On the other hand, curcumin has been previously reported to govern a number of cellular target involved in regulation of apoptosis (Kumar et al. 2016). Moreover, p38 MAPK has been shown to be involved in apoptosis by acting both upstream and downstream of caspases cascade (Zarubin and Han 2005). In this study, downregulation of p38 MAPK activation by curcumin enhanced the cisplatin-induced cytotoxicity in NSCLC cells. However, the detailed molecular mechanism of curcumin-downregulated p38 MAPK activity upon cisplatin exposure and effects of curcumin and its combination with cisplatin on apoptosis in NSCLC cells were under our investigation.
XRCC1 is a key mediator of SSBs DNA repair, which includes both base excision repair and nucleotide excision repair mechanisms (Tebbs et al. 1999; Moser et al. 2007). XRCC1 was found to identify and bind to cisplatin-induced DNA interstrand crosslinks (Zhu and Lippard 2009). In this study, downregulation of XRCC1 by p38 MAPK inhibitor or curcumin played a role in enhancing cisplatin-induced cytotoxic effects in NSCLC cells. A recent study indicated that in HepG2 hepatocellular cancer cells, depletion of XRCC1 resulted in hypersensitivity to cisplatin chemotherapy (Zhang et al. 2010). Moreover, inhibition of XRCC1 expression by irinotecan leads to an increase in the sensitivity of resistant cells to cisplatin in gastric cancer (Xu et al. 2014).
Taken together, we report first that curcumin has a synergistic cytotoxic effect with cisplatin in NSCLC cells through suppression of XRCC1. Downregulation of XRCC1 expression by curcumin may inhibit the repair of platinum-DNA adduct in NSCLC cells. We suggest that decreasing XRCC1 expression may enhance the therapeutic effect of cisplatin in patients with NSCLC. Although further study is required to evaluate the effect of curcumin, cisplatin, and their combination in vivo, the concept of curcumin combined with cisplatin seems to present a strategy for the treatment of NSCLC.
Abbreviations
- CFA:
-
colony-forming ability
- XRCC1:
-
X-ray repair cross-complement group 1
- siRNA:
-
small interfering RNA
- MAPK:
-
mitogen-activated protein kinase
- NSCLC:
-
non-small cell lung cancer
References
Brem R, Hall J (2005) XRCC1 is required for DNA single-strand break repair in human cells. Nucleic Acids Res 33:2512–2520
Chen YR, Tan TH (1998) Inhibition of the c-Jun N-terminal kinase (JNK) signaling pathway by curcumin. Oncogene 17:173–178
Cheng Y, Chang LW, Tsou TC (2006) Mitogen-activated protein kinases mediate arsenic-induced down-regulation of survivin in human lung adenocarcinoma cells. Arch Toxicol 80:310–318
Chuang SM, Wang LH, Hong JH, Lin YW (2008) Induction of Rad51 protein levels by p38 MAPK decreases cytotoxicity and mutagenicity in benzo[a]pyrene-exposed human lung cancer cells. Toxicol Appl Pharmacol 230:290–297
Du B, Jiang L, Xia Q, Zhong L (2006) Synergistic inhibitory effects of curcumin and 5-fluorouracil on the growth of the human colon cancer cell line HT-29. Chemotherapy 52:23–28
Duarte VM, Han E, Veena MS, Salvado A, Suh JD, Liang LJ, Faull KF, Srivatsan ES, Wang MB (2010) Curcumin enhances the effect of cisplatin in suppression of head and neck squamous cell carcinoma via inhibition of IKKbeta protein of the NFkappaB pathway. Mol Cancer Ther 9:2665–2675
Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G (2012) Molecular mechanisms of cisplatin resistance. Oncogene 31:1869–1883
Han SS, Keum YS, Seo HJ, Surh YJ (2002) Curcumin suppresses activation of NF-kappaB and AP-1 induced by phorbol ester in cultured human promyelocytic leukemia cells. J Biochem Mol Biol 35:337–342
Kelland L (2007) The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer 7:573–584
Kudo K, Gavin E, Das S, Amable L, Shevde LA, Reed E (2012) Inhibition of Gli1 results in altered c-Jun activation, inhibition of cisplatin-induced upregulation of ERCC1, XPD and XRCC1, and inhibition of platinum-DNA adduct repair. Oncogene 31:4718–4724
Kumar G, Mittal S, Sak K, Tuli HS (2016) Molecular mechanisms underlying chemopreventive potential of curcumin: current challenges and future perspectives. Life Sci 148:313–328
Massarelli E, Herbst RS (2006) Use of novel second-line targeted therapies in non-small cell lung cancer. Semin Oncol 33:S9–16
Montopoli M, Ragazzi E, Froldi G, Caparrotta L (2009) Cell-cycle inhibition and apoptosis induced by curcumin and cisplatin or oxaliplatin in human ovarian carcinoma cells. Cell Prolif 42:195–206
Moser J, Kool H, Giakzidis I, Caldecott K, Mullenders LH, Fousteri MI (2007) Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol Cell 27:311–323
Pae HO, Jeong SO, Zheng M, Ha HY, Lee KM, Kim EC, Kim DH, Hwang SY, Chung HT (2009) Curcumin attenuates ethanol-induced toxicity in HT22 hippocampal cells by activating mitogen-activated protein kinase phosphatase-1. Neurosci Lett 453:186–189
Persons DL, Yazlovitskaya EM, Cui W, Pelling JC (1999) Cisplatin-induced activation of mitogen-activated protein kinases in ovarian carcinoma cells: inhibition of extracellular signal-regulated kinase activity increases sensitivity to cisplatin. Clin Cancer Res 5:1007–1014
Pfister DG, Johnson DH, Azzoli CG, Sause W, Smith TJ, Baker S Jr, Olak J, Stover D, Strawn JR, Turrisi AT, Somerfield MR (2004) American Society of Clinical Oncology treatment of unresectable non-small-cell lung cancer guideline: update 2003. J Clin Oncol 22:330–353
Reuter S, Eifes S, Dicato M, Aggarwal BB, Diederich M (2008) Modulation of anti-apoptotic and survival pathways by curcumin as a strategy to induce apoptosis in cancer cells. Biochem Pharmacol 76:1340–1351
Rosell R, Lord RV, Taron M, Reguart N (2002) DNA repair and cisplatin resistance in non-small-cell lung cancer. Lung Cancer 38:217–227
Rudin CM, Yang Z, Schumaker LM, VanderWeele DJ, Newkirk K, Egorin MJ, Zuhowski EG, Cullen KJ (2003) Inhibition of glutathione synthesis reverses Bcl-2-mediated cisplatin resistance. Cancer Res 63:312–318
Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, Johnson DH (2002) Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 346:92–98
Shishodia S, Sethi G, Aggarwal BB (2005) Curcumin: getting back to the roots. Ann N Y Acad Sci 1056:206–217
Siddik ZH (2003) Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 22:7265–7279
Siddiqui-Jain A, Bliesath J, Macalino D, Omori M, Huser N, Streiner N, Ho CB, Anderes K, Proffitt C, O’Brien SE, Lim JK, Von Hoff DD, Ryckman DM, Rice WG, Drygin D (2012) CK2 inhibitor CX-4945 suppresses DNA repair response triggered by DNA-targeted anticancer drugs and augments efficacy: mechanistic rationale for drug combination therapy. Mol Cancer Ther 11:994–1005
Silvestri GA, Rivera MP (2005) Targeted therapy for the treatment of advanced non-small cell lung cancer: a review of the epidermal growth factor receptor antagonists. Chest 128:3975–3984
Tebbs RS, Flannery ML, Meneses JJ, Hartmann A, Tucker JD, Thompson LH, Cleaver JE, Pedersen RA (1999) Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Dev Biol 208:513–529
Tebbs RS, Thompson LH, Cleaver JE (2003) Rescue of Xrcc1 knockout mouse embryo lethality by transgene-complementation. DNA Repair 2:1405–1417
Tsai MS, Weng SH, Kuo YH, Chiu YF, Lin YW (2011) Synergistic effect of curcumin and cisplatin via down-regulation of thymidine phosphorylase and excision repair cross-complementary 1 (ERCC1). Mol Pharmacol 80:136–146
Usanova S, Piee-Staffa A, Sied U, Thomale J, Schneider A, Kaina B, Koberle B (2010) Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol Cancer 9:248
Weir NM, Selvendiran K, Kutala VK, Tong L, Vishwanath S, Rajaram M, Tridandapani S, Anant S, Kuppusamy P (2007) Curcumin induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by modulating Akt and p38 MAPK. Cancer Biol Ther 6:178–184
Weng SH, Tsai MS, Chiu YF, Kuo YH, Chen HJ, Lin YW (2012) Enhancement of mitomycin C-induced cytotoxicity by curcumin results from down-regulation of MKK1/2-ERK1/2-mediated thymidine phosphorylase expression. Basic Clin Pharmacol Toxicol 110:298–306
Xu W, Wang S, Chen Q, Zhang Y, Ni P, Wu X, Zhang J, Qiang F, Li A, Roe OD, Xu S, Wang M, Zhang R, Zhou J (2014) TXNL1-XRCC1 pathway regulates cisplatin-induced cell death and contributes to resistance in human gastric cancer. Cell Death Disease 5:e1055
Zarubin T, Han J (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res 15:11–18
Zhang R, Niu Y, Zhou Y (2010) Increase the cisplatin cytotoxicity and cisplatin-induced DNA damage in HepG2 cells by XRCC1 abrogation related mechanisms. Toxicol Lett 192:108–114
Zhu G, Lippard SJ (2009) Photoaffinity labeling reveals nuclear proteins that uniquely recognize cisplatin-DNA interstrand cross-links. Biochemistry 48:4916–4925
Acknowledgments
We thank Dr. Tsui-Chun Tsou, Dr. Show-Mei Chuang, and Dr. Jia-Ling Yang for providing us with expression plasmids for transfection.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Grant support
This study was funded by grants from the Ministry of Science and Technology, Taiwan, Grant MOST 104-2314-B-415-002 (Y-W. Lin) and Ditmanson Medical Foundation Chia-Yi Christian Hospital Research Program (R104-039).
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Supplementary Fig. S1
Curcumin enhanced the cisplatin-induced cytotoxic effects of human fibroblasts HFW cells. Cisplatin and curcumin were combined at a ratio of 1:2 or 1:4 and the MTS assay was used to analyze cell viability. The results (mean ± SEM) were from three independent experiments. **P < 0.01, using the Student’s t test for the comparison between cells treated with cisplatin or curcumin alone or with curcumin–cisplatin cotreatment. (JPEG 756 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Tung, CL., Jian, YJ., Chen, JC. et al. Curcumin downregulates p38 MAPK-dependent X-ray repair cross-complement group 1 (XRCC1) expression to enhance cisplatin-induced cytotoxicity in human lung cancer cells. Naunyn-Schmiedeberg's Arch Pharmacol 389, 657–666 (2016). https://doi.org/10.1007/s00210-016-1235-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-016-1235-5