
Abstract
Since previous in vitro experiments revealed that nicotine can impair endothelial intercellular communication via the downregulation of connexin43 (Cx43), we wanted to find out which nicotinic acetylcholine receptors are involved in the molecular mechanism of communication failure. Cultured human endothelial cells were exposed to 1 μM nicotine for 5 days. Intercellular communication was measured using dye transfer study with/without subtype-specific nicotinic acetylcholine receptor (nAChR) inhibitors. Reverse transcriptase (RT)-PCR was used to further investigate the regulation of nAChR subtypes. Electron microscopy together with MAP LC3-II western blot was used to investigate possible autophagy processes. In cultured human endothelial cells, nicotine decreased the Cx43 protein amount as shown by western blot and immunohistochemistry; however, together with an unaltered mRNA expression as shown by RT-PCR. The nicotine-induced Cx43 downregulation functionally impaired intercellular dye transfer, which could be prevented by mecamylamine, κ-bungarotoxin, lobeline, and dihydro-β-erythroidine but not α-bungarotoxin, indicating that the nAChR subtypes α4β2 and α3β2 but not α7 are involved in signal cascade. RT-PCR analysis revealed that nicotine exposure resulted in the upregulation of α3 and β4 and the downregulation of α4-nAChR, while α7- and β2-nAChR-mRNA expressions remained unaltered. Furthermore, nicotine increased total protein ubiquinylation and proteasome activity as was shown by immunohistochemistry and peptide degradation analysis. Evidence of enhanced autophagic processes was assured by the occurrence of autophagic vacuoles in transmission electron microscopy and enhanced formation of MAP LC3-II in western blot. Reduced intercellular endothelial communication together with programmed cell death helps to explain the toxic effect of nicotine leading to endothelial dysfunction. The nAChR involved in the impairment of intercellular communication seem to be α4β2 and α3β2 but not α7.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acetylcholine (ACh) and its receptors are among the best characterized neurotransmitter/receptor systems (Conti-Tronconi et al. 1994; Changeux 1995; Lindstrom 1995; Albuquerque et al. 1997). Two principal forms are to be distinguished: muscarinic acetylcholine receptors (mAChRs) and nicotinic acetylcholine receptors (nAChRs). Besides, in nerve cells, nAChRs were found in several other cell types such as keratinocytes (Grando et al. 1995), epithelial cells (Conti-Fine et al. 2000; Wessler et al. 1998), and endothelial cells (Macklin et al. 1998). In contrast to nerve cells, the physiological role of nAChRs in endothelial cells is not well understood yet. Since the endothelial cells also can synthesize and secrete ACh, an autocrine or paracrine mechanism of the intra- or intercellular signalling of ACh has been suggested (Sastry and Sadavongvivad 1979; Wessler et al. 1995; Wessler and Kirkpatrick 2008). In endothelial cells, it is well known that muscarinic AChRs control no release and, at least in parts, prostacyclin release. These muscarinic AChRs belong to the class of G-protein-coupled receptors and are probably of the M3 subtype in endothelial cells (for review, see Dhein et al. 2001). In contrast, there is only very little known about nicotinic ACHRs which belong to the class of ligand-gated ion channels, which possess a permeability for Na+, Ca++, or K+, depending on the isoform. Moreover, almost nothing is known about their role in the endothelial physiology and pathophysiology.
Two different principal subtypes of nAChR are known: (1) the “muscle type” which is expressed in the motor endplate and involved in the transfer of electrical signals, and (2) the “neuronal type” which was recently found in nerve cells and seems to play a role by the maintenance of cell-to-cell contacts (Wessler et al. 1998). The nAChR is a multisubunit ligand-gated ion channel and consists of five subunits (Bertrand and Changeux 1995), for example in the muscle type, two α-subunits, 1 β-subunit, 1 γ-subunit, and 1 δ-subunit are found. Among these nine different types of α-subunits and three different types of β-subunits can be discriminated to (α2–α10) and (β2–β4) (McGehee and Role 1995), respectively. From these subunits, nAChR of different stoichiometries can be composed. Thus, in neuronal nAChR, other stoichiometries are found, such as (α4)2(β2)3, (α3)2(α5)1(β4)1(βX)1, or (α7)5 (Le Novère et al. 2002; Thany et al. 2007). The subunits α2–α6 are able to assemble together with β-subunits in heteropentamers, while the subunits α7, α9, and α10 form only functional homopentamers or α-heteropentamers (Couturier et al. 1990; Elgoyhen et al. 2001; Khiroug et al. 2002; Sgard et al. 2002). The composition of the subunits in the receptor determines the ligand specificity, binding affinity, cation permeability, and channel kinetics (Buisson et al. 2000). An important point about these receptors is that they desensitize in a subtype-specific extend with chronic nicotine stimulation (Giniatullin et al. 2005). Thus, it can be assumed that in chronic nicotine exposure, other nAChRs are involved than in acute stimulation.
In the last years, we could show that nicotine affects endothelial cells and angiogenesis via changes in the expression of the endothelial gap junction proteins (Haussig et al. 2008; Dhein et al. 2011). However, only very little is known about the nAChR subtypes mediating the effects of nicotine on intercellular communication and on their physiological role. Therefore, the aim of the present study was to find out which nAChR subtypes are involved in the effect of nicotine on endothelial intercellular communication.
Methods
The study was approved by the local Institutional Ethical Committee and conformed to the declaration of Helsinki.
Cell culture of endothelial cells
Human umbilical vein endothelial cells (HUVECs) were isolated from fresh umbilical cords using collagenase IV as described (Morawietz et al. 1999). In order to minimize variations of the primary cultures, the isolated endothelial cells from different umbilical cords were pooled and cultured in the medium M199 containing Earle’s salts, 100 mg/L l-glutamine, 25 mmol/L HEPES (Gibco, Karlsruhe, Germany), 10% (vol/vol) fetal calf serum (Biochrom AG, Berlin, Germany), 16.7 mg/L endothelial cell growth supplement (C. C. Pro, Neustadt, Germany), 100,000 U/L penicillin, and 100 mg/L streptomycin (Gibco, Karlsruhe, Germany) at 37°C and 5% CO2. Besides controls, endothelial cells were treated with 1 μmol/L nicotine (Sigma, Taufkirchen, Germany) with/without inhibitors over 5 days. Each day, the medium with/without nicotine/inhibitors was replaced.
Immunohistochemistry
HUVECs grown on coverslips were washed with Tyrode’s solution (10 mmol/L HEPES, pH 7.4; 135 mmol/L NaCl; 4 mmol/L KCl; 2 mmol/L CaCl2; 1 mmol/L MgCl2; 0.33 mmol/L NaH2PO4), fixed with 4% formaldehyde/PBS for 15 min, washed again with PBS for 10 min, and permeabilized with 0.05% (vol/vol) Tween 20/PBS two times each for 10 min. After washing with PBS for 10 min, nonspecific binding sites were saturated using 2% (wt/vol) BSA/PBS for 30 min. For Cx43 expression analysis, cells were incubated with rabbit anti-Cx43 antibody (Sigma, Taufkirchen, Germany) overnight at 4°C, washed with 0.05% (vol/vol) Tween 20/PBS two times each for 10 min and with PBS for 10 min, and incubated with Alexa Fluor 488-conjugated goat anti-rabbit antibody for 30 min. After washing out with 0.05% (vol/vol) Tween 20/PBS two times each for 10 min and PBS for 10 min, nuclei were stained with 0.5 μg/mL DAPI (Roche Diagnostics, Mannheim, Germany). Coverslips were mounted upside down in Dako Glycergel containing 25 μg/mL 1,4-diacabicyclo[2,2,2]octane. The Cx43 expression was analysed using a fluorescence microscope (Axioplan 2; Zeiss) equipped with video camera and image analysis system (Axiovision; Zeiss).
To assess a possible induction of apoptosis, we investigated the nuclear translocation of apoptosis-inducing factor (AIF) (Candé et al. 2002; Zhang et al. 2004) using rabbit anti-AIF antibody (Santa Cruz, Santa Cruz, USA) overnight at 4°C and an HRP-labelled goat anti-rabbit antibody (Sigma, Taufkirchen, Germany) as the secondary antibody (1 h, 21°C) followed by tyramide signal amplification (TSA Biotin System, Perkin Elmer, Boston, USA) and 3,3′-diaminobenzidine (DAB) staining (Roth, Karlsruhe, Germany), according to the manufacturer’s protocol. We evaluated the number of positively stained nuclei related to the total number of endothelial cells on the basis of evaluating 760 cells.
For analysis of ubiquitinylated proteins, HUVECs were incubated with anti-mono- and anti-poly-ubiquitinylated protein antibodies (1:100; Biomol, Plymouth Meeting, PA, USA) overnight at 4°C. After washing with 0.05% (vol/vol) Tween 20/PBS and with PBS, the primary antibody was detected with peroxidase-labeled goat anti-rabbit IgG (Sigma) diluted 1:200 for 30 min. After rinsing with 0.05% (vol/vol) Tween 20/PBS and with PBS, cells were incubated in ACE-positive high sensitivity substrate chromogen (DakoCytomation) for 20 min and washed with pure aqua for 5 min. Nuclei were stained with Mayer’s haemalaun (Dr. K. Hollborn & Sons, Leipzig, Germany). After rinsing with pure aqua, coverslips were mounted upside down in Dako Glycergel, and cells were analysed using microscope (Axioplan 2; Zeiss) equipped with video camera and image analysis system (Axiovision; Zeiss).
Western blot
HUVECs were washed with Tyrode’s solution, subsequently collected by scraping in lysis buffer (20 mmol/L Na2HPO4 2 H2O, pH 7.4; 150 mmol/L NaCl; 2 mmol/L MgCl2 6 H2O; 0.1% (vol/vol) Nonidet P40; 10% (vol/vol) glycerol; 1% (vol/vol) Triton X100; 1 mg/mL aprotinin; 1 mg/mL leupeptin; 1 μmol/L okadaic acid; 10 mmol/L phenylarsine oxide; 10 mmol/L cantharidin; 10 mmol/L Na-orthovanadate; 10 μg/mL pepstatin A; 100 μmol/L PMSF; 10 mmol/L NaF; 20 mmol/L Na-pyrophosphate), and homogenized by ultrasonification. Thirty micrograms protein of each sample were mixed 1:4 with loading buffer (300 mmol/L Tris/HCl, pH 6.8; 2.8% (vol/vol) β-mercaptoethanol; 40% (vol/vol) glycerol; 140 mmol/L SDS; 0.02% (wt/vol) bromophenol blue), denatured for 5 min at 95°C, separated using SDS-PAGE, and transferred to a nitrocellulose membrane. The nonspecific binding sites were blocked with 5% (wt/vol) milk powder in Tris-buffered saline (TBS-T) buffer (10 mmol/L Tris/HCl, pH 7.3; 500 mmol/L NaCl; 0.2% (vol/vol) Tween 20), and then the membrane was treated with rabbit anti-Cx43 (1:5,000) and rabbit anti-MAP LC3 antibody (1:4,000; Santa Cruz Biotechnology, Santa Cruz, Ca, USA) overnight at 4°C. After rinsing, the primary antibody was detected with peroxidase-labeled goat anti-rabbit IgG diluted 1:5,000 for 1 h and by using Uptilight HRP Blot Chemiluminescent Substrate (KMF, Lohmar, Germany) according to the manufacturer’s instructions, and Kodak XOmat AR films. After rinsing the nitrocellulose membranes, GAPDH protein level was detected with mouse anti-GAPDH antibody (1:5,000; Acris, Herford, Germany) and rabbit anti-mouse IgG (1:5,000; Sigma) to equalize the Cx43 protein expression, and MAP LC3-I and MAP LC-II occurrences, respectively. Protein expressions were quantified using AIDA Image Analyser software (Raytest, Berlin, Germany). The unstimulated control was set to 100%.
Quantitative RT-PCR
Total RNA from the HUVECs was isolated using TRIzol Reagent (Sigma) according to the manufacturer’s recommendation. DNA remnants were degraded using 5 U/μL DNase I (RNase-free; Roche Diagnostics, Mannheim, Deutschland) for 10 min. Subsequently, DNase I was deactivated by heating to 65°C for 10 min. Two hundred fifty nanograms RNA were transcripted using Omniscript RT Kit (Qiagen, Hilden, Deutschland) according to the manufacturer’s instructions. PCRs were carried out in LightCycler® (Roche Diagnostics). For analysis of Cx43 and α3-AChR-subunit expressions, specific primers (Cx43, forward 5′-TTG CTG CGA ACC TAC ATC AT-3′; reverse 5′-ATG ATA TTC AAG GCC AGG GA-3′; NM000165; 701–927 bp; α3-AChR, forward 5′-GAA GGT GAC CCT GTG CAT TT-3′; reverse 5′-GGG GTT CTG TAG TGC ACG TT-3′; NM000743; 999–1,184 bp) were used. cDNA of 12.5 ng was mixed with the following PCR reagents: 0.25 mg/mL BSA, each 0.2 μmol/L primers, 1× buffer without MgCl2; 2.5 mmol/L MgCl2, each 0.2 mmol/L dNTPs, 1 U Taq-DNA polymerase (Invitrogen, Karlsruhe, Germany); and 1× SYBR green (Sigma). The following cycling conditions were applied: denaturation at 95°C for 10 s, annealing at 60°C for 10 s, and extension at 72°C for 20 s (40 cycles).
For expression studies of α4-, α7-, β2-, and β4-nAChR subunits, specific fluorescein-labeled ProbeLibrary probes with appropriate primers and LightCycler® TaqMan® Master-Mix (Roche Diagnostics) were used according to the manufacturer’s recommendation. The following cycling conditions were applied: denaturation and Taq-DNA-polymerase activating at 95°C for 10 min, and then 40 cycles of denaturation at 95°C for 10 s, annealing at 60°C for 30 s, and extension at 72°C for 1 s.
The mRNA expressions were normalized using 18S rRNA expression with specific primers (forward 5′-TAG AGG GAC AAG TGG CGT TC -3′; reverse 5′-TGT ACA AAG GGC AGG GAC TT-3′; U13369; 5,101–5,355 bp) and the following conditions: denaturation at 95°C for 10 s, annealing at 62°C for 10 s, and extension at 72°C for 20 s (30 cycles). The unstimulated control was set to 100%.
Dye transfer
Endothelial cells were grown on coverslips and stimulated with and without 1 μmol/L nicotine and specific inhibitors against nAChR subtypes like 1 μmol/L mecamylamine (inhibits all nAChR subtypes; Sigma), 10 nmol/L α-bungarotoxin (inhibits α7-AChR; Sigma), 100 nmol/L κ-bungarotoxin (inhibits α3-AChR; Biotoxins Inc., St. Cloud, FL, USA), 5 nmol/L cytisine (inhibits β2-AChR; Tocris Cookson Inc., Ellisville, MO, USA), 10 nmol/L lobeline (inhibits α4β2-AChR; Tocris Cookson Inc.), and 100 nmol/L dihydro-β-erythroidine (inhibits α4β2 and α3β2-AChR; Sigma), or for the inhibition of muscarinic receptors 1 μmol/L atropine (Sigma) for 5 days. The dye transfer assay using the fluorescent dye Lucifer Yellow was described recently (Haussig et al. 2008). Briefly, coverslips with cells were transferred to a 1 mL organ bath superfused with Tyrode’s solution (135 mmol/L NaCl, 4 mmol/L KCl, 2 mmol/L CaCl2, 1 mmol/L MgCl2, 0.33 mmol/L NaH2PO4, 10 mmol/L HEPES, 10 mmol/L glucose; pH 7.4) at 37°C. Dye coupling and transfer experiments were performed after 30–60 min nicotine washout. Subsequently, cells superfused with Tyrode’s solution under nicotine-free conditions were patched using glass pipettes of 5 MΩ filled with “intracellular” solution containing 140 mmol/L KCl, 4 mmol/L MgCl2, 0.06 mmol/L CaCl2, 5 mmol/L EGTA, 3.1 mmol/L Na2ATP, 5 mmol/L Na2creatinephosphate, 10 mmol/L HEPES, pH 7.1, and 0.5% Lucifer Yellow (Sigma, Taufkirchen, Germany). After break-in and establishing the whole cell configuration (seal resistance >3 GΩ), cells were always kept at a holding potential of −40 mV and were injected with Lucifer Yellow (we used always the same injection pressure and the same Lucifer Yellow concentration, i.e., 0.5%). The first cell was usually stained with Lucifer Yellow directly after injection within a second. Subsequently, the dye diffuses through gap junctions in neighbored and coupled cells within some minutes (Ransom and Sontheimer 1992). The dye transfer in the adjacent cells was measured by means of fluorescence microscopy (excitation, 430 nm; emission, 535 nm) 5 min after the dye injection, assessing the number of communication by the use of computer-assisted image analysis.
Proteasome activity assay
Chymotrypsin-like, trypsin-like, and peptidylglutamyl-peptide, hydrolysing activities of proteasomes, were analysed using fluorogenic peptides N-succinyl-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin (Suc-LLVY-AMC; Biomol), benzonyl-Val-Gly-Arg-7-amino-4-methyl-coumarin (Bz-VGR-AMC; Biomol), and benzyloxycarbonyl-Leu-Leu-Clu-7-amino-4-methylcoumarin (Z-LLE-AMC; Biomol). HUVECs were washed with Tyrode’s solution, subsequently harvested, and analysed as described (Tsukamoto et al. 2006). Twenty micrograms protein were incubated with and without 0.5 μmol/L MG132 (an inhibitor of proteasome activity; Sigma) in proteasome activity buffer (50 mmol/L Tris-HCl, pH 8.0; 0.5 mmol/L EDTA; 40 μmol/L Suc-LLVY-AMC, Bz-VGR-AMC, and Z-LLE-AMC, respectively). The hydrolysing activities of proteasomes were obtained over 20 min using FLUOstar OPTIMA (BMG, Jena, Germany) with excitation at 380 nm and emission at 440 nm. The proteasome activity values were calculated from the AMC calibration curve.
Transmission electron microscopy
HUVECs were grown on Thermanox coverslips (Nunc, Inc., Naperville, IL, USA) and prepared for transmission electron microscopy like previously described (Duerrschmidt et al. 2006). Ultrathin sections were examined in a Zeiss EM 10 (Zeiss, Jena, Germany).
Statistics
The data of each experiment are shown as mean ± SEM. Statistical analysis was performed with Student’s t test and ANOVA procedure using Bonferroni’s method (SigmaStat, Jandel Scientific, San Rafael, CA). Differences were considered statistically significant if p < 0.05.
Results
Reduced protein amount of Cx43 is unaffected of mRNA expression regulation
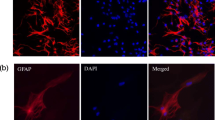
The finding of an impaired Cx43 protein expression opened the hypothesis that nicotine may influence the Cx43 regulation by reduced transcription. To prove this, we investigated Cx43 protein and Cx43-mRNA expression simultaneously in cultured endothelial cells using western blot and reverse transcriptase (RT)-PCR. We found in our cell culture model a decreased total Cx43 protein amount under nicotine exposure using western blot analysis (p < 0.05; Fig. 1a, b). However, to our surprise, Cx43-mRNA expression was not altered by nicotine (105 ± 6% of control, n = 8; Fig. 1c). Immunohistochemical analysis revealed that the reduction in Cx43 protein in these cells was particularly seen in the cell membranes of the endothelial cells (Fig. 1d). While under control conditions, 45.7 ± 6.3% of the membrane length was positively stained for Cx43; after nicotine exposure, this membrane-Cx43-immunopositivity was significantly decreased to 9.3 ± 2.8% (p < 0.05). Thus, we made the hypothesis that this reduction might have functional consequences (see “Dye transfer studies show reduced intercellular communication due to nicotine”).

Nicotine reduced the Cx43 protein amount but did not affect the Cx43-mRNA expression. HUVECs were cultured with and without 1 μmol/L nicotine over 5 days. Total RNAs and proteins was isolated from the endothelial cells and analysed using western blot (n = 4; p < 0.05 vs. control; a and b) and RT-PCR (n = 8; c). Moreover, Cx43 protein from the control HUVECs and from the HUVECs exposed to nicotine for 5 days was investigated using immunohistochemistry. The nuclei were stained using DAPI (bars = 20 μm; d). Please note, the lower expression of Cx43, in particular, in the membranes
Dye transfer studies show reduced intercellular communication due to nicotine
Dye injection into the endothelial cells normally resulted in a positive dye transfer to 17.3 ± 0.6 cells. This was significantly reduced after 5 days nicotine exposure down to 12.8 ± 2.1 cells (p < 0.05) (Figs. 2 and 3a), which could be completely prevented by cotreatment with the nAChR-antagonist mecamylamine (18.8 ± 1.6 cells, p < 0.05) (Fig. 3a), opening the question which nAChR subtype might be involved (see “The nAChR subtypes α4β2 and α3β2 seem to play a role in the signal cascade”).

Shows an original dye transfer experiment. Cultured HUVECs were transferred to an organ bath and clamped using a patch clamp pipette. After break-in, Lucifer yellow was injected into the cell, and the dye was transferred to the adjacent cells within 5 min. Nicotine was carefully washed out before the experiment was started


Effects of nicotine and AChR inhibitors on metabolic coupling in the HUVECs as assessed using Lucifer Yellow dye injection (see “Methods” for details; for original dye transfer image, see Fig. 2). HUVECs were cultured for 5 days with/without 1 μmol/L nicotine in the absence or presence of the nicotinic nAChR-antagonist mecamylamine (1 μmol/L; a), the muscarinic AChR-antagonist atropine (1 μmol/L; b), the subtype-specific nAChR-antagonists dihydro-β-erythroidine (α3/β2, α4/β2; 100 nmol/L; c), cytisine (β2; 5 nmol/L; d), lobeline (α4/β2; 10 nmol/L; e), κ-bungarotoxin (α3; 100 nmol/L; f), α-bungarotoxin (α7; 10 nmol/L; g). Data are given as mean ± SEM of n = 6 experiments for each bar in each panel. SEM standard error of the mean
The nAChR subtypes α4β2 and α3β2 seem to play a role in the signal cascade
Special AChR subtypes can be activated by nicotine and might be important for signal transduction. The intercellular communication was determined by dye transfer experiments using the fluorescent Lucifer Yellow in cells exposed to nicotine for 5 days with/without cotreatment with the subtype-specific nAChR-antagonists α-bungarotoxin (α7), κ-bungarotoxin (α3), lobeline (α4/β2), dihydro-β-erythroidine (α3/β2, α4/β2), and cytisine (β2), and with the mAChR-antagonist atropine. While atropine had no effect on the nicotine effects (Fig. 3b), there was some effect of atropine alone, which also reduced intercellular metabolic coupling. Moreover, the nicotinergic reduction in intercellular communication could be significantly antagonized by dihydro-β-erythroidine, by cytisine, by lobeline, by κ-bungarotoxin, but not by α-bungarotoxin (see Fig. 3c–g).
When nicotine or related agonists are continuously applied, nAChRs may become desensitized (Katz and Thesleff 1957; Quick and Lester 2002). Since nicotine binds with a high affinity to α4β2, and this subtype typically desensitizes completely for nicotine, a shift of subtype-specific effects during chronic nicotine stimulation may be assumed (Giniatullin et al. 2005). Using RT-PCR, we therefore investigated the presence of nAChR-subtype mRNA before and after 5 days nicotine exposure. Five days nicotine exposure resulted in a significant decrease in α4-nAChR-mRNA expression, while β2- and α7-nAChR-mRNA expressions remained unchanged, and α3- and β4-nAChR-mRNA expressions were slightly but significantly increased (Fig. 4).

RT-PCR results for the expression of subtype-specific nAChR-mRNA in the human endothelial cells cultured for 5 days with/without 1 μmol/L nicotine. Data are given as mean ± SEM of n = 6 experiments. Significance is indicated by an asterisk (p < 0.05). αβ
Proteasome studies
Immunohistochemical investigation of cultured endothelial cells showed enhanced ubiquitinated protein presence if cells were exposed to nicotine (Fig. 5a). To investigate whether this may indicate enhanced apoptosis, we assessed the nuclear translocation of AIF. On the basis of 760 cells, we found positively stained nuclei in 3.1 ± 0.25% of the cells in the control series and 3.3 ± 0.35% of the nicotine-treated cells, and the difference being not significant (Fig. 5b). Since ubiquitination typically is preceding proteasomal degradation, we further investigated whether the degradation of typical proteasome test peptides was also enhanced after nicotine exposure. This peptide degradation study revealed enhanced peptide degradation with particularly and significantly enhanced chymotrypsin-like hydrolytic activity, which was nearly doubled (p = 0.026) (Fig. 6).

a Ubiquitination of proteins in the HUVECs cultured for 5 days with/without 1 μmol/L nicotine. Mono- and poly-ubiquitinated proteins are stained red. b Nuclear translocation of apoptosis-inducing factor after 5 days culture with/without 1 μmol/L nicotine. AIF is stained brown. Nuclear translocation (arrows) was similar in both groups

Degradation of fluorogenic proteasome test peptides for chymotrypsin-like, trypsin-like, or peptidylglutamyl hydrolytic activity (for methodological details, see “Methods”). Data are given as proteasomal enzyme activity for the turnover of the test peptides in (nmol/L/min/mg protein) as mean ± SEM of n = 10 experiments. Significant changes are indicated by an asterisk (p < 0.05)
Autophagy typically leads to the conversion of microtubule-associated protein 1 light chain 3-I (MAP LC3-I) to its phosphatidylethanolamine-conjugated form, named MAP LC3-II (McLeland et al. 2011). In our present study, accordingly, we found that the amount of MAP LC3-II was significantly increased after nicotine exposure, while MAP LC3-I was slightly decreased (Fig. 7).

Formation of the autophagy marker LC3-II in the HUVECs cultured for 5 days with/without 1 μmol/L nicotine. The upper panel shows an original western blot. The lower panel gives the quantitative data as mean ± SEM of n = 8 experiments. Significant changes are indicated by an asterisk (p < 0.05)
ELMI studies
Since these results indicated that possibly nicotine exposure could enhance autophagy, we investigated the cells for the occurrence of vacuoles using transmission electron microscopy. Electron microscopy revealed that multilamellar vacuoles of about 0.5-μm diameter occurred after nicotine exposure, probably representing autophagic vacuoles (Fig. 8).

Transmission electron microscopy showing autophagic vacuoles in an endothelial cell exposed for 5 days to 1 μmol/L nicotine in comparison to a nonexposed cell (control)
Discussion
Chronic (5 days) nicotine exposure resulted in a downregulation of Cx43 expression and reduced metabolic coupling between endothelial cells. The antagonization of the nicotine effects by mecamylamine but not by atropine indicates that the effect is transduced via a nicotinic acetylcholine receptor and not via a muscarinic AChR. Since, furthermore, the nicotine effects regarding the functional (dye transfer) level could be antagonized by dihydro-β-erythroidine, one could assume the involvement of α3/β2 or α4/β2. The antagonization by cytisine further supports the hypothesis of β2-nAChR involvement. Regarding the α-nAChR subunits, the lack of effect of α-bungarotoxin is against a role of α7-nAChR, while the effectiveness of κ-bungarotoxin further indicates a contribution of α3-nAChR. The involvement of α4/β2, as evident from the effect of dihydro-β-erythroidine, is further supported by the observation of inhibitory effects of lobeline, which is known to inhibit α4/β2. Since it is known that nicotine exposure leads to desensitisation (Katz and Thesleff 1957; Quick and Lester 2002) and in particular to fast α4/β2 desensitization (for review, see Giniatullin et al. 2005), the contribution of α3/β2 might be higher. It is necessary to point out that the term desensitization refers to a reduced functional response, which occurs in α7-nAChR within milliseconds, in non-α7-nAChR within seconds (α4/β2), or in α3/β4-nAChR being only moderate. Changes in nAChR protein expression have been shown only partially to contribute to these desensitizing effects (Giniatullin et al. 2005). Accordingly, we only found minor changes in nAChR expression, with small (however significant) increases in α3- and β4-nAChR expressions, and a small decrease in α4-nAChR expression (Fig. 4). It must be stated that for a desensitization study additional binding experiments would be necessary, which was out of the scope of our present investigation.
However, since our functional inhibitor data show significant antagonization of intercellular communication in the pattern described above, we assume that both α4/β2 and α3/β2 but not α7-nAChR are involved. This is in some contradiction to the results of Heeschen et al. (2002) who found that nicotine exerts effects on endothelial cells via α7-nAChR in a way that could be inhibited by α-bungarotoxin. However, these authors investigated the inhibitory effect of nicotine on angiogenesis and used 12–24 h exposure times instead of 5 days as in our present study on connexin43 expression in 2D-confluent endothelial cells. Thus, one may conclude that the antiangiogenetic effects of nicotine are transduced via α7-nAChR, while the inhibitory effects on intercellular communication, as investigated in our study, are mediated via other nAChR subtypes, namely α4/β2- and α3/β2-nAChR.
Regarding the mechanism of the decrease in Cx43 and intercellular communication, we found reduced Cx43 protein expression but unchanged Cx43-mRNA. This indicates that the negative regulation of Cx43 expression by nicotine seems to be posttranscriptional and may either involve changed translational efficacy, posttranslational degradation, or both. The present data show that nicotine increases proteasomal protein degradation as became obvious from the enhanced degradation of test proteins and from the conversion of the microtubule associated protein LC3-I to its phosphatidylethanolamine-conjugated form LC3-II, a step which typically is involved in autophagy (McLeland et al. 2011). Thus, nicotine enhances autophagy, so that one may assume that the Cx43 downregulation is at least partially linked to enhanced protein degradation. However, it must be taken into account that we did not show specific Cx43 degradation but the general protein degradation and autophagic processes. To show specific Cx43 degradation, radioactive pulse-chase experiments would be necessary, which was technically not possible. However, our results are in good accordance with Tsai et al. (2004) who showed that nicotine indeed can enhance Cx43 degradation.
Conclusions
The results of our study indicate that nAChR participates in the regulation of the human endothelial cell function. This opens the possibility that nAChR may play a, yet widely unknown, role in vascular and endothelial physiology and pathophysiology. Interestingly, others described the existence of choline-acetyl-transferase in endothelial cells (Kirkpatrick et al. 2001) and that these cells can form acetylcholine (Kawashima et al. 1990), and, moreover, the existence of muscarinic receptors elicitating nitric oxide release is long known. Our finding that atropine alone also reduced to some extend the metabolic coupling (Fig. 3b) might be interpreted as indicative of endothelial acetylcholine formation which in presence of the muscarinic antagonist atropine then solely acts on nicotinic receptors, thus imitating the nicotine effects. However, this needs further investigation.
Thus, taken together, these data indicate the possibility of the existence of an autocrine cholinergic system in the endothelium with the nAChR playing a role in the regulation of intercellular endothelial communication via α4/β2- and α3/β2-nAChR.
Abbreviations
- Cx43:
-
Connexin43
- nAChR:
-
Nicotinic acetylcholine receptor
References
Albuquerque EX, Alkondon M, Pereira EF, Castro NG, Schrattenholz A, Barbosa CT, Bonfante-Cabarcas R, Aracava Y, Eisenberg HM, Maelicke A (1997) Properties of neuronal nicotinic acetylcholine receptors: pharmacological characterization and modulation of synaptic function. J Pharmacol Exp Ther 280:1117–1136
Bertrand D, Changeux JP (1995) Nicotinic receptor: an allosteric protein specialized for intercellular communication. Semin Neurosci 7:75–90
Buisson B, Picard F, Bertrand D (2000) Neuronal nicotinic acetylcholine receptors: from biophysical properties to human diseases. In: Clementi F, Gotti C, Fornasari D (eds) Neuronal nicotinic receptors. Springer, Heidelberg, pp 271–299
Candé C, Cohen I, Daugas E, Ravagnan L, Larochette N, Zamzami N, Kroemer G (2002) Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie 84:215–222
Changeux JP (1995) Thudichum medal lecture. The acetylcholine receptor: a model for allosteric membrane proteins. Biochem Soc Trans 23:195–205
Conti-Fine BM, Navaneetham D, Lei S, Maus AD (2000) Neuronal nicotinic receptors in non-neuronal cells: new mediators of tobacco toxicity? Eur J Pharmacol 393:279–294
Conti-Tronconi BM, McLane KE, Raftery MA, Grando S, Protti MP (1994) The nicotinic acetylcholine receptor: structure and autoimmune pathology. Crit Rev Biochem Mol Biol 29:69–123
Couturier S, Bertrand D, Malter JM, Hernandez MC, Bertrand S, Miller N, Valera S, Barkas T, Ballivet M (1990) A neuronal nicotinic acetylcholine receptor subunit (α7) is developmentally regulated and forms a homomeric channel blocked by α-bungarotoxin. Neuron 5:847–856
Dhein S, van Koppen C, Brodde OE (2001) Muscarinic receptors in the mammalian heart. Pharmacol Res 44:161–182
Dhein S, Gaertner C, Ziegelhöffer B, Mohr FW (2011) Angiogenesis can be regulated by intercellular communication via gap junctions. Thorac Cardiovasc Surg 59(suppl1):S93
Duerrschmidt N, Zabirnyk O, Nowicki M, Ricken A, Hmeidan FA, Blumenauer V, Borlak J, Spanel-Borowski K (2006) Lectin-like oxidized low-density lipoprotein receptor-1-mediated autophagy in human granulosa cells as an alternative of programmed cell death. Endocrinology 147:3851–3860
Elgoyhen AB, Vetter DE, Katz E, Rothlin CV, Heinemann S, Boulter J (2001) Alpha10: a determinant of nicotinic cholinergic function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci USA 98:3501–3505
Giniatullin R, Nistri A, Yakel L (2005) Desensitization of nicotinic ACh receptors: shaping cholinergic signaling. Trends Neurosci 28:371–378
Grando SA, Horton RM, Pereira EF, Diethelm-Okita BM, Gorge PM, Albuquerque EX, Conti-Fine BM (1995) A nicotinic acetylcholine receptor regulating cell adhesion and motility is expressed in human keratinocytes. J Invest Dermatol 105:774–781
Haussig S, Schubert A, Mohr F-W, Dhein S (2008) Sub-chronic nicotine exposure induces intercellular communication failure and differential down-regulation of connexins in cultures human endothelial cells. Atherosclerosis 196:210–218
Heeschen C, Weis M, Aicher A, Dimmeler S, Cooke JP (2002) A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J Clin Invest 110:527–536
Katz B, Thesleff S (1957) A study of the ‘desensitization’ produced by acetylcholine at the motor end-plate. J Physiol 138:63–80
Kawashima K, Watanabe N, Oohata H, Fujimoto K, Suzuki T, Ishizaki Y, Morita I, Murota S (1990) Synthesis and release of acetylcholine by cultured bovine arterial endothelial cells. Neurosci Lett 119:156–158
Khiroug SS, Harkness PC, Lamb PW, Sudweeks SN, Khiroug L, Millar NS, Yakel JL (2002) Rat nicotinic ACh receptor alpha7 and beta2 subunits co-assemble to form functional heteromeric nicotinic receptor channels. J Physiol 550:425–434
Kirkpatrick CJ, Bittinger F, Unger RE, Kriegsmann J, Kilbinger H, Wessler I (2001) The non-neuronal cholinergic system in the endothelium: evidence and possible pathobiological significance. Jpn J Pharmacol 85:24–28
Le Novère N, Corringer PJ, Changeux JP (2002) The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J Neurobiol 53:447–56
Lindstrom JM (1995) Nicotinic acetylcholine receptors. In: North RA (ed) Ligand- and voltage-gated ion channels. Handbook of Receptors and Channels. CRC, Boca Raton, pp 153–175
Macklin KD, Maus ADJ, Pereira EFR, Albuquerque EX, Conti-Fine BM (1998) Human vascular endothelial cells express functional nicotinic acetylcholine receptors. J Pharmacol Exp Ther 287:435–439
McGehee DS, Role LW (1995) Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol 57:521–46
McLeland CB, Rodriguez J, Stern ST (2011) Autophagy monitoring assay: qualitative analysis of MAP LC3-I to II conversion by immunoblot. Methods Mol Biol 697:199–206
Morawietz H, Rueckschloss U, Niemann B, Duerrschmidt N, Galle J, Hakim K, Zerkowski HR, Sawamura T, Holtz J (1999) Angiotensin II induces LOX-1, the human endothelial receptor for oxidized low-density lipoprotein. Circulation 100:899–902
Quick MW, Lester RAJ (2002) Desensitization of neuronal nicotinic receptors. J Neurobiol 53:457–478
Ransom BR, Sontheimer H (1992) Cell–cell coupling demonstrated by intracellular injection of the fluorescent dye Lucifer Yellow. In: Kettenmann H, Grantyn R (eds) Practical electrophysiological methods. Wiley-Liss, New York, pp 336–342
Sastry BV, Sadavongvivad C (1979) Cholinergic systems in non-nervous tissues. Pharmacol Rev 30:65–132
Sgard F, Charpantier E, Bertrand S, Walker N, Caput D, Graham D, Bertrand D, Besnard F (2002) A novel human nicotinic receptor subunit, alpha10, that confers functionality to the alpha9-subunit. Mol Pharmacol 61:150–159
Thany SH, Lenaers G, Raymond-Delpech V, Sattelle DB, Lapied B (2007) Exploring the pharmacological properties of insect nicotinic acetylcholine receptors. Trends Pharmacol Sci 28:14–22
Tsai CH, Yeh HI, Tian TY, Lee YN, Lu CS, Ko YS (2004) Down-regulating effect of nicotine on connexin43 gap junctions in human umbilical vein endothelial cells is attenuated by statins. Eur J Cell Biol 82:589–595
Tsukamoto O, Minamino T, Okada K, Shintani Y, Takashima S, Kato H, Liao Y, Okazaki H, Asai M, Hirata A (2006) Depression of proteasome activities during the progression of cardiac dysfunction in pressure-overloaded heart of mice. Biochem Biophys Res Commun 340:1125–1133
Wessler I, Kirkpatrick CJ (2008) Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol 154:1558–1571
Wessler I, Bender H, Haerle HK, Kirdorf G, Klapproth H, Reinheimer T, Ricny J, Schniep-Mendelson KE, Racké K (1995) Release of [3H]acetylcholine in human isolated bronchi: effect of indomethacin on muscarinic autoinhibition. Am J Respi Crit Care Med 151:1040–1046
Wessler I, Kirkpatrick CJ, Racké K (1998) Non-neuronal acetylcholine, a locally acting molecule, widely distributed in biological systems: expression and function in humans. Pharmacol Ther 77:59–79
Zhang W, Li D, Mehta JL (2004) Role of AIF in human coronary artery endothelial cell apoptosis. Am J Physiol Heart Circ Physiol 286:H354–358
Acknowledgments
We thank J. Craatz and S. Krabbes for their excellent technical assistance. This work was supported by a grant given by the Medical Faculty, University of Leipzig (Germany), to N.D.
Author information
Authors and Affiliations
Corresponding author
Additional information
Nicole Duerrschmidt and Anja Hagen contributed equally to this work.
Rights and permissions
About this article
Cite this article
Duerrschmidt, N., Hagen, A., Gaertner, C. et al. Nicotine effects on human endothelial intercellular communication via α4β2 and α3β2 nicotinic acetylcholine receptor subtypes. Naunyn-Schmiedeberg's Arch Pharmacol 385, 621–632 (2012). https://doi.org/10.1007/s00210-012-0738-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-012-0738-y