
Abstract
Drug–receptor interactions are traditionally quantified in terms of affinity and efficacy, but there is increasing awareness that the drug-on-receptor residence time also affects clinical performance. While most interest has hitherto been focused on slow-dissociating drugs, D2 dopamine receptor antagonists show less extrapyramidal side effects but still have excellent antipsychotic activity when they dissociate swiftly. Fast dissociation of clozapine, the prototype of the “atypical antipsychotics”, has been evidenced by distinct radioligand binding approaches both on cell membranes and intact cells. The surmountable nature of clozapine in functional assays with fast-emerging responses like calcium transients is confirmatory. Potential advantages and pitfalls of the hitherto used techniques are discussed, and recommendations are given to obtain more precise dissociation rates for such drugs. Surmountable antagonism is necessary to allow sufficient D2 receptor stimulation by endogenous dopamine in the striatum. Simulations are presented to find out whether this can be achieved during sub-second bursts in dopamine concentration or rather during much slower, activity-related increases thereof. While the antagonist’s dissociation rate is important to distinguish between both mechanisms, this becomes much less so when contemplating time intervals between successive drug intakes, i.e., when pharmacokinetic considerations prevail. Attention is also drawn to the divergent residence times of hydrophobic antagonists like haloperidol when comparing radioligand binding data on cell membranes with those on intact cells and clinical data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Drug–receptor interactions are traditionally quantified in terms of affinity and efficacy only, and most preclinical screening studies still probe ligand–receptor interactions in those terms. Yet, there is increasing awareness that the time period over which they reside at their receptor (also denoted as the target residence time of the drug) is also able to affect their clinical performance. This led to drug candidates being screened for their binding kinetics, and, to this end, a number of interesting principles have been advanced in recent review papers (Swinney 2004, 2006a, b, 2008, 2009; Copeland et al. 2006; Tummino and Copeland 2008; Copeland 2010; Zhang and Monsma 2009, 2010; Vauquelin et al. 2006; Vauquelin and Van Liefde 2006; Vauquelin and Charlton 2010). Among them, a distinction should be made about whether the drug–target complex is a source of adverse events or not. Most of the cases that were enumerated in those reviews dealt with situations where this is not the case. A typical example is that for the AT1 angiotensin II receptor antagonists, of which an optimal clinical outcome is assumed to require continuing, high levels of target occupancy (Kjeldsen et al. 2010; Eklind-Cervenka et al. 2011). However, there are some situations in which a long residence time might have unfavorable or even detrimental consequences (such intrinsic adverse events are also denoted as “mechanism-based toxicity”) (Swinney 2006b; Copeland et al. 2006; Zhang and Monsma 2009). This is certainly the case for agonists which by virtue of their long-lasting binding and/or presence usually trigger receptor desensitization, sequestration, and even downregulation (Laruelle 2000; Lipworth 2002). Yet, it may also show up for slowly dissociating antagonists by virtue of their propensity to block the access of their receptor to its natural messenger. This is likely to be the case for the first-generation neuroleptics (further named “typical” antipsychotics) used in clinical therapy to attenuate psychotic episodes that are associated with neurological disorders such as schizophrenia (Seeman et al. 1975, 1976; Creese et al. 1976).
The dopamine hypothesis
Schizophrenia is considered to be a neuro-developmental condition that affects 0.8–1% of the population. Its symptoms and signs are generally classified as positive (delusions, hallucinations, psychosis, and paranoia) and negative (lack of emotional expression, social withdrawal, and cognitive deficits) (van Os and Kapur 2009). Antipsychotic drugs were proposed to act as pharmacological antagonists of the neurotransmitter dopamine, and a good correlation between their clinical potency and their affinity for the dopamine D2 receptor was observed (Creese et al. 1976; Seeman et al. 1975, 1976; Seeman 1992; Kapur and Mamo 2003). While different classes of antipsychotic drugs could effectively alleviate psychotic symptoms, non-dopaminergic drugs alone were not capable of doing so (Sanger 2004), and dopamine-enhancing drugs were shown to elicit psychotogenic effects (Angrist and Van Kammen 1984; Lieberman et al. 1987; Wilson et al. 1998). This further consolidated the “dopamine theory” (Van Rossum 1967) where hyperactive dopamine transmission was proposed to be responsible for the positive symptoms.
Four major dopaminergic pathways and five dopamine receptor subtypes exist. The mesocortical and mesolimbic (projecting to the nucleus accumbens, hippocampus, and amygdala) pathways are involved in cognitive and emotional functions, respectively (Mogenson et al. 1980, 1988; Csernansky et al. 1991; Amalric and Koob 1993). The tubo-hypophyseal pathway inhibits prolactin secretion from the pituitary gland (Caron et al. 1978), and the nigrostriatal pathway (projecting from the substantia nigra to the dorsal striatum) is involved in movement and contains the highest density of dopaminergic neurons (Bédard et al. 1969; Björklund et al. 1970; Fluxe et al. 1974). The receptors can be divided into D1-like (D1 and D5) and D2-like (D2, D3, and D4), and the D2 receptors themselves comprise at least two splice variants: The D2Short isoform has been proposed to act as a presynaptic inhibitory autoreceptor, and D2Long (with an extra 29 amino acid sequence in the third intracellular loop) is located postsynaptically (Kebabian and Calne 1979; Wilson et al. 1998; Dal Toso et al. 1989; Usiello et al. 2000; Wang et al. 2000; Lindgren et al. 2003). This different location could be the consequence of the higher expression levels of D2Short in dopaminergic neurons (Jomphe et al. 2006). A longer splice variant, D2Longer, has also been found in the striatum of post-mortem brains from some individuals (Seeman et al. 2000), and three single amino-acid mutation variants have been identified as well (Gejman et al. 1994).
Better knowledge of the involvement of these pathways and receptor subtypes in the multiple manifestations of schizophrenia has prompted a reformulation of the classical dopamine hypothesis. It has been speculated that negative and cognitive signs may arise from diminished release of dopamine in the frontal cortex. In addition, the emergence of psychotic states is linked to intermittent elevations in subcortical dopamine transmission and resulting D2 receptor hyperstimulation (Davis et al. 1991; Laruelle 2003; Weinberger and Laruelle 2001; Guillin et al. 2007). Interestingly, neuroimaging studies reveal that baseline occupancy of striatal D2 receptors by dopamine is also higher in patients with schizophrenia compared with matched control subjects (Abi-Dargham et al. 2000; Frankle et al. 2004) and that there is a better correlation between the antipsychotic effect of drugs like risperidone, olanzapine, and aripiprazole and their degree of D2 receptor occupancy in the ventral striatum than in cortical and thalamic regions (Agid et al. 2007; Kegeles et al. 2008). These recent findings point to an important role of the mesolimbic striatum in the antipsychotic response of these drugs.
D2 receptor antagonist binding: affinity and kinetics
By acting as high-affinity antagonists at D2 receptors, first-generation or typical antipsychotics such as haloperidol and chlorpromazine should effectively prevent dopaminergic transmission. Hence, they should exert antipsychotic effects provided that they block a sufficiently high proportion of those receptors in limbic structures of the brain. In this respect, early neuroimaging studies led to the general picture that typical antipsychotics start to show clinical efficacy at 60–70% receptor occupancy (Farde et al. 1992; Seeman 2002). Because of the only limited “signal-to-noise” ratio of the early positron emission tomography (PET) and single-photon emission computed tomography (SPECT) radiotracers, this value was obtained for the striatum where D2 receptors are most concentrated (Kessler et al. 1993). Yet, these drugs display only a narrow therapeutic window. When they occupy more than 80% of the striatal D2 receptors, extrapyramidal side effects including Parkinson-like symptoms (tremor, rigidity, and bradykinesia), akathisia, dystonia, and tardive dyskinesia (Farde and Nordström 1993; Farde 1996; Raleigh 1996), as well as chronic hyperprolactinemia (Petty 1999) start to show up. These side effects result in poor drug compliance and early discontinuation of the therapy and are thought to emerge when postsynaptic D2 receptors on spiny neurons in the striatum (Hauber 1998), and those on lactotrope cells of the anterior pituitary gland (Ben-Jonathan and Hnasko 2001) experience too little stimulation by endogenous dopamine.
Based on radioligand competition binding studies on isolated cell membranes, this insufficient stimulation by dopamine was initially linked to the high affinity of typical antipsychotics for D2-receptors (Seeman et al. 1996, 1997a). However, this strict interpretation can be called into question since the very affinity concept has only physical meaning if equilibrium conditions apply, and, under those conditions, receptor occupancy is not only dictated by the affinity but also by the concentration of the drug. In other words, when equilibrium conditions apply, low-affinity antipsychotic agents should be able to occupy as many receptors as high-affinity agents, provided that they are given at a sufficiently high dose (Kapur and Seeman 2000). Yet, in the striatum where the concentration of endogenous dopamine is susceptible to dynamic fluctuations (Kawagoe et al. 1992; Koepp et al. 1998; Cragg and Rice 2004; Rice and Cragg 2008; Salimpoor et al. 2010; Dreyer et al. 2010), such equilibrium can hardly be reached and maintained, and this is especially so in the case of slowly dissociating antagonists.
A very similar situation is artificially created in organ bath experiments (Gaddum et al. 1955; Leff and Martin 1986), and the theoretical background for the observed phenomena is well understood (Vauquelin et al. 2002a,b; Kenakin et al. 2006). In those experiments, tissues are habitually pre-equilibrated with the antagonist before their challenge with agonist and measurement of the evoked response. In case of competitive antagonism, the extent of this response will not only be determined by antagonist and agonist concentrations and affinities but also by the time lapse between agonist administration and the measurement of the response. If this time lapse exceeds the antagonist’s dissociation t 1/2 sufficiently well, most of the antagonist–receptor complexes will have dissociated so that the involved receptors will have become prone to agonist stimulation. However, if this time lapse is too short, only a small fraction of the concerned receptor molecules will have become agonist-responsive.
Based on these very principles, Seeman and coworkers subsequently proposed (Seeman and Tallerico 1998, 1999; Kapur and Seeman 2000) that it is not the affinity of the antipsychotic agent but rather how fast it comes off the postsynaptic D2 receptors that determines their responsiveness to a transient increase in striatal dopamine concentration. Typical, high-affinity antipsychotics were assumed to dissociate slowly, and above 80% receptor occupancy, they should no longer permit sufficient D2 receptor stimulation by transient surges in striatal dopamine concentration to prevent the emergence of extrapyramidal symptoms. Alternatively, antipsychotics with lower affinity, and thus with a shorter dissociation t1/2 , should allow sufficient D2 receptor stimulation. The underlying link between affinity and dissociation rate was based on the fact that, for reversible bimolecular interactions, the equilibrium dissociation constant of a ligand (K D, an often-used parameter which is an inverse measure of the ligand’s affinity) is the ratio of the ligand’s dissociation (k off) and association (k on) rate constants (i.e., K D = k off/k on), and the contention that antipsychotics exhibit more or less the same k on for D2 receptors (Kapur and Seeman 2000; Tresadern et al. 2011). Taken together, these considerations imply that the affinity of an antipsychotic agent should closely reflect its dissociation t1/2 . In agreement therewith, radioligand binding experiments with rat striatal membranes revealed that typical antipsychotics display high affinity for D2 receptors and dissociate slowly therefrom, while clozapine, the prototype of the second-generation antipsychotics, shows low affinity and fast dissociation. However, while low-affinity antipsychotics are intended to dissociate swiftly, the issue whether the liberated D2 receptors are effectively stimulated will also depend on the concentration of endogenous dopamine. Low-affinity antipsychotics should therefore permit more pronounced D2 receptor stimulation in areas with high levels of endogenous dopamine, like the striatum, than in those with lower levels of dopamine (Seeman et al. 1996, 1997a). This should prevent the appearance of extrapyramidal symptoms while still preserving antipsychotic efficacy.
Atypical antipsychotics
Clozapine, the first of the second-generation antipsychotic drugs, was introduced into clinical practice in the 1970s. When given at the usual therapeutic doses of 300 to 600 mg per day, clozapine was found to exert an antipsychotic effect with minimal extrapyramidal side effects and prolactin secretion in schizophrenic patients (Seeman 2002; Miyamoto et al. 2005). The low incidence of such side effects encouraged the patients to comply with treatment, and this was an important basis for the clinical superiority of clozapine. The term “atypicality” was introduced to highlight this significant disconnection between the dosage of clozapine needed to bring about its antipsychotic action and the extrapyramidal signs (Hippius 1989; Arnt and Skarsfelt 1998; Kapur and Seeman 2001). However, it became evident in the early 1990s that clozapine has a whole spectrum of therapeutic effects that are different from those of the earlier, typical antipsychotics (Baldessarini and Frankenburg 1991; Hippius 1999; Meltzer 2002). In this respect, it was especially salient for clozapine to be effective in some of the patients whose positive symptoms were refractory to the classical antipsychotics (Kane et al. 1988a, b; Hagger et al. 1993). The beneficial clinical profile of clozapine has encouraged the development of additional D2 receptor binding antipsychotics but with varying pharmacological profiles for other receptors (Wilson et al. 1998; Abi-Dargham and Laruelle 2005; Seeman 2011). Yet, rather than categorizing these newer drugs as “atypical”, some authors prefer the term “novel” or “second/new-generation” or even to abandon the use of general denominations altogether (Abi-Dargham and Laruelle 2005).
Nevertheless, there is still a widespread consensus for the term “atypical” to apply when drugs exert antipsychotic activity without producing extrapyramidal symptoms at the usual doses in clinical therapy (Kinon and Lieberman 1996; Remington and Kapur 2000; Seeman 2002). Many theories have been proposed to explain atypicality. Those that invoked a pharmacological basis drew attention to the higher affinity of clozapine for several other receptors than for D2 receptors (Schotte et al. 1996; Arnt and Skarsfelt 1998; Meltzer 2002; Sanger 2004). In this respect, D4 receptors (Seeman et al. 1997; Jardemark et al. 2002) and serotonin 5-HT2A receptors (Meltzer 1999; Weiner et al. 2001) were subject to special attention. Yet, several objections have been raised against these propositions. For example, despite their full 5-HT2A receptor occupy at clinically relevant doses, distinct antipsychotics do not trigger extrapyramidal side effects to the same extent (Kapur and Seeman 2000; Olson 2005), and the threshold D2 receptor occupancy at which such side effects emerge does not depend on their 5-HT2A antagonistic profile (Frankle et al. 2004). In this respect, D2 antagonists with highly unfavorable 5-HT2A/D2 receptor affinity ratio may also display excellent antipsychotic activity (Langlois et al. 2010; Schmidt et al. 2010). Also, D4 receptor-selective antagonists have not shown clinical efficacy for the treatment of schizophrenia either (Kramer et al. 1997). An alternative explanation banked on a more physiological basis and proposed that atypical antipsychotics manifest mesolimbic versus striatal selectivity (Kinon and Lieberman 1996; Pilowsky et al. 1997; Xiberas et al. 2001; Kessler et al. 2006; Grunder et al. 2006), but ex vivo autoradiographic and neuroimaging studies by others (Schotte et al. 1996; Mukherjee et al. 2001; Talvik et al. 2001) failed to confirm such regional differences. Whatever the outcome, this debate may be overshadowed by more recent observations linking the occupancy of D2 receptors in the mesolimbic striatum to the amelioration of psychoses in schizophrenia (Agid et al. 2007; Kegeles et al. 2008). In our opinion, the most compelling explanation for atypicality of antipsychotic drugs deals with molecular aspects of their D2 receptor interaction. For those that display low affinity for D2 receptors, like clozapine and quetiapine, rapid dissociation is likely to be important (Kapur and Seeman 2000, 2001; Seeman 2002; Farah 2005). For others, like aripiprazole, it is their proficiency to produce mild D2 receptor stimulation (i.e., with low intrinsic efficacy) that constitutes the most evident explanation (Burris et al. 2002; Jordan et al. 2007a,b; Tadori et al. 2009).
While the partial agonism of aripiprazole has been extensively documented, it is amazing that, since the initial radioligand binding studies with [3H]-clozapine and [3H]-quetiapine by Seeman and coworkers (Seeman and Tallerico 1999; Kapur and Seeman 2000; Seeman 2005) and a functional experiment (Coldwell et al. 1999), very little experimental work has been performed till recently to corroborate their findings and especially to provide further insight in the time frame during which their dissociation takes place (Dyhring et al. 2010; Langlois et al. 2010; Packeu et al. 2010b,c; Tresadern et al. 2011). Yet, such kinetic data and simulations therewith are important to our understanding of the implications of “the fast dissociation proposal” at the physiological level. More precisely, they may give us better insight into the ability of these drugs to discriminate between fluctuations in synaptic and extrasynaptic dopamine concentrations, to link up with pharmacokinetics, and also to get a better insight about the effect of receptor dissociation rates on fluctuations in D2 receptor blockade between consecutive drug intakes.
In vitro studies: prospects and limitations
The fast dissociation proposal for clozapine and quetiapine can, in principle, be verified experimentally by in vitro radioligand binding studies as well as by functional experiments involving D 2 receptor activity measurements. A large panoply of D 2 receptor sources and assay techniques are presently available to this end. In this respect, it is of note that functional assays to investigate ligand–D 2 receptor interactions are quite often carried out on intact cell systems (either naïve cells expressing native D 2 receptors or recombinant cell lines expressing wild-type or mutated D 2 receptors, other proteins or even hybrids between D 2 receptors and other proteins). In contrast, D 2 receptor-dedicated radioligand binding assays have hitherto almost exclusively been carried out on membrane preparations. Although the intact cell approach is certainly feasible, only a handful of D 2 receptor binding studies have hitherto been performed on intact cells (Itokawa et al. 1996; Kim et al. 2001; Vauquelin and Packeu 2009; Packeu et al. 2008, 2010a, b, c). Compared with the binding experiments, functional assays yield less direct information about the molecular characteristics of antagonist–receptor interactions, and they are potentially also more difficult to interpret due to the possible interference of phenomena like receptor desensitization/internalization and cellular amplification of the receptor-evoked response (this last phenomenon is also named “receptor reserve” and “spare receptors”).
Recombinant cell systems are now also increasingly used for the investigation of D2 receptors. A paramount advantage of this approach is that the parent cells can be used as negative controls (at least when they do not express D 2 receptors by themselves), so that other receptor, other receptor-subtype, and even receptor-unrelated phenomena can be dissociated from those that are connected to the receptor of interest. This is of particular interest for D2 receptors since most of its drugs also display high affinity for one or more of the other dopamine receptor subtypes and even unrelated G protein-coupled receptors (GPCRs). Yet, the use of recombinant cell systems may also shed light on phenomena with uncertain physiological relevance so that the obtained results need to be appraised with due caution. Among others, D2 receptors are often expressed in cell lines from foreign species and/or with artificial signaling cascades. The receptors are also often over-expressed in such cell lines. This may engender phenomena like constitutive receptor activity (drawing excessive attention on inverse agonism), the emergence of earlier conceptualized D1–D2 receptor heterodimers (Seeman et al. 2006; Faron-Górecka et al. 2008) and for excessively high agonist potencies (even in case of adenylate cyclase activity inhibition) and efficacies (i.e., maximal levels of response) of partial agonists (Black and Leff 1983; Brink et al. 2000).
In general, it can be said that the simplest sources such as cell membranes provide the most direct information with respect to drug–receptor interaction mechanisms while the most complex sources such as intact tissues provide the most relevant information from the physiological and clinical point of view (Vauquelin and Charlton 2010). Yet, the functional approach can be considered to be more relevant from the clinical perspective since it is not receptor occupancy per se, but rather the resulting decline in D 2 receptor activation by endogenous dopamine that counts. As each approach is subject to inherent advantages but also to handicaps and limitations, it is preferable to confront information from a wide array of approaches.
Radioligand binding
From the technical point of view, it is definitely more convenient to perform radioligand binding on membrane preparations than on intact cells. Moreover, phenomena like binding of hydrophobic radioligands to internalized receptors (Itokawa et al. 1996; Guo et al. 2010) and other intracellular acidic compartments (Rayport and Sulzer 1995) do not constitute a source of concern. This may explain why nearly all radioligand binding studies on D2 receptors have been carried out on membrane preparations from tissues like striatum or, more recently, from cells expressing recombinant receptors.
To investigate the rate by which a drug dissociates from its receptor, the most straightforward procedure is to acquire it in a radiolabelled form (denoted as radioligand) and to monitor the time-wise decline of its specific (i.e., receptor) binding. In these experiments, the receptor-containing membranes are preincubated with the radioligand and the dissociation phase is initiated by adding a large excess of (the same or preferentially another) unlabelled drug. This will prevent or at least limit the formation of new radioligand–receptor complexes and/or rebinding phenomena (Vauquelin and Szczuka 2007; Vauquelin and Charlton 2010; Vauquelin 2010). Different variants of this procedure are depicted in Fig. 1. Such experiments were performed by Seeman and coworkers to investigate the rate by which [3H]-clozapine and [3H]-quetiapine dissociate from D2 receptors. Upon addition of a large excess of raclopride (10 μM), these radioligands were found to dissociate from rat striatal membranes with a t 1/2 of 30 and 14 s, respectively (Kapur and Seeman 2000). This is appreciably faster than the dissociation t 1/2 of haloperidol (42 min) under the same conditions. In agreement therewith, [3H]-clozapine and [3H]-quetiapine were both found to dissociate with a t 1/2 of about 15 s from membranes prepared from human D2Long receptor-expressing recombinant Sf9 insect cells upon addition of 100 μM raclopride (Seeman 2005). Similar dissociation experiments with radiolabelled antipsychotics were recently also presented by Langlois et al. (2010). While these experiments are likely to produce the most direct information about a drug’s dissociation rate, it has to be conceded that the low affinity of clozapine and quetiapine for D2 receptors constitutes an inherent handicap with this approach. Accordingly, additional information gathered by alternative experimental approaches should be highly welcome if we wish to obtain a more reliable insight into the binding kinetics of clozapine and other antipsychotics.

Different procedures to study the dissociation rate of a radioligand. Left panels: timing of experimental manipulations; right panel: expected observations. Methods 1 and 2 start with preincubating the receptors with radioligand, a brief wash (method 1) or no wash (method 2) followed by addition of medium only. In method 2, a large excess of medium is added to dilute the radioligand concentration substantially. Under both conditions, radioligand rebinding is not abated, and the resulting curves may give rise to (at first sight) only partial radioligand dissociation. This latter observation is sometimes interpreted in terms of receptor heterogeneity and/or non-competitivity. More information about rebinding phenomena is provided in (Vauquelin and Szczuka 2007; Vauquelin and Charlton 2010; Vauquelin 2010). Methods 3 and 4 start with preincubating the receptors with radioligand, a brief wash (method 3) or no wash (method 4) followed by washout in the presence of receptor-saturating concentration of unlabelled ligand. The unlabelled ligand will occupy all the receptor sites that were liberated by the radioligand and thereby effectively prevent radioligand rebinding (method 3) and/or further binding (method 4). The genuine dissociation is expected to follow monoexponential curve. Data points are simulated for a radioligand that binds reversibly to its receptor according to the Law of Mass Action (with equations 1, 3, 5 and 6 in Table 3, where A stands for “radioligand”). Parameters are k 1A = 1.108 M−1 min−1; k −1A = 0.0057 min−1, initial [AR] = 30% of [R tot] at equilibrium and initial a[R]k 1A = 1.22 in case of rebinding
Fortunately, and contrary to the belief that individual drugs need to be radiolabelled for quantifying their dissociation rate (Sanger 2004), this information can also be obtained indirectly by methods that rely on the ability of unlabelled drugs to affect the binding of a competitive radioligand (Packeu et al. 2010c). This allows the use of radioligands with high affinity and selectivity for D2 receptors along with little non-specific binding. In this respect, the co-incubation-based method proposed by Motulsky and Mahan (1984) has been applied successfully in the case of some receptors (Dowling and Charlton 2006; Heise et al. 2007). Yet, at least to our knowledge, such experiments have not yet been performed in the case of D2 receptors. However, the dissociation properties of unlabelled drugs can also be obtained by other indirect procedures that start with preincubating the receptor-bearing biological material with a high concentration of unlabelled drug (to occupy the vast majority of receptors) and terminate by incubating with radioligand in fresh medium. Different variants do exist, and the two that most closely resemble the direct radioligand dissociation experiments mentioned above comprise “one-step” or “multi-step” “washout” episodes during which the originally bound drug is allowed to dissociate (Fig. 2, methods 1 and 2, respectively). Radioligand is then finally added to quantitate the amount of unbound receptors (i.e., those from which the drug did already dissociate).

Different multi-step procedures to study the dissociation rate of unlabelled drugs. Methods 1 and 2 (i.e., “one-step” and “multi-step” washout”methods) start with preincubating the receptors with a high concentration of unlabelled drug (to occupy the vast majority of receptors), followed by washout in fresh medium once but for variable periods of time (method 1) or in a variable amount of consecutive fixed-time incubations and this every time in fresh medium (method 2) and, to terminate, by a short-lasting incubation with a fixed concentration of radioligand. The drug’s dissociation rate could be calculated from the exponential recovery of radioligand binding (when plotted vs. the total washout time). Method 3 (i.e., “delayed association” method) differs from the previous ones by keeping the washout time as short as possible and by (finally) incubating the receptors with a fixed concentration of radioligand for increasing periods of time. The drug’s dissociation rate can be estimated by comparing such obtained radioligand association curves with those obtained without drug pretreatment or calculated by the equation provided by Malany et al. (2009). The number of time periods could be reduced to only one for the purpose of high-throughput screening (Tresadern et al. 2011). Method 4 (i.e., “2-step” competition method) comprises a preincubation with increasing concentrations of drug, no wash (pathway A at bifurcation) or a brief wash (pahway B at bifurcation) and a final fixed-time incubation with a single concentration of radioligand. Information that can be obtained from the so-generated curves is presented in Fig. 3a. Common to these approaches is the requirement to exchange the medium swiftly at distinct phases of the experiment. This is most easily done in binding studies on intact plated cells or in ex vivo binding studies (in where drug-treated animals are sacrificed and “washout” and binding is performed on brain slices) as well as in functional assays such as the traditional “organ bath” experiments with intact tissues and related experiments with intact plated cells (Ojima et al. 1997; Morsing et al. 1999; Fierens et al. 1999). In those cases, the drug to be investigated is an antagonist, and the amount of unbound receptors at the end of the “washout” episode is quantified by the gain in functional response when an agonist is finally added
Such “multi-step” washout experiments (Fig. 2, method 2) were already reported more than 25 years ago (Leysen and Gommeren 1984) to compare the dissociation rates of unlabelled antipsychotics (but not including clozapine and quetiapine) from D2 receptors from rat striatal membranes. The authors introduced an elegant approach to minimize the “medium-exchange” hurdle, namely, to perform a filtration after the preincubation with unlabelled drug and to perform successive 5-min washouts with fresh medium on the membranes that remained attached to those filters. The final incubation with [3H]-haloperidol as radioligand was performed in the same way. This approach has recently also been adopted by Tresadern et al. (2011) for high-throughput screening studies aimed at comparing the rate by which antipsychotics and other experimental drugs dissociate from human D2Long receptor-expressing recombinant CHO cell membranes. Since the unlabelled ligands continue to dissociate during the final incubation with radioligand, it is preferable to keep this incubation time minimal. In an effort to optimize this incubation time, Tresadern et al. (2011) performed preliminary experiments according to a “delayed association” binding paradigm (Fig. 2, method 3) wherein the membranes were preincubated with unlabelled clozapine, quetiapine, and a few other antipsychotics at concentrations corresponding to four to eight times their IC50 (i.e., occupying well over 80% receptors). The membranes were then briefly washed by filtration and (while still present on the filters) incubated with [3H]-spiperone for time periods ranging between 1 and 10 min. Compatible with a fast dissociation of clozapine and quetiapine, the specific binding of [3H]-spiperone reached already about 70% of the control level (i.e., binding to membranes that were not exposed to drug before) after as little as 1 min incubation. However, rather than the expected further time-wise increase, the binding of [3H]-spiperone remained steady for as long as 10 min. Interestingly, a similar plateau level was recently also observed when plated human D2Short as well as D2Long receptor-expressing recombinant CHO cells were exposed to clozapine, briefly washed, and then incubated for increasing periods of time with [3H]-raclopride (Packeu et al. 2010a). Based on the similar behavior of the slow-dissociating antagonist spiperone (Packeu et al. 2008), it was initially suggested that the clozapine–D2 receptor complexes dissociate slowly as well. However, ensuing experiments according to an original two-step competition binding paradigm revealed that this plateau level emanated from the emergence of a new equilibrium between the binding of [3H]-raclopride and clozapine that got released from the cells (Packeu et al. 2010c).
This two-step competition binding paradigm (Fig. 2, method 4) comprised a preincubation of plated human D2Long receptor-expressing recombinant CHO cells with an increasing concentrations of an unlabelled drug, no wash (for pathway A) or a brief wash (for pathway B) and a final incubation with a single concentration of [3H]-raclopride for a fixed, 30 min time period. This approach not only permits a determination of the drug’s affinity and dissociation rate but also whether or not a sizable amount of drug is released from the membranes or cells during the binding process. As shown in Fig. 3a, this approach permits a determination of the drug’s affinity (from curve A) and dissociation rate and to estimate the extent of drug released from the membranes/cells during the final incubation with radioligand (by comparing the upward and rightward shift of curve B versus curve A, respectively). A situation in where both curves overlap took place for the potent hydrophobic D2 receptor antagonists spiperone, haloperidol, and (+)-butaclamol. This is likely to reflect very slow drug dissociation. An upward shift of curve B (corresponding to a lesser maximal inhibition by the drug) was only partial for unlabelled raclopride, indicating that its dissociation t 1/2 is in the same range as the incubation time. On the other hand, the upward shift was nearly maximal for sulpiride, indicating that the dissociation t 1/2 of this hydrophilic antagonist (Rayport and Sulzer 1995) is appreciably (≥4 times) less than the incubation time. Finally, a fourfold parallel rightward shift of curve B was observed with clozapine and control experiments based on alternative binding paradigm (Packeu et al. 2010c) revealed that this shift was due to the release of clozapine from the cells during the incubation with radioligand. Simulations further revealed that such parallel rightward shift of curve B could only take place if all the initially D2 receptor- bound clozapine got the opportunity to dissociate within this 30-min incubation period. In other words, these experiments revealed that the dissociation t 1/2 of clozapine is well below 30 min, i.e., the duration of the final incubation with [3H]-raclopride. To narrow down this evaluation, a new series of two-step competition binding experiments with clozapine have now been carried out under the same conditions as in Packeu et al. (2010c), except that the incubation time was reduced to 5 min. A similar parallel rightward shift of curve B took place here also (Fig. 3b). This suggests that the dissociation t 1/2 of clozapine–D2Long receptor complexes is even appreciably less than 5 min at 37°C and also that the partitioning between free and membrane/cell- associated clozapine is a rapid reversible process.

Estimation of the clozapine dissociation rate by the two-step competition method. The experimental procedure is shown in Fig. 2 (method 4). The preincubation is with unlabelled ligand is followed by no wash (closed square, pathway A) or a brief wash (open square, pahway B). a Simulated curves by the two-step competition method: expected observations for a fast-dissociating (t 1/2 = 6.9 min, left side) and a slow-dissociating (t 1/2 = 138 min, right side) drug when preincubation and incubations last 30 min. No (top) or 20% (bottom) of the initially added unlabelled drug takes place during the incubation with radioligand. Data points are simulated for drugs and radioligands that bind competitively to one-site bimolecular reaction obeying the Law of Mass Action (with equations 1, 2, and 4 in Table 3, where A stands for “drug” and D for “radioligand”). Other parameters are k 1A and k 1D = 1.108 M−1 min−1; k −1D = 0.1 min−1; radioligand concentration, 2 nM. b Clozapine dissociation from intact D2Long receptor expressing CHO cells by the two-step competition method. The experiments are similar to those reported by Packeu et al. (2010c) except that the incubation with radioligand last much less. In short, cells were preincubated for 30 min at 37°C with increasing concentrations of clozapine, briefly washed or not and then incubated for only 5 min with 2 nM [3H]-raclopride. Data refer to specific binding expressed as percentage of control binding (i.e., specific binding to naïve cells) and are presented as means±SEM of three individual experiments with three determinations each. Competition curves are generated by non-linear regression analysis by GraphPad Prism based on a one-site model
That these latter experiments were performed with intact D2 receptor-expressing cells is of importance. Indeed, contrary to results from several prior studies on membranes, specific binding of [3H]-spiperone and [3H]-raclopride to those cells embraced the same amount of sites, likely to be located at the cell surface (Packeu et al. 2008). Moreover, from the physiological perspective, it is only in intact cells that D2- and other membrane-associated receptors reside in a natural environment. Yet, some important characteristics of this environment like the presence of guanosine triphosphate (GTP) in the cytoplasm and non-alike redox potentials and ion concentrations at each face of the plasma membrane (i.e., reducing conditions only at the cytoplasmic face and, among others, a 14-fold higher Na+ concentration at the external face) are lost when cells or tissues are homogenized (Vauquelin and Charlton 2010). Evidence is accumulating that this may affect antagonist binding characteristics of D2 receptors and other GPCRs. Indeed, it was recently observed that [3H]-spiperone dissociates much faster from human D2 receptors when the binding studies were performed on membranes instead of the corresponding, intact recombinant Chinese hamster ovary (CHO) cells (Vauquelin and Packeu 2009). Quite similar distinctions have also been reported for GPCRs (Fierens et al. 2002; Hara et al. 1998; Smith et al. 2006), and it is most striking that leaky cells already had the faculty to behave membrane-like (Verheijen et al. 2004; Vauquelin and Packeu 2009). Finally, compared with membranes, plated cells mimic much better the micro-anatomic complexity of tissues (Spivak et al. 2006; Grießner et al. 2009; Vauquelin and Charlton 2010). Indeed, those cells are separated by clefts that somewhat mimic neuronal synapses and other interstitial spaces whose walls and macromolecular content hinders the free diffusion of ligands to and from their receptors (Spivak et al. 2006; Cragg and Rice 2004; Hrabctová and Nicholson 2004).
Last but not least, the standard interpretation of data obtained from binding experiments involving the active participation of a radioligand and an unlabelled drug is based on the premise that both bind according to the law of mass-action (i.e., via a bimolecular process) to the same, or at least overlapping binding sites (to ensure competitive interactions). In this respect, a recent molecular modeling study (Kalani et al. 2004) suggests that the criterion of competitivity applies to several classes of D2 receptor binding ligands, at least if the receptors are acting as monomers. Indeed, class I, clozapine-like bulky antagonists were found to bind to the agonist binding pocket of human D2 receptors and class II antagonists like haloperidol, spiperone, and sulpiride bound to a somewhat different but still overlapping pocket.
However, D2 receptor binding studies reveal a more complex picture. One of the most striking examples of non-standard behavior was documented by Seeman and coworkers. Although assay conditions (like temperature and buffer/medium composition) may vary considerably from one laboratory to another, the compilation of a large number of reports revealed that, contrary to most other antipsychotics, the K D of clozapine was too low to account for the implication of D2 receptors in its antipsychotic action (Seeman 1992). The calculation of these K D values was based on competition binding studies with [3H]-spiperone, and it was discovered soon afterward that the very nature of the radioligand was at the origin of this discrepancy. Indeed, D2 receptor competition binding experiments with membranes from pig anterior pituitary tissue and human D2Long receptor-expressing recombinant CHO cells unveiled a positive correlation between the K D of clozapine and the propensity of the radioligand to incorporate/partition in the membrane, i.e., the K D decreased in the following order: [3H]-nemonapride > [3H]-spiperone > [3H]-raclopride (Seeman 1995; Seeman and Van Tol 1995; Seeman and Kapur 1997; Seeman et al. 1997). This finding was also of major importance in neuroimaging studies as it could explain the apparently lower occupancy of D2 receptors by clozapine in human striatum when PET scans were performed with [18 F]-fluoroethylspiperone, [11 C]-N-methylspiperone, and [18 F]-methylspiperone than with [11 C]-raclopride as tracer. Similarly, striatal D2 receptor occupancy by [11 C]-raclopride was reduced in quetiapine-treated subjects, while [11 C]-N-methylspiperone binding was refractory (Hagberg et al. 1998).
Radioligand binding studies with plated human D2Short as well as D2Long receptor-expressing recombinant CHO cells (Packeu et al. 2008) confirm these initial findings by Seeman and colleagues. Whereas potent hydrophobic D2 receptor antagonists like spiperone, haloperidol, and (+)butaclamol competed with almost the same K D with both radioligands, the K D values of raclopride and clozapine were consistently higher when [3H]-spiperone was used instead of [3H]-raclopride (Packeu et al. 2008, 2010a). Simulations argued against the potential involvement of kinetic issues, and additional experiments shed light on the marked non-competitive nature between the binding of spiperone and raclopride (Packeu et al. 2010b). Most strikingly, raclopride failed to adequately delay the association of [3H]-spiperone even though it already occupied nearly all of its specific sites, and in co-incubation experiments, the maximal binding of [3H]-raclopride was markedly depressed in presence of a sub-maximal concentration of spiperone (Packeu et al. 2010b). In contrast to spiperone, clozapine behaves as a competitive antagonist in similar [3H]-raclopride saturation binding experiments (Fig. 4). The above findings are not compatible with the molecular modeling data by Kalani et al. (2004). However, they could potentially be explained if D2 receptors function as dimers (Seeman et al. 1992; Ng et al. 1996; Armstrong and Strange 2001). This allows the existence of two interconnected (allosteric) binding sites with different pharmacological specificity (Packeu et al. 2010b).

Effect of clozapine on [3H]-raclopride saturation binding to intact D2Long receptor expressing CHO cells. Cells were co-incubated for 30 min at 37°C with increasing concentrations of [3H]-raclopride either in the absence (closed circle) or presence (open circle) of 200 nM clozapine. Saturation binding data (specific binding) of a representative experiment with three determinations is presented under the form of Scatchard plots. The plots were generated by linear regression analysis. Insert: effects of spiperone (open triangle, non-competitive) and raclopride itself (filled triangle, competitive) on [3H]-raclopride saturation binding under the same experimental conditions (adapted from Packeu et al. (2010b). For the sake of clarity, the plots a represented here are only assigned by a symbol, i.e., without the individual data points
Taken together, in agreement with the initial direct [3H]-clozapine dissociation experiments on cell membranes (Kapur and Seeman 2000; Seeman 2005), indirect “delayed association” and two-step competition binding experiments on intact cells suggest that clozapine dissociates swiftly (i.e., dissociation t 1/2 ≤ 1 min) from D2 receptors. However, each binding paradigm has distinct advantages and disadvantages. While direct dissociation experiments provide the most direct information about a drug’s dissociation rate, radioligands with low affinity for their receptor are likely to display a high degree of non-specific binding, especially if they are able to partition within the membrane. Low affinity is less of a problem in the “delayed association” and two-step competition binding experiments. Yet, partitioning and subsequent release of unlabelled drugs could interfere with the interpretation of results obtained by the former while, for the latter, the incubation times can hardly be reduced to a range that is a low as the estimated dissociation t 1/2 of clozapine and quetiapine. It is therefore of interest to explore whether functional assays offer additional or maybe even better opportunities to estimate their D2 receptor dissociation t 1/2.
Functional assays
Although G o is the preferred D 2 receptor binding partner and also the most abundant G protein in the brain, all three Gi proteins can be recognized as well (Sternweis and Robishaw 1984; Jiang et al. 2001; Gazi et al. 2003a; Nickolls and Strange 2004). The activated Gα and liberated βγ subunits are able to regulate a large variety of signal transduction pathways (Huff 1996; Jiang et al. 2001). Additionally, evidence is emerging that D 2 receptors are also able to trigger cellular events by interacting with other proteins like β-arrestin in a G protein-independent way (Pierce and Lefkowitz 2001; Beaulieu et al. 2005). In primary cell cultures as well as in cell lines that either express native or recombinant D 2 receptors, this allows their activation/signaling to be monitored by different in vitro biochemical readouts, not only to estimate the intrinsic efficacy of a drug (and to classify it as a full agonist, partial agonist, antagonist, or inverse agonist) but also its potency either directly (agonist dose–response curves) or indirectly (antagonist inhibition curves). Although less solicited, such functional experiments can also provide information about antagonist binding kinetics.
Numerous findings indicate that D 2 receptor-mediated responses depend on the cell-specific expression level of Gi/o proteins as well as on the repertoire of available effector proteins. This explains why D 2 receptors may trigger different, even opposite, responses in distinct cell types (Vallar et al. 1990; Kanterman et al. 1991). In this respect, a major distinction can be made between cells of neuroendocrine and mesenchymal origin (Ghahremani et al. 1999). In neurons, D 2 receptor stimulation is likely to trigger “inhibitory” pathways resulting in diminished excitability (Albin et al. 1989; Gerfen 1992), gene transcription, and proliferation. A well-known pathway comprises consecutive inhibition of the adenylyl cyclase activity, cell membrane hyperpolarization (via potassium channel activation), and decrease in the cytoplasmic calcium concentration ([Ca2+]i, via L-type calcium channel inhibition). In agreement therewith, nearly all striatal effects of D 2 receptor activation can be linked to decreased adenylyl cyclase activity (Sibley 1995). On the other hand, in cells of mesenchymal descent like fibroblasts, D 2 receptor activation is likely to trigger “stimulatory” pathways culminating in enhanced gene transcription and cell proliferation and comprising the consecutive activation of the phospholipase C-β enzyme, intracellular calcium mobilization, and activation of the mitogen-activated protein (MAP) kinase cascade.
Among the alternative factors that may influence the D2 receptor response mechanisms, it has been proposed that, in spite of their similar pharmacologic profile (Packeu et al. 2010a), the D2Short and D2Long isoforms can couple to different G proteins/transduction pathways and thereby exert different physiological roles in neurons (Wang et al. 2000; Centonze et al. 2002; Lindgren et al. 2003; Jomphe et al. 2006). Recent co-expression experiments also suggest that D1 and D2 receptors are able to form heterodimers (Faron-Górecka et al. 2008), and this could offer an explanation for the ability of D2 receptors to increase [Ca2+]i when D1 receptors are co-activated (Lee et al. 2004). Such heterodimerization could be of physiological relevance since anatomical studies point at the co-expression of both receptor subtypes in striatal neuron populations (Surmeier et al. 1996; Aizman et al. 2000). Finally, the very nature of the agonists could also play a role. Indeed, different agonists have been proposed to stabilize unique receptor conformations with different abilities to couple to G proteins and downstream effectors, a process denoted as “agonist directed-trafficking” (Kenakin 1996, 2003; Nickolls and Strange 2004). This could lead to divergent results when comparing the in vitro functional characteristics of D2 receptor agonists and partial agonists in different assays and even hinder the detection of the latter in certain screening studies (Lawler et al. 1999; Jordan et al. 2007a).
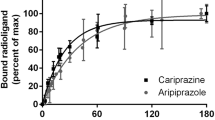
Estimates of drug potency and intrinsic activity (for partial agonists like aripiprazole) are particularly sensitive to the D2 receptor density (Burris et al. 2002; Tadori et al. 2009), and the degree of cellular amplification that the generated signal underwent is likely to have an impact as well ((Black and Leff 1983; Kenakin 1993; Vauquelin and von Mentzer 2007). This phenomenon, commonly denoted as “receptor reserve”, is associated with a leftward shift of agonist (like dopamine) dose–response curves and/or an upward shift in case of partial agonists (like aripiprazole). This paves the way to exacerbated agonist potencies and/or intrinsic activities. Receptor reserve unmistakably plays an important physiological role in vivo. In this regard, differences in “receptor reserve” have been repeatedly invoked to explain the higher sensitivity of presynaptic D2 receptors to dopamine as their postsynaptic counterparts (Meller et al. 1987, 1988; Carlsson and Carlsson 2006). Yet, for in vitro functional assays, it interferes with the correct analysis of agonist–D2 receptor interactions in terms of affinity and intrinsic efficacy (i.e., the ability of the agonist to generate/favor a stimulus or, in other words, an active receptor conformation; Furchgott 1966) and even of antagonist–D2 receptor interactions.
In this respect, high levels of receptor expression and attempts to boost cellular amplification of the response have been linked to the occurrence/detection of constitutive D2 receptor activity (i.e., spontaneous receptor activity under basal conditions) and the propensity of typical antagonists like haloperidol and sometimes even of clozapine to dose-dependently decrease that activity (i.e., to act as “inverse agonists”) (Nilsson and Eriksson 1993; Hall and Strange 1997; Wilson et al. 2001; Gazi et al. 2003b; Akam and Strange 2004; Roberts et al. 2004a; Roberts and Strange 2005; Burstein et al. 2005; Masri et al. 2008). However, this phenomenon is not readily observed at a lower receptor expression and/or when the response is measured at the level of naïve G protein activation, i.e., early on in the response pathway (Gardner et al. 1996; Malmberg et al. 1998; Vanhauwe et al. 1999; Gilliland and Alper 2000; Wilson et al. 2001; Pauwels et al. 2001a; Jordan et al. 2007b; Masri et al. 2008).
Additionally, receptor reserve will also interfere with the evaluation of antagonist potencies (when based on dose–inhibition curves, Tadori et al. 2009), but this can be circumvented by “Schild-plot” experiments in where agonist dose–response curves are generated in the presence of different concentrations of antagonist (Arunlakshana and Schild 1959). Provided that both are competitive and that the so-obtained dose–response curves reflect a mass-action equilibrium, the antagonists will trigger a dose-dependent rightward shift of those curves from which antagonist potencies can be calculated (Arunlakshana and Schild 1959; Vauquelin et al. 2002a, b). Yet, such equilibrium situations may be difficult to achieve. One paradigm is to perform co-incubation experiments (Fierens et al. 1999), but this may lead to an underestimate of the potency of some antagonists (i.e., when the antagonist–receptor interaction was unable to reach equilibrium at the time the response is measured). Especially for the classical organ-bath experiments, this provides a good rationale for the widespread trend to preincubate tissues plated cells with antagonists (to allot them sufficient time to reach equilibrium binding) before their challenge with agonist (Leff and Martin 1986).
Interestingly, this latter paradigm allows the antagonist’s dissociation rate to be estimated in the absence of receptor reserve (Vauquelin et al. 2002a,b; Kenakin et al. 2006; Charlton and Vauquelin 2010). Indeed, fast-dissociating competitive antagonists may liberate the receptors swiftly enough to make all of them accessible to the agonist so that new mass-action equilibrium can be reached at the time the response is measured. This allows the agonist to fully overcome receptor blockade provided that its concentration is high enough. Such antagonists are classified as “surmountable” and will typically produce a rightward shift of the agonist’s dose–response curve. At the other extreme, antagonists that bind irreversibly will not permit subsequently administered agonists to bind to/stimulate the affected receptors whatever their concentration and incubation time (Furchgott 1966). They are rightfully denoted as “insurmountable” as they will only trigger a dose-dependent reduction of the maximal response. Most antagonists, however, will bind reversibly but too slow to allow equilibrium to be fully attained when the response is measured. These antagonists will produce a mixed-type inhibition (Fig. 5), and their dissociation rate can be estimated from the decline in the maximal response (i.e., the lesser the decline the faster the dissociation; Kenakin et al. 2006). In fact, this estimate relies on how much re-equilibration can be achieved in the time allotted for observing the agonist response. This principle is very much the same as for the earlier described two-step competition binding experiments (Fig. 2, method 4) but with this particularity that the agonist concentration can be raised sufficiently high to outwin the competition with the antagonist (under equilibrium conditions) so that no intermediate wash-step is required. Unfortunately, the situation is much more complicated in the case of pronounced receptor reserve. This is clearly illustrated by the elegant study by Burris et al. (2002) in where the irreversible D2 receptor antagonist N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ) (Hamblin and Creese 1983) produced a dose-dependent rightward shift of the dopamine dose–response curve instead of depressing the maximal response. However, since it allows receptor reserve to be detected, the use of EEDQ can be turned to our advantage in control experiments.

Insurmountable behavior of competitive antagonists in functional experiments: effect of the antagonist’s dissociation rate and duration of the subsequent incubation with agonist. To observe this phenomenon, receptors have to be preincubated with a fixed concentration of antagonist and then incubated with increasing concentrations of agonist (timing of experimental manipulations shown in the left panel) after which a response is measured and plotted under the form of a agonist concentration–response curve (right panels). Data points are simulated (with equations 1, 2, and 4 in Table 3, where A stands for “antagonist” and D for “agonist”) for a situation in where the agonist’s response is linearly proportional to its receptor occupancy at the time of the measurement (i.e., the receptor occupancy–response relationship is not biased by cellular amplification of the signal, commonly referred to as “receptor reserve”). The agonist concentration–response curves are generated by non-linear regression analysis by GraphPad Prism based on a one-site model. Of note is that insurmountable antagonism could also point at allosteric interactions. More explicit information about insurmountable antagonism can be found in the following review articles (Vauquelin et al. 2002a, b; Kenakin et al. 2006). For the present simulation studies, receptors are preincubated for 90 min with a fast-dissociating (closed circle, t 1/2 = 6.9 min) and a slow-dissociating (open circle, t 1/2 = 138 min) antagonist at concentrations corresponding to ten times their K D and then incubated for 3 min (top right panel) or 30 min (bottom right panel) with agonist. While the fast dissociating antagonist is clearly insurmountable after 3 min (i.e., the maximal response is well below that of the response generated by naïve receptors (open triangle)), it becomes nearly completely surmountable after 30 min incubation with agonist (i.e., mainly a rightward shift of the dose–response curve). The maximal response is more depressed after preincubation with the slow-dissociating antagonist, and this depression is only little abated after 30 min incubation. Other parameters—k 1A = 1.108 M−1 min−1; k 1D = 1.109 M−1 min−1; and k −1D = 1 min−1
Taken together, functional responses are more problematic to interpret than radioligand binding data because of the potential interference of receptor reserve. Unfortunately, as appropriate controls relative to this matter have almost never been included in functional experiments with D2 receptors in past studies, the occurrence of receptor reserve can merely be deduced from the potency of dopamine to generate a given response (typically when the EC50 of dopamine is closer to the nanomolar than the micromolar range). On the other hand, functional assays also offer some advantages over radioligand binding. They are usually carried out in intact cells, and for the purpose of better pinpointing the dissociation t 1/2 of allegedly fast-dissociating D2 receptor antagonists like clozapine, some of the responses arise very swiftly. Incidentally, this swiftness also minimizes the interference of phenomena like receptor desensitization and internalization. Tables 1 and 2 summarize the most conspicuous assay techniques that have been exploited in distinct D2 receptor research domains. It appears that each assay is endowed with a distinct set of assets and limitations that have to be taken into account when tackling the issue of fast antagonist dissociation.
The most common tests in D2 receptor pharmacology are [35S]GTPγS binding, cAMP production (Table 1), and the measurement of transient rises in [Ca2+]i (Table 2). In the [35S]GTPγS binding assay, the labeled nucleotide effectively replaces guanosine diphosphate (GDP) in the GTP/GDP exchange process, an essential step in receptor-mediated G protein activation (Oldham and Hamm 2008). Positive is the very low degree of receptor reserve. This is likely related to the addition of GDP whose prime goal is to reduce non-D2-receptor-related [35S]GTPγS binding but which will also uncouple activated receptors from their G proteins (Gilliland and Alper 2000; Roberts et al. 2004b). On the other hand, the long incubation time and the fact that the experiments are carried out on cell membranes rather than on intact cells precludes the use of this technique for the present purpose. Dopamine is well known to inhibit cyclic adenosine monophosphate (cAMP) production in intact cells (but to this end, the product must first be stimulated with compounds like forskolin). Assay times down to 5 min are attainable (Kanterman et al. 1991), but the degree of receptor reserve is usually too pronounced to be useful. The high D2 receptor levels in those assays are likely to contribute to this pronounced receptor reserve (Burris et al. 2002; Tadori et al. 2009). Of particular interest is that, based on dopamine dose–response curves in the presence of different concentrations of EEDQ, Burris et al. (2002) calculated that the dopamine binds D2 receptor with an equilibrium dissociation constant, K A (Furchgott and Bursztyn 1967; Agneter et al. 1991), of 178 nM.
Transient rises in cytosolic calcium concentration (i.e., [Ca2+]i transients), can be conveniently measured spectrophotometrically, and with appropriate precautions (Charlton and Vauquelin 2010), it is well adapted to pharmacological screening purposes. D2 receptor stimulation has repeatedly been recorded by this technique in fibroblasts (Table 2), and βγ subunits are likely to intervene. Yet, as D2 receptors are inherently only less inclined to trigger robust [Ca2+]i transients in the usual recombinant cell lines (i.e., CHO and HEK-293), the signal is often “boosted” by co-expressing them with Gαq-like proteins that are known to display little receptor-preference (Gα15 and Gα16), with chimeric G proteins (Gαqi5 and Gαqo5, i.e., Gαq proteins containing the five carboxyl-terminal amino acids from Gαi and Gαo, respectively) or even to directly express fusion proteins in where Gαqo5 is covalently tethered to the carboxy terminus of the D2 receptor (Table 2). A major advantage of the [Ca2+]i transients is that, at maximally effective dopamine concentrations, the peak level is attained swiftly (within less than 20 s) and that the subsequent decline is swift as well. This implies that in Schild-type experiments in where the cells are pretreated with a receptor-saturating concentration of antagonist, only the very fast-dissociating ones do not produce a substantial decline of the maximal response. The inhibitory effect of clozapine preincubation on dopamine-mediated [Ca2+]i transients have successively been reported by Pauwels et al. (2001b), Moreland et al. (2004), and Dyhring et al. (2010). In all three studies, the low EC50 of dopamine suggests the existence of a pronounced receptor reserve. Although only two dopamine concentrations were used in the first study, the data suggest that clozapine produced a rightward shift of the dopamine dose–response curve. Yet, in light of the arguably large receptor reserve, the small size of this shift (<10-fold) precludes its unequivocal interpretation in terms of clozapine dissociation kinetics. The same consideration applies to the second study wherein only a single dopamine concentration was used.
Instead, Dyhring et al. (2010) investigated the effect of clozapine in presence of a wide range of dopamine concentrations. Here, clozapine was clearly shown to produce a concentration-dependent rightward shift of the dopamine dose–response curve without apparently decreasing the maximal response. At the highest clozapine concentration used, the shift was so large (i.e., dopamine EC50 from 5 to 700 nM after preincubation with 10 μM clozapine) that it can hardly be interpreted in terms of receptor reserve only. At least, if one assumes that the K A of dopamine does not largely exceed the value found by Burris et al. (2002) (i.e., 178 nM), this surmountable behavior of clozapine is compatible with a dissociation t 1/2 in the range of seconds. Interestingly, quetiapine showed similar behavior in those experiments.
Alongside, these authors (Dyhring et al. 2010; Pettersson et al. 2010) also compared the dissociation rates of clozapine, quetiapine, and selected “dopamine stabilizers” in an alternative experimental setting that is reminiscent to the earlier evoked multi-step/intermediate “washout” radioligand binding procedures depicted in Fig. 2. In short, antagonist-pretreated cells were briefly washed and incubated for variable periods of time with fresh medium before the final exposure to a single concentration of dopamine and measurement of the evoked [Ca2+]i transient. These experiments revealed that the calcium response was fully restored within 5 min in case of quetiapine while the response still only attained 25% of the control level after 30–120 min exposure of clozapine-pretreated cells to the new medium. At first glance, such findings could reflect differences in the dissociation rate of both antipsychotics but, upon closer inspection, alternative interpretations also come to light. First, in the case of a pronounced receptor reserve and an elevated agonist concentration (here dopamine was present at 30 times its EC50), the response may already be fully restored after liberation of a limited fraction of the D2 receptors present. Hence, when exclusively based on this experimental paradigm, it cannot be conclusively deduced that quetiapine–D2 receptor complexes dissociate swiftly. Second, it is remarkable that the “washout” and “Schild-type” paradigms utilized by Dyhring et al. (2010) should lead to distinct points of view when the results for clozapine are interpreted in terms of dissociation only (i.e., much slower dissociation under “washout” conditions). Yet, based on the information gathered from the two/multistep radioligand binding experiments (Packeu et al. 2010a, c; Tresadern et al. 2011), it is quite plausible that the stagnating dopamine response in the “washout” paradigm reflects the formation of a new mass-action equilibrium between the binding of the dopamine and clozapine that got released from cellular stores.
To get a better insight into this important issue, we have now also investigated the behavior of clozapine in related Schild-type experiments, but this time under conditions wherein the interpretation of the [Ca2+]i transients is not plagued by receptor reserve. To this end, we made use of the fact that the recombinant CHO cells (kindly donated by Dr. M. Detheux, Euroscreen s.a., Gosselies, Belgium) that were used in the earlier binding studies on D2Short receptors (Packeu et al. 2008) also expressed Gα16 and the calcium reporter apoaequorin (Robert et al. 2000). In line with the involvement of a single population of receptors, dopamine produced steep dose–response [Ca2+]i curves (Fig. 6). However, in contrast with the other studies presented in Table 2, the EC50 was quite high and in the same range as the K A of dopamine found by Burris et al. (2002) (i.e., 80–170 vs. 178 nM, respectively). Most importantly, rather than producing a leftward shift of the dopamine dose–response curve, preincubation of the cells with EEDQ produced here a concentration-dependent depression of the maximal response (Fig. 6). Both characteristics argue against the existence of a sizable receptor reserve for the intervening signaling pathway. Similar to the results presented by Dyhring et al. (2010), we here also found that pretreatment of the cells with clozapine produced a rightward shift of the dose–response curve, along with a small depression of the maximal response (Fig. 6b). This quasi-surmountable behavior provides additional evidence in favor of the very fast dissociation of clozapine–D2 receptor complexes.

Effect of clozapine (top panel) and EEDQ (bottom panel) preincubation on dopamine-mediated calcium transients in D2Long receptor-expressing CHO-AEQ cells. The CHO-AEQ cell line (kindly donated by Dr. M. Detheux; Euroscreen s.a., Gosselies, Belgium) expresses also Gα16 (to allow the stimulation of the phospholipase-C β/calcium signal transduction pathway by D2 receptors) and apo-aequorin (which, in the presence of co-elenterazine, will emit light at 466 nm when exposed to Ca2+ (Pinton et al. 2000)). To start the experiments, cells suspended in HEPES-buffered DMEM containing 0.1% BSA and 0.25 mM ascorbic acid were first incubated for 4 h with 5 μM coelenterazine h (Gentaur, Kampenhout, Belgium) and then diluted to a density of 105 cells/ml. Top panel: Cells were preincubated for 30 min at 37°C with medium only (closed square) or containing 0.2 μM (open circle) or 2 μM (closed circle) clozapine. One hundred microliters of the cell suspension was then injected into wells (white 96-well plates from Greiner Bio-one, Wemmel, Belgium) containing 100 μl dopamine at the indicated final concentrations. The luniniscence at 466 nm was measured during 80 cycles (200 ms each) immediately thereafter using a multi-well reader (Infinite M200, Tecan Benelux Group Ltd., Mechelen, Belgium). Signals (luminiscence above background, from a representative experiment and expressed in arbitrary units) in the presence of 100 μM dopamine either alone or in presence of 0.2 or 2 μM clozapine (curves 1 to 3, respectively) are shown in the insert. Signals in the main panel are expressed as area under the curve (AUC) luminiscence above background and expressed as percent of control (i.e., maximal signal in presence of dopamine only). The dopamine concentration–response curves are generated by non-linear regression analysis by GraphPad Prism based on a one-site model. Bottom panel: Cells were preincubated for 30 min at 37°C with medium only (closed square) or containing 1 μM (open circle) or 10 μM (closed circle) of the irreversible D2 receptor antagonist EEDQ (N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (Hamblin and Creese 1983)) and then challenged with different concentrations of dopamine as above. As expected for irreversible D2 receptor blockade in a cellular background devoid of “receptor reserve”, EEDQ produced a concentration-dependent decline in the maximal dopamine response. In contrast, 2 μM clozapine produced already a tenfold rightward shift of the dopamine concentration–response curve (corresponding to a K i for clozapine of about 0.2 μM) but only an about 25% reduction in the maximal response. In support of this only limited insurmountable effect, a large portion of the receptors was already stimulated by a receptor-saturating concentration of dopamine (or, in other words, liberated from clozapine) when the signal reached its peak level (i.e., within 3 s., insert of top panel). The dissociation t 1/2 of clozapine is therefore likely to be less than 3 s. More precise estimations are impeded by the transient nature of the signal and the complex nature of the equations that were proposed to delineate such signals (Christopoulos et al. 1999; Charlton and Vauquelin 2010)
On the other hand, D2 receptor agonists like quinpirole have also been shown to trigger a very fast and quite long-lasting decrease in the basal intracellular calcium concentration in primary cultures of rat mesencephalic neurons expressing recombinant D2Short and D2Long receptors (Table 2) (Jomphe et al. 2006). Although the maximal decline barely exceeded 30% and despite the labor-intensiveness of the assay, this experimental avenue could also be of potential interest for the purpose of detecting fast antagonist dissociation. Indeed, such functional experiments lend themselves to be performed in the same way as the delayed radioligand association experiments (Fig. 2), and pending the absence of receptor reserve, the data can be processed similarly. Likewise, D2-receptor-stimulated K+ channel opening and L-type Ca2+ channel inhibition (Table 1) proceed very fast as well, and it could therefore also be worthwhile to explore the benefits and limitations of the thereto dedicated electrochemistry-oriented assays. In this respect, a rapid restoration the dopamine-mediated L-type Ca2+ channel inhibition was shown to take place in sulpiride-pretreated intact medium spiny neurons following the removal of this antagonist (Hernandez-Lopez et al. 2000). This finding is compatible with the fast dissociation of sulpiride–D2 receptor complexes (Packeu et al. 2010a), but, in the absence of information about the degree of receptor reserve that is inherent to the assay, it does not constitute solid proof thereof. Finally, a number of additional biochemical techniques, like the measurement of agonist-evoked transient extracellular acidification, arachidonic acid release and MAP kinase activation have also been solicited for characterizing drug–D2 receptor interactions and/or the thereby elicited intracellular response pathways (Table 1). Among them, the extracellular acidification process seems to be the least susceptible to receptor reserve. This response is clearly transient, and although its evolution is appreciably slower than for the [Ca2+]i transients discussed above, the first measurement can already be carried out after 1 min in current microphysiometers. Also, the ability to perform repetitive measurements (spaced in time by the length of a cycle) allows the transient nature of the response to be followed. The D2 receptor-mediated increase in extracellular acidification is likely to be caused by the transport of protons by a Na+/H+ antiporter, and while it is readily observed in recombinant rat C6 glioma and CHO cells (Neve et al. 1992; Chio et al. 1994; Coldwell et al. 1999; Vanhauwe et al. 1999), other cell types may respond differently (Ganz et al. 1990). By using this technique, clozapine was reported to behave surmountably in a cellular background with only little receptor reserve (Coldwell et al. 1999). Important here is that measurements were routinely done 4.5 min after agonist addition, so that it can only be deduced that the dissociation t 1/2 of clozapine is likely to be less than this time period. Yet, there is room for improvement (such as performing measurements faster, i.e., after less and/or shorter cycles) to further narrow down the time frame during which clozapine–D2 receptor complexes have largely dissociated.
Taken together, the surmountable behavior of clozapine in fast responsive experimental systems with minimal receptor reserve confirms that clozapine–D 2 receptor complexes dissociate very swiftly, i.e., with a dissociation half-life likely to be in the range of seconds. Besides the more or less conventional assays discussed above (summarizes in Tables 1 and 2), fluorescence-based assays have recently been developed to monitor molecular processes that immediately follow GPCR activation. In this respect, it is known that the GTP/GDP exchange at the Gα subunit will trigger its physical segregation from the βγ subunits. While the previously mentioned [35S]GTPγS binding assay takes advantage of the first process, the latter process can, for example, be followed over time by measuring the energy transfer between bioluminescently tagged Gα and fluorescently tagged βγ (i.e., bioluminiscence resonance energy transfer (BRET)) (Turu et al. 2009). In addition, activated D 2 receptors are also able to bind non-G proteins like β-arrestin 2 very swiftly, and time-wise evolution of this binding process can be followed measuring the energy transfer between fluorescently tagged receptors and β-arrestin 2 molecules (i.e., fluorescence resonance energy transfer (FRET)) (Masri et al. 2008). These three assays are unlikely to engender significant receptor reserve, but, whereas [35S]GTPγS binding is usually done on cell membranes and requires a quite long incubation time, the latter can be performed on intact cells, and consecutive measurements on the same preparation can be made within very brief intervals. Similar to the previously mentioned [Ca2+]i transients, the signal rises also rapidly in the latter assays, but the maximum can be maintained over a prolonged time period. As thereto-dedicated fluorimeters become more and more omnipresent, such BRET- and FRET-based assays could greatly facilitate the study of antagonist dissociation kinetics not only for D2 receptors but also for GPCRs in general.
Simulations
The atypical character of clozapine and quetiapine has been proposed to emanate from their fast dissociation rate from D2 receptors. This kinetic property was initially discovered by investigating the dissociation of the labeled drugs from D2 receptors in cell membrane preparations. The sparse results from alternative, intact cell-based approaches confirm that their dissociation half-lives should not exceed a few seconds. Hence, there is presently a general consensus that clozapine and quetiapine dissociate very swiftly from D2 receptors. Simulations constitute a useful means of obtaining a better understanding about the link between the atypicality of clozapine and quetiapine and their fast dissociation from D2 receptors. Yet, seeing the complex nature of dopamine transmission, it is presumptuous to provide a full link between the in vitro and in vivo situation. Indeed, incorporating phenomena like the existence of autoreceptors with negative feedback on dopamine release, intra- and extrasynaptically located receptors, dopamine-mediated receptor internalization, and difference in receptor reserve between pre- and postsynaptic receptors should render the outcome of those simulations dependent on too many ill-known variables. Accordingly, we will use simplified systems with the major aim to illustrate how antagonist dissociation rates affect (1) D2 receptor occupancy by dopamine at time scales at which transient rises in the dopamine concentration takes place and (2) their own D2 receptor occupancy under in vivo-like conditions at time scales covering the interval between consecutive drug intakes.
Impact of the antipsychotic dissociation rate on D2 receptor occupancy by dopamine
Such simulations have to take account of the characteristics of striatal D2 receptors (like affinity and neuroanatomical localization) as well as of changes in the synaptical and extrasynaptical dopamine concentration (like amplitude and duration). In this respect, neuroimaging and subcellular localization studies provide interesting information at the in vivo level. At the other end of the “ladder of complexity” (Vauquelin and Charlton 2010), in vitro experiments on simple systems like cell membranes (widely used and also allowing ex vivo approaches) and intact cell lines (the simplest way to investigate “in live” receptor behavior without being bothered by the structural complexity of tissues) provide information at the molecular and cell physiological level. The most salient observations are briefly addressed hereunder.
Positron emission tomography (PET) and single photon emission computed tomography (SPECT) neuroimaging studies (Ichise et al. 2001) have traditionally focused on the striatum because of their much higher D2 receptor content than other brain areas. Because of the link between the striatal D2 receptor occupancy by antipsychotics and the relief of positive symptoms (Agid et al. 2007; Kegeles et al. 2008), as well as extrapyramidal side effects, focus has been restricted to this brain region. A first observation is that already part of the striatal D2 receptors are occupied by dopamine under baseline conditions. Indeed, acute depletion of endogenous dopamine upon a bolus administration of reserpine or the tyrosine hydroxylase inhibitor AMPT causes a significant increase in the D2 receptor binding potential (which reflects the receptor density in a brain region of interest) of the antagonist radiotracers [11 C]-raclopride and [123I]-IBZM in different species (Ginovart et al. 1997; Laruelle et al. 1997a,b; Abi-Dargham et al. 2000; Frankle et al. 2004; Narendran et al. 2004, 2005). Based on these observations, the baseline occupancy of D2 receptors in the striatum in antipsychotic drug-free humans and nonhuman primates was calculated to be in the 10% to 30% range. Based on this method, Abi-Dargham et al. (2000) and Frankle et al. (2004) estimated the occupancy of striatal D2 receptors by endogenous dopamine to be 16–19% in medication-free schizophrenics as compared with 9% in healthy controls. This finding lends support to the proposition that schizophrenia is associated with increased striatal D2 receptor stimulation.
Neuroimaging studies further suggest that the interaction between striatal D2 receptors and endogenous dopamine is not a simple homogenous process. Indeed, increasing the endogenous dopamine concentration to very high levels by intravenous administration of the psychostimulant d-amphetamine produces less decline of the baseline [11 C]-raclopride and [123I]-IBZM signals than that of agonist radiotracers in nonhuman primates, cats, and mice (Narendran et al. 2004, 2005; Seneca et al. 2006; Ginovart et al. 2006; Skinbjerg et al. 2010). This behavior endorses a model wherein the antagonist tracers bind to all D2 receptors alike while dopamine and agonist tracers prefer a subpopulation thereof. Such D2 receptor heterogeneity is likely to be related to their presence in distinct “environments”. Three major sources of heterogeneity have been advanced in this context. They are briefly commented below.
First of all, Kortekaas et al. (2004) evoked potential differences in the occupancy level of synaptic and extrasynaptic D2 receptors by endogenous dopamine. In this respect, striatal D2 receptors are well known to be located postsynaptically on medium spiny neurons as well as presynaptically on neurons from the cerebral cortex and the substantia nigra (Sesack et al. 2003, 1994). In addition, D2 receptor heterogeneity has also been perceived at the subcellular level. Electron micrographs of immunostained rat neostriatal D2 receptors indeed revealed their location in synaptic structures as well as in extrasynaptical regions (Yung et al. 1995; Hersch et al. 1995). Yet, no information about their relative abundance in both locations could be provided, and present opinions about which population is physiologically active in the striatum are sometimes even diametrically opposite (Rice and Cragg 2008; Miyake et al. 2010). In this respect, issues like release, uptake, and diffusion will greatly affect the free dopamine concentration and time-wise changes thereof in those distinct locations (Kawagoe et al. 1992; Garris et al. 1994; Cragg and Rice 2004; Schultz 2007). In this respect, simulations by Cragg and Rice (2004) suggest that, following a quantal release at a single synapse, the dopamine concentration should experience brief pulses of high amplitude in the synaptic space and less pronounced but longer-lasting bursts at some distance outside. Despite its complexity, this spatiotemporal “landscape” of dopamine is likely to permit distinct functions thereof (Schultz 2007). Indeed, dopamine could act as a classical neurotransmitter to produce fast impulse responses to reward when its concentration only reaches sufficiently high levels in the synapse and/or in the perisynaptic regions. On the other hand, dopamine could act as a neuromodulator to produce tonic enabling of postsynaptic motor systems if sufficiently high concentrations are continually present or can be temporarily reached over a wide spatial range outside the synapse. Such “volume transmission” of dopamine (Agnati et al. 2010) has recently been invoked to simulate the average, time-related exposure of D2 receptors in an idealized block of striatal tissue (Dreyer et al. 2010). Phasic and tonic firing of the neurons, the diffusion of dopamine out of the synapse, and its reuptake by the dopamine transporters was taken into account to simulate bursts and pauses in dopamine concentration.
Next, functional D2 receptor heterogeneity could arise from the ability of dopamine and other agonists to desensitize and internalize D2 receptors as part of their recycling process (Laruelle 2000; Paspalas et al. 2006; Marchese et al. 2008). In this respect, different ligands have unequal ability to reach/bind to internalized D2 receptors. For example, spiperone has much higher propensity to do so than raclopride while dopamine and the benzamide sulpiride hardly cross the cell membrane (Rayport and Sulzer 1995; Itokawa et al. 1996; Laruelle 2000; Guo et al. 2010). This was attributed to differences in hydrophobicity and other physicochemical characteristics of the drugs in question. Yet, contrary to initial suspicions, observations with wild-type and β-arrestin-3-knockout mice (which are incapable of internalizing D2 receptors), suggest that little internalization takes place upon the usual “short-term” (30 min) exposure of striatal D2 receptors to increased levels of dopamine in neuroimaging experiments (Skinbjerg et al. 2010). It is only appreciably later that such internalization is likely to take place (Laruelle 2000; Skinbjerg et al. 2010). However, the fast binding of β-arrestin 2 by activated D 2 receptors in recombinant HEK293 cells (Masri et al. 2008) suggests that such receptors may become desensitized (i.e., unable to interact with G proteins; Luttrell and Lefkowitz 2002) at a much faster pace. Hence, the ability of intravenous amphetamine to reduce the binding of the agonist radiotracers more than the binding of antagonists like [11 C]-raclopride in typical neuroimaging studies could reflect receptor desensitization and, as commented upon below, a thereto inherent decline in agonist binding potential.
Finally, biphasic D2 receptor agonist vs. radiolabelled antagonist competition binding curves are commonplace when working with cell membranes. This unveils the occurrence of two D2 receptor populations/states with the same antagonist affinity but with either low or high agonist affinity (designated as D High2 and D Low2 , respectively) (Sibley et al. 1982; Malmberg et al. 1998; Gazi et al. 2003a). This state of affairs is traditionally explained by the “ternary complex” model (De Lean et al. 1980; further extended by Samama et al. 1993) wherein GPCRs adopt a high agonist affinity state/conformation when they couple to their cognate G proteins. This model also explains why a larger proportion D High2 sites are observed with agonist vs. agonist radioligand/radiotracer competition experiments (Malmberg and Mohell 1995; Seeman et al. 2003; Skinbjerg et al. 2009). Although such functionally distinct D2 receptor populations are also likely to co-exist in vivo (see concluding chapter), it is still unclear whether the coupling-prone population also displays high affinity for dopamine in a living cell. On the one hand, the absence of D High2 sites in some studies on intact cells (Sibley et al. 1983; Skinbjerg et al. 2009) was explained by the fact that, due the presence of GTP, the ternary complex only constitutes a transient intermediate so that it never accumulates to an extent measurable in binding assays (Oldham and Hamm 2008). On the other hand, biphasic dopamine/[3H]domperidone (a D2 agonist) competition curves were observed with rat adenoma anterior pituitary cells (Seeman 2008) and, as presently shown in Fig. 7, dopamine/[3H]raclopride competition curves are also biphasic for D2Long receptor-expressing CHO cells. Whether this reflects differences in G protein binding capability and related phenomena like desensitization remains to be firmly established but receptor internalization is unlikely to be at stake. Indeed, the same competition binding profile is also observed with cells that were pretreated with 10 μM phenylarsine oxide, a condition which effectively prevents the internalization of the insulin-regulated aminopeptidase enzyme in CHO cells with only minimal cell death (Demaegdt et al. 2008, 2011). Similar studies by others have also led to the conclusion that D2 receptors do not internalize easily (Barton et al. 1991; Itokawa et al. 1996; Skinbjerg et al. 2009; Guo et al. 2010). Hence, internalization was not taken into account for the present simulations.

Dopamine/[3H]-raclopride competition curves in human D2Long receptor-expressing CHO cells. Naïve cells (closed circle) or cells pretreated for 10 min with 10 μM phenylarsine oxide (open circle) were co-incubated for 30 min at 37°C with 2 nM [3H]-raclopride and increasing concentrations of dopamine. Data refer to specific binding expressed as percentage of control binding (i.e., specific binding to naïve cells, phenylarsine oxide has no sizable effect thereon) and are presented as means±SEM of (five to six individual experiments with three determinations each). Competition curves are generated by non-linear regression analysis by GraphPad Prism. Date fitted best with a two-site model with distinct affinity for dopamine, i.e., D2 receptors in naïve cells were composed of high affinity sites with K i = 410 nM (52% of the specific binding) and low affinity sites with K i = 11 μM. Phenylarsine oxide (10 μM) has earlier been shown to effectively prevent internalization phenomena in CHO cells without causing seizable cell death (Demaegdt et al. 2008, 2011). D2 receptor internalization is an unlikely cause for the biphasic shape of the competition binding curve of dopamine since it is not noticeably affected by phenylarsine oxide
The occupancy of D2 receptors by dopamine was calculated by using a K D value of 10 nM in the aforementioned simulations (Cragg and Rice 2004; Dreyer et al. 2010). This reflects the affinity of dopamine for the D High2 sites in membrane preparations, but, as outlined above, dopamine–D2 receptor interactions are likely to proceed with lower affinity in vivo. In this respect, several authors have opted for affinity values that were higher by about one order in magnitude (i.e., K i = 100–160 nM) in order to fit the baseline occupancy of striatal D2 receptors in neuroimaging studies with recorded free dopamine concentrations (Ginovart et al. 1997; Laruelle et al. 1997b; Abi-Dargham et al. 2000; Frankle et al. 2004). Incidentally, these values are close to the K i of dopamine for the D High2 sites in the present study on intact CHO cells (410 nM, Fig. 7); its EC50 for eliciting calcium transients without receptor reserve in the same cells (80–170 nM, Fig. 6) as well as with its K A (178 nM) obtained by Burris et al. (2002) based on the effect of EEDQ on cAMP responses. Hence, experiments on intact cells and in vivo seem to converge to a value closer to 100 nM for functionally active D2 receptors. This latter value has been retained for the present simulations.
The ability of antagonists with different dissociation rates to protect D2 receptors against transient increases in dopamine concentration has previously been briefly explored. Olson (2005) explored their ability to suppress single spikes (lasting 100 ms) and bursts (lasting 300 ms) while Kapur and Seeman (2001) explored the effect on a 5-min surge in dopamine concentration. Such surges have indeed been observed to last from seconds to several minutes (Kawagoe et al. 1992; Koepp et al. 1998; Salimpoor et al. 2010). These simulations were based on the premises that dopamine and the radioligand bind to a single site at the receptor according to a bimolecular process (i.e., binding follows the law of mass-action) and that they are competitive with one another (i.e., that their binding is mutually exclusive such as in the case of overlapping binding sites). Especially since modeling studies indicate that clozapine and dopamine occupy closely the same binding pocket at the D2 receptor (Kalani et al. 2004), the present simulations will be based on the same premises.
The differential equations that govern time-wise changes in receptor occupancy by its ligands (see below) are usually integrated prior to the actual calculations, and this implies a number of “shortcuts” to be made to avoid the use of utterly long equations. For example, the integrals used by Olson (2005) only evoked the antagonist dissociation rate (implying dopamine association and dissociation and antagonist association to be instantaneous). In contrast, the present simulations are based on repeatedly, simultaneously implementing (typically 1,000 times per second) the underlying differential equations (1, 2, and 4 in Table 3) over very small time intervals (d(t)) as previously described (Vauquelin et al. 2001). To allow very fast binding kinetics, the association rate constant for dopamine (k 1D = 5.108 M−1 s−1) was set close to the maximal limit allowed by 3D diffusion of a ligand from the liquid phase to its membrane-associated receptor (Rhodes et al. 1985). Based on a K D of 100 nM, the dissociation rate constant for dopamine (k −1D) was 50 s−1. However, such fast binding properties do not constitute a necessary condition for fast responses to be generated if the cellular amplification of the signal is sufficiently high and if the response in “shut off” at a step downstream to receptor activation (Charlton and Vauquelin 2010).
Two antagonist concentrations were selected to model their impact on [DR]. At the lowest concentration, the antagonist occupied 60% of the receptors at the end of a sufficient number of low activity cycles for equilibrium to be reached. This level constitutes the lowest threshold at which efficacious antipsychotic action is observed. At the highest concentration, 80% of the receptors were occupied under the same conditions. At this level, extrapyramidal side effects and increased prolactin release start to appear. Further translation of [DR] into physiological responses was not attempted, especially because this requires the introduction of even more, often ill-known, variables such as those dealing with phenomena like functional receptor heterogeneity (see below), desensitization, internalization, and cellular amplification of the signal.
Based on recent simulations by Dryer et al. (2010), we first emulated time-related changes in (spatial-averaged) D2 receptor occupancy by dopamine in a block of striatal tissue (Fig. 8). The free dopamine concentration was be modeled as a train of successive cycles, each one with a “burst” (due to phasic dopamine release) and a subsequent “pause” (due to tonic release). In a situation of “low activity”, the bursts ([D] = 100 nM) lasted 200 ms, and the pauses (with [D] = 15 nM) lasted 1 s. Dopamine concentrations were allowed to rise and fall sharply for matter of convenience. These low activity periods are likely to be interspersed with attention- and task-related increases in the dopamine levels. Such increases could be several-fold and last from seconds to minutes (Koepp et al. 1998; Kapur and Seeman 2001). For the first series of simulations, the “high activity” situation (denoted as “Hi 1”) comprised a 300-ms burst with [D] = 250 nM and a subsequent 900 ms pause with [D] = 30 nM.

Simulated time-related changes in D2 receptor occupancy by dopamine (solid line) and antagonist (dotted line). Simulated data (with equations 1, 2, and 4 in Table 3, where A stands for “antagonist” and D for “dopamine”) are expressed in percent of control, i.e., receptor occupancy in the absence of antagonist. Cycles lasted 1.2 s and comprised a burst and subsequent pause. In a situation of “low activity” (denoted as “L”), the bursts ([D] = 100 nM) lasted 200 ms and the pauses (with [D] = 15 nM) lasted 1 s. For the “high activity” situation (denoted as “Hi 1”) the bursts ([D] = 250 nM) lasted 300 ms and the pauses (with [D] = 30 nM) lasted 900 ms. Dopamine concentrations were allowed to rise and fall sharply for matter of convenience. Antagonist concentrations are set to occupy 60% (left panels) or 80% (right panels) of the receptors after a sufficient number of low activity cycles for equilibrium to be reached. Antagonist dissociation t 1/2 values are 230 s (top panels), 6.9 s (mid panels), and 0.23 s (bottom panels). Only the cycles of the “H 1” high activity period are numbered. Other parameters—k 1A = 1.107 M−1 s−1; k 1D = 5.108 M−1 s−1 and k −1D = 50 s−1. Provided that the k 1A/[A] ratio is kept constant, varying k 1A between 1.106 and 1.108 M−1 s−1 (resp. tenfold lower and higher than the k 1A for [3H]-raclopride in intact cell binding studies (Packeu et al. 2010a)) yield the same outcome (data not shown). This “flexibility” allows the exploration of very fast and slow antagonist dissociation behavior while still remaining within reasonable K D(A) limits
Under control conditions (i.e., without antagonist), burst and pause [DR] values followed closely the changes in free dopamine concentration (Fig. 8). As shown in the top panel, a slow dissociating/“tight binding” antagonist (i.e., with a dissociation t 1/2 of 230 s that is well above the time-lapses for bursts and pauses) produced a concentration-dependent drop in [DR] without affecting the shape of the bursts in each individual cycle. Alternating between burst and pauses and between low and high (“Hi 1”) activity conditions had only minimal effect on the receptor occupancy by the antagonist but produced an immediate, corresponding change in [DR] values. A different profile emerges in case of a very fast dissociating/“loose binding” antagonist (i.e., with a dissociation t 1/2 of 230 ms, i.e., within the range of the time-lapse of a burst, lower panels of Fig. 8). Here, a time-wise increase in [DR] is readily perceptible within each individual burst. The corresponding drop in [AR] further suggest that binding of dopamine and the antagonist tends to reach a new mass-action equilibrium within the very short time lapse of such burst. In other words, this antagonist shows a time-wise gain in surmountability during the time lapse of a burst. An additional phenomenon also comes to light when the antagonist’s dissociation t 1/2 (i.e., 6.9 s, mid-panels of Fig. 8) is close to the time-lapse of a high activity period. While the dissociation of such antagonists is too slow to permit sizable surmountable behavior during individual bursts, a modest, time-wise increase in [DR] (and corresponding decrease in [AR]) is perceptible during the high activity period.
Using the same simulation paradigm, this latter phenomenon was examined in more detail by comparing the behavior of broader assortment of antagonists in “Hi 1” and also in an even higher activity condition (denoted as “Hi 2” with 300 with [D] = 750 nM for 300 ms and [D] = 90 nM for 900 ms pauses). The evolution of [DR]av (i.e., the time-averaged [DR] in each cycle) during a 45-cycle high activity period is shown in the top panels of Fig. 9 for 60% and 80% initial receptor occupancy by the antagonists, respectively.

Simulated time-related changes in D2 receptor occupancy by dopamine during “L” and “Hi 1” periods (see legend of Fig. 8) as well as during a period with even higher activity (“Hi 2”, with bursts ([D] = 750 nM) lasting 300 ms and with pauses (with [D] = 90 nM) lasting 900 ms). Simulated data are expressed in percent of control, i.e., receptor occupancy in the absence of antagonist. Antagonist concentrations are set to occupy 60% (left panels) or 80% (right panels) of the receptors after a sufficient number of low activity (L) cycles for equilibrium to be reached. Antagonist dissociation t 1/2 values are 230 s (closed diamond), 69 s (open triangle), 23 s (closed triangle), 6.9 s (open circle), 2.3 s (closed circle), 0.69 s (open sqare), and 0.12 s (closed square). Top panels. Simulations are cycle-based such as in Fig. 1, but the data are presented as average receptor occupancy per cycle of 1.2 s (denoted as [DR]av), and for the sake of clarity, the plots are only assigned by a symbol, i.e., without the individual data points. For “Hi 1”, these symbols are given vertically (by lack of space available) in the same order as the plots. E: [DR]av at equilibrium is obtained by non-linear regression analysis of the [DR]av values of consecutive cycles by GraphPad Prism according to a monoexponential association paradigm is equal to the value obtained by equation 8 in Table 3. Bottom panels. Same situations and representation as above but the simulations are simplified as they are only based on the average [D] values of each period, i.e., without paying attention to bursts and pauses. Curves are only shown in case of the three slowest dissociating antagonists
In the presence of all antagonists, [DR]av increased abruptly at the onset of the “high activity” periods. This abrupt increase is at least partly due to the increased occupancy of remaining free receptors by dopamine, and, as expected, it is most pronounced when only 60% of the receptors are initially occupied by the antagonist and for “Hi 2”. For the tighter binding antagonists, this initial event was clearly followed by a slower, mono-exponential increase in [DR]av, and the t 1/2 thereof was positively related to the dissociation t 1/2 of the antagonist in question. This increase reflects the time-wise gain in the antagonist’s surmountability during the high activity period. Eventually, the same maximal [DR]av will be attained for all antagonists if enough time is given for a new equilibrium between antagonist and dopamine binding to be reached. This phenomenon is most perceptible when the antagonist’s dissociation t 1/2 is in the same range as the time lapse of that period; too slowly dissociating antagonists (t 1/2 ≥ 230 s) remain nearly fully insurmountable during the entire high activity period while too swiftly dissociating antagonists (t 1/2 ≤ 2.3 s) are already fully surmountable after a few cycles. When the antagonists dissociate even faster, [DR]av start to increase even further at the very onset of the “high activity” periods. This additional boost can be ascribed to the surmountable antagonist behavior during the bursts themselves. Yet, this boost is only modest here because of the only limited contribution of the bursts to [DR]av.
Interestingly, the increase in [DR]av is of much larger amplitude in “Hi 2” in comparison to the “Hi 1” situation, and this is valid for both levels of initial antagonist-occupancy. This means that it is only when the dopaminergic output is sufficiently high that antagonists with different dissociation rates may bring about sizable differences in [DR]av during a high activity period. Very much the same outcome is obtained when simulations are based on a simpler model in where the concentration of free dopamine [D] is no longer subject to bursts and pauses. Here, [D] merely represents the time-averaged concentration of dopamine in the “low”- and both “high-activity” periods (i.e., 29, 85, and 255 nM, respectively). The so-simulated evolution of [DR] during a long high activity period is shown in the bottom panels of Fig. 9 for the three slowest-dissociating antagonists only. Two comparisons between the simulated [DR] (bottom panels) and [DR]av (top panels) values merit attention. First, the “gain in surmountability”- related increases in [DR] and [DR]av proceed with the same rate. This process is consistently faster than the antagonists’ dissociation rate and also depends on the initial level of antagonist occupancy (only shown for [DR] in Table 4). Second, the maximal [DR] values (i.e., when all antagonists have become surmountable) represent an overestimation of [DR]av but correspond to the values predicted for mass-action-type equilibrium binding of two competitive ligands (Equation 8 in Table 3).
Taken together, the present simulations could contribute to a better understanding of the implications of “the fast dissociation proposal” at the physiological level. More precisely, they may give us better insight into the ability of these drugs to discriminate between fluctuations in synaptic and extrasynaptic dopamine concentrations. The overall emerging picture is that antagonists only permit increased D2 receptor simulation during surges in dopamine concentration if their dissociation t 1/2 is in the same range or less than the duration of those surges. When credit is given to the dissociation half-lives of these atypical antipsychotics that were obtained by radioligand binding experiments on cell membranes (Kapur and Seeman 2000; Seeman 2005; Langlois et al. 2010), the present simulations reveal that increased D2 receptor stimulation by dopamine will take place at time scales ranging from seconds to minutes such as during high activity periods (Kapur and Seeman 2001). If the low incidence of side effects like extrapyramidal symptoms and increased prolactin release is indeed related to surmountable D2 receptor blockade at these time scales, one could expect a gradual variation in the “atypicality” of different D2 receptor antagonists. In agreement therewith, all seven D2 receptor antagonists that result in low or negligible parkinsonism (remoxipride, clozapine, quetiapine, norclozapine, perlapine, S-(-)-amisulpiride, and amoxapine) have been found to dissociate from the D2 receptor with half-lives ranging between 13 and 66 s (Seeman 2005, 2011). On the other hand, clozapine also displays surmountable antagonism when measuring its capacity to dampen transient rises in the dopamine-generated cytosolic calcium concentration in intact cell systems (Dyhring et al. 2010 and Fig. 6). Based on these findings, it is not excluded that clozapine dissociates even faster than the estimates that were obtained by radioligand binding. At the present level of investigation, it is still not clear yet whether this is indeed the case. Finally, extra D2 receptor stimulation during individual peaks in dopamine concentration becomes even more improbable since it should imply extremely fast dissociation of clozapine and quetiapine, i.e., with half-lives in the millisecond range such as already evoked by Olson (2005).
Impact of the antipsychotic’s dissociation rate on its D2 receptor occupancy between successive intakes
At time scales in the order of hours, the atypical character of antipsychotics like clozapine and quetiapine could also be linked to the transient nature of their D2 receptor occupancy at the system level (Kapur and Seeman 2001). This is clearly illustrated by a neuroimaging study with a subject who received a typical daily dose (350 mg) of clozapine. While the striatal D2 receptor occupancy was reasonably high (71%) 1 to 2 h after the last administration, it declined to 55% at 12 h and to 26% at 24 h (Jones et al. 2000). Similar transient occupancy has also been observed for clozapine in primates and for quetiapine in humans (Gefvert et al. 1998; Kapur et al. 2000; Suhara et al. 2002). This may explain why psychotic relapses of patients on clozapine and quetiapine occur soon after withdrawal of the antipsychotic (Alphs and Lee 1991; Seeman and Tallerico 1999). In sharp contrast therewith, a classical antipsychotic like haloperidol will maintain substantial D2 receptor occupancy over 24 h and even well beyond (Karbe et al. 1991; Nordström et al. 1992). This long-lasting residual occupancy by haloperidol may also account for the very slow rate of psychotic relapse in patients after its withdrawal (Baron et al. 1989; Alphs and Lee 1991). Consistent with these differences in the duration of D2 receptor blockade at the system level, long-term use of clozapine and quetiapine has not been found to trigger sizable D2 receptor upregulation while haloperidol did (Burt et al. 1977; Lidow and Goldman-Rakic 1994, 1997; Kapur and Seeman 2001; Seeman et al. 2006).
Initial neuroimaging studies with [11 C]-raclopride also revealed that, at doses known to be effective in routine clinical settings, clozapine and quetiapine occupied a smaller fraction of the D2 receptor than the typical antipsychotics. Yet, when the usually long lag time between the last intake of such drugs and the challenge with radiotracer is taken into account (typically 12 h, Tauscher et al. 2002), these findings can easily be explained by differences in the duration of striatal D2 receptor occupancy. Long lag times between intake and measurement (usually 12–24 h) have also been held responsible for the common belief that atypical antipsychotics do not elevate plasma prolactin levels. In this respect, Kapur et al. (2000) and Turrone et al. (2002) indeed observed that clozapine and quetiapine are able to provoke a modest increase in the plasma prolactin level in humans in the 1–5-h period after the last intake but that baseline values are returned by the time of the next dose. In the same vein, even atypical antipsychotics are now considered to be capable of producing extrapyramidal side effects when their D2 receptor occupancy exceeds the 80% mark (Farde et al. 1992; Kapur et al. 1999; Seeman 2002).
The ability of clozapine and quetiapine to only occupy over 60% of the striatal D2 receptors for a limited period of time (Gefvert et al. 1998; Kapur et al. 2000; Jones et al. 2000) and yet to be clinically efficacious has led to the proposal that transient D2 receptor occupancy is already sufficient to this end. In other words, this “transient D2 hypothesis” implies that continuous high occupancy at every hour of every day by these D2 receptor antagonists is not required for inducing or maintaining an adequate antipsychotic response (Kapur et al. 2000; Seeman 2011). Recent findings with a novel fast-dissociating D2 receptor antagonist, JNJ-37822681, are in line with hypothesis (Schmidt et al. 2010).
Contemporary mechanism-based pharmacokinetic/pharmacodynamic (PK/PD) models consider that, in a living system, the efficacy and duration of drug action not only depends on its PK properties (including its extent and rate of absorption, its distribution between blood plasma and the “effect compartment” wherein the therapeutic target resides, its metabolism and its excretion) but also on its PD properties (binding affinity, binding kinetics and efficacy) (Mager et al. 2003; Ploeger et al. 2009; Vauquelin and Charlton 2010). Although the peak concentration of a free drug in the effect compartment (here in the extracellular fluid in the striatum) and its elimination t 1/2 therefrom play an important role in its effect duration, an additional contribution of the drug–target dissociation t 1/2 is also to be expected at least if this dissociation is slow enough (Vauquelin and Van Liefde 2006; Tummino and Copeland 2008). While little contribution of this pharmacodynamic component is to be expected in case of the very fast dissociating atypical antipsychotics like clozapine and olanzapine, it is of interest to find out until what dissociation t 1/2 this pharmacodynamic component remains quiescent. This prompted us to compare the time-dependent D2 receptor occupancy by antagonists with different dissociation t 1/2 during a 24-h period following their administration in a system with open compartments, i.e., wherein drugs are able to enter and leave over time. To this end, the concentration of free antagonist in the receptor-containing compartment was simulated to undergo a time-wise increase and a subsequent decline during this 24 h period according to Equation 7 in Table 3. The rate constants for its uptake and elimination were set to stand for t 1/2 values of 1 and 6 h, respectively. This reproduces quite fairly the striatal D2 receptor occupancy profile of clozapine such as observed in humans and primates (Suhara et al. 2002; Seeman 2002). Values of interest here corresponded to 60% and 80% D2 receptor occupancy at equilibrium in the absence of dopamine, respectively.
The simulated binding behavior of antagonists with different dissociation t 1/2 during a 24-h period is depicted in the top panels of Fig. 10. A biphasic pattern is clearly observed for all the antagonists investigated. Yet, it is noteworthy that receptor occupancy by those antagonists declines at a slower pace than their free concentration and this even when their dissociation t1/2 is in the sub-second range. Moreover, this delay is even more pronounced at the highest (i.e., 80%) theoretical occupancy level. These phenomena can readily be explained by a combination of the high initial receptor occupancy level and the hyperbolic ligand concentration–receptor occupancy relationship (i.e., a trait of the sigmoidal ‘E max’ model in pharmacokinetics) (Meibohm and Derendorf 1997; Szczuka et al. 2009). Hence, high local antagonist concentrations in the effect compartment will already lead to a longer-lasting receptor blockade by itself. It is also of interest to note that, under the same conditions, antagonists with a wide range of dissociation t 1/2 values (i.e., from 0.2 s till as high as 40 min) display very much the same time-related receptor occupancy profile. This contrasts with the dissimilar capability of such antagonists to prevent dopamine binding during bursts and even during high activity periods such as depicted in Figs. 8 and 9. When their free concentration has already substantially declined at the moment of the subsequent intake (such after 24 h in the present case), such antagonists will also show very similar recurrent binding profiles (shown for day 4 in the bottom panels of Fig. 10).

Simulated time-related changes in D2 receptor occupancy by antagonists, [AR], during a 24 h inter-dose period after the first intake (day 1, top panels) and after 4 days daily intake (bottom panels). Simulated data are expressed in percent of total receptor occupancy. All receptors are free at the start. After administration, the concentration of free antagonist [A] in the receptor-containing compartment (curve in bold) evolves with time according to equation 7 in Table 3 (where A stands for “antagonist”) with k up = 0.0115 min−1 and k el = 0.0019 min−1. Parameter A’ was adjusted to obtain peak values of [A] corresponding to 60% (left panels) and 80% (right panels) D2 receptor occupancy at equilibrium after the first intake. The bold black curve in all panels refers to [A] (normalized for its maximal concentration to be at the same level as its receptor occupancy at equilibrium). [AR] vs. time curves are simulated (with equations 1 and 3 in Table 3, where A stands for “antagonist”) for antagonists with dissociation t 1/2 values of 0.23 s (closed square), 38 min (open square), 6.3 h (closed circle), and 19 h (open circle). Other parameters—k 1A = 6.108 M−1 min−1
It is only when the antagonist’s dissociation t 1/2 nears its elimination t 1/2 that a further delay in its receptor occupancy becomes perceptible. This delay is positively related to the antagonist’s dissociation t 1/2 and has previously also been observed in simulation studies in where the free antagonist concentration was only allowed to decline exponentially instead of exhibiting the present biphasic pattern (Vauquelin and Van Liefde 2006; Vauquelin and Charlton 2010). However, this further delay comes at the expense of a deferred and also less pronounced peak occupancy level. Indeed, because of the high affinity of such very slow dissociating antagonists, the desired level of receptor occupancy (at equilibrium) only requires a minute amount of free antagonist to be present. Yet, this goes along with their slow association (since the pseudo-first-order association rate constant is positively related to the ligand concentration, Charlton and Vauquelin 2010), and this will eventually lead to an outspoken non-equilibrium situation wherein the binding only culminates when the concentration of free antagonist has already commenced to decline. This phenomenon also constitutes a handicap for very high affinity radiotracers for which the lack of equilibrium within the timeframe of the neuroimaging experiment does not allow accurate determination of D2 receptor availability in the striatum (Olsson and Farde 2001). Finally, repeated daily intake of such slowly dissociating antagonists will increase their receptor occupancy during a 24-h period and decrease the variability thereof within that period as well, albeit without reaching the same maximal receptor occupancy as the faster dissociating antagonists (bottom panels of Fig. 10).
The present simulations give us better insight about the impact of receptor dissociation kinetics at the systemic level after single and consecutive drug intakes. They reveal that the pharmacokinetic properties like the drug’s elimination t 1/2 at the effect compartment as well as the maximal level of receptor occupancy by that drug exert a preeminent influence on how long the receptor occupancy can exceed a certain level between consecutive dosings. In this respect, clozapine and quetiapine have been found to experience fast distribution between the plasma and the brain and also to experience fast elimination thereof (Hartvig et al. 1986; Kapur et al. 2000; Suhara et al. 2002). Although the partitioning of clozapine within the membrane could be held responsible for its higher concentration in the brain, it will apparently not delay its elimination thereof (Rayport and Sulzer 1995; Olsen et al. 2008; Packeu et al. 2010c). This is in agreement with intact cell studies in where the partitioning between free and membrane/cell-associated clozapine was found to be a rapid reversible process (Packeu et al. 2010c). Along with their pharmacokinetic properties, the fast dissociation of clozapine and quetiapine will contribute to a fast fluctuating D2 receptor occupancy in vivo. However, the present simulations also reveal that antipsychotics with even considerable slower dissociation characteristics should also be able to bring about comparable transient D2 receptor occupancy at the system level as long as their dissociation t 1/2 is less than their elimination t 1/2. This implies that, if atypicality was merely to be related to fluctuations in receptor occupancy at the systemic level, clozapine and quetiapine should need to possess some rather unique pharmacokinetic characteristics. If so, one could invoke higher peak occupancy values, slower elimination t 1/2 from brain tissue, longer-lasting retention in cellular stores, and/or a higher propensity for rebinding phenomena in case of the other antipsychotics.
Interestingly, because of the bell-shape of the time-related occupancy curves, beneficial levels of occupancy (≥60%) are bound to last longer than the detrimental ones (≥80%). In this respect, as evidenced by the only temporary and modest elevation in the plasma prolactin levels by clozapine, this 80% threshold should only be transcended briefly with the usual dosage (300–400 mg daily, Suhara et al. 2002), if at all. Indeed, it should be a reminder that the clinical dose range of clozapine is severely limited by its propensity to trigger agranulocytosis (Idanpaan-Heikkila et al. 1977).
Concluding remarks
Because of their more rapid dissociation from D2 receptors than other antipsychotics, it is now widely accepted that clozapine and quetiapine allow more extensive and frequent access of endogenous dopamine to the receptor and this even when they are administered at high dosages. To illustrate this assertion, the present simulations provide some overall insight into the influence of the antagonist’s dissociation rate on fluctuations of their D2 receptor occupancy between daily intakes and, at shorter time frames, on the D2 receptor occupancy by dopamine under burst and pause conditions as well as during low and high activity periods. These simulations suggest that the low propensity of clozapine and quetiapine to elicit extrapyramidal side effects and chronic hyperprolactinemia are related to a rather unique combination of pharmacodynamic (i.e., their ability of to allow increased D2 receptor stimulation by endogenous dopamine during high activity periods) and pharmacokinetic (i.e., fast fluctuating D2 receptor occupancy between successive doses) properties. Yet, this combination also precludes the traditional “beneficial” 60% receptor occupancy threshold to be permanently exceeded at every hour of every day. Despite of this, clozapine, quetiapine, as well as the recently described highly selective and fast-dissociating D2 receptor antagonist, JNJ-37822681, are able to induce and maintain an adequate antipsychotic response under such conditions (Gefvert et al. 1998; Kapur et al. 2000; Jones et al. 2000; (Langlois et al. 2010; Schmidt et al. 2010). Further work should certainly be beneficial to understand why such transient D2 receptor occupancy is already clinically efficacious and, more generally, for which pathologies this principle is also applicable.
However, fast dissociation of antagonist–D2 receptor complexes in cell membrane preparations does not preclude long-lasting occupancy under in vivo conditions. This note of caution is clearly illustrated by the discrepant behavior of haloperidol. First of all, the t 1/2 by which [3H]-haloperidol dissociates from D2 receptor-containing cell membranes was found to vary considerably from one study to another (e.g., 72 s (Langlois et al. 2010), 7 min (Leysen and Gommeren 1984), and 42 min (Kapur and Seeman 2000)), even though these dissociation events were all recorded in the presence of a large excess of unlabelled ligand. While this experimental approach is generally considered to produce the most direct information about a drug’s dissociation rate, the present example illustrates that the outcome could be very reactive to the experimental conditions. A second, even larger discrepancy comes to light when comparing these in vitro results with those of neuroimaging studies in humans. Here, the occupancy of striatal D2 receptors by haloperidol was found to undergo only little decline during a 24-h post-intake period (Karbe et al. 1991; Nordström et al. 1992). A similar discrepancy is also observed for spiperone. While [3H]-spiperone dissociates manifestly from D2 receptors in cell membrane preparations (e.g., t 1/2 ~ 30 min from recombinant CHO cell membranes at 37°C), related radiotracers like [11C]-N-methylspiperone bind almost irreversibly in vivo (Vauquelin and Packeu 2009; Ichise et al. 2001).
As outlined more extensively in the “Radioligand binding” section, binding studies on intact recombinant CHO cells unveiled at least part of the phenomena that may be responsible for this discrepant behavior. First, it was found that [3H]-spiperone dissociated more slowly from the cells than from the membranes thereof (Vauquelin and Packeu 2009). [3H]-Spiperone was also found to bind quasi-irreversibly to those cells if its “rebinding” was unchecked by the addition of an excess of unlabelled ligand and similar rebinding properties could also explain the quasi-irreversible binding of unlabelled haloperidol and (+)butaclamol (Packeu et al. 2008, 2010c). Interestingly, D2 receptor antagonists like raclopride and clozapine could only prevent [3H]-spiperone rebinding when present at unexpectedly high concentrations (Packeu et al. 2010b). Dopamine is likely to perform poorly as well, as evidenced by recent competition binding studies on intact CHO cells in where dopamine “competed” with [3H]-spiperone binding with a Ki of only 21 μM (N = 4, data not shown) as well as by the insensitivity of in vivo [11 C]-N-methylspiperone binding to dopamine depletion (Laruelle 2000). This corroborates with the results from recent competition binding studies on intact CHO cells The possible existence of allosteric interactions was already alluded by Laruelle et al. (2000), and Packeu et al. (2008) further proposed that, whereas raclopride should approach its binding site at the D2 receptor via the classical diffusion in the medium, spiperone should rather gain access to the D2 receptor via the membrane bilayer and then reach its binding site by diffusing within the receptor’s α–helical transmembrane domains (Fig. 11). Such approach has also been proposed for other receptors (reviewed in Vauquelin and Packeu 2009), and it even constitutes major element of the “diffusion microkinetic” model (Anderson 1993; Anderson et al. 1994) that was proposed to explain a long-lasting bronchodilatory action of the hydrophobic β2 adrenergic receptor agonist salmeterol, despite of its rapid dissociation from these receptors (Fig. 11). Additionally, hydrophobic butyrophenones like haloperidol, spiperone, and butaclamol could also extensively bind to the plasma membrane by “lipophilic solvation” and undergo “acidothropic uptake” in intracellular compartments (Rayport and Sulzer 1995; Laruelle et al. 2000). Slow release of these antagonists from these storage compartments could then also contribute to their long-lasting D2 receptor occupancy in vivo.

Potential similarity between the binding profile of haloperidol, spiperone, clozapine, raclopride, sulpiride, and dopamine to D2 receptors and the binding profile of beta2 adrenergic receptor ligands. More information is provided in Anderson (1993), Anderson et al. (1994), Szczuka et al. (2009), and Packeu et al. (2010b, c). The benzodiazepines are likely to partition in the membrane first and then reach their binding site at the D2 receptor by lateral diffusion between the membrane lipids and the receptor’s alpha-helical transmembrane domains. On the other hand, clozapine is likely to undergo reversible partitioning but still needs to reach the receptor via 3D diffusion within the fluid phase. Raclopride, sulpiride, and dopamine will essentially remain in the fluid phase
Finally, schizophrenia is now also widely accepted to be associated with increased striatal D2 receptor stimulation. In this respect, Frankle et al. (2004) advanced that, to be clinically efficacious in schizophrenia, antipsychotics should have to reduce D2 receptor occupancy by dopamine to a lower level than that observed in healthy subjects. The thereto dedicated calculations were based on the observed differences in striatal D2 receptor occupancy by endogenous dopamine in medication-free schizophrenics and in healthy controls (Abi-Dargham et al. 2000; Frankle et al. 2004) and took account of the clinically effective occupancy levels by antipsychotics in general. Inherent to this conclusion is the premise that the extent of D2 receptor occupancy and the thereby evoked response are similarly related in both populations. Yet, this may not be the case. Indeed, there is now solid evidence that only part of the D2 receptors are functionally active in vivo and that primary neuronal defects and drug insults that are able to elevate their level also trigger psychosis (Seeman et al. 2006; Seeman 2011). This is at variance with the initial assertion by De Lean et al. (1980) that, in cell membrane preparations, all the GPCRs present should be able to couple their cognate G proteins without discrimination. Yet, other investigators have rather endorsed the existence of two non-interconvertible receptor populations. Among others, it was argued that the limited proportion of GPCRs that reside in the high affinity (i.e., G protein-coupled) state is hardly compatible with the observed molar excess of G proteins (~100-fold) (Neubig et al. 1985; Ransnäs and Insel 1988). Moreover, based the ability of the alkylating reagent N-ethylmaleimide to trigger quasi-irreversible agonist binding to Gs-coupled β-adrenoceptors (Korner et al. 1982; Vauquelin and von Mentzer 2007), the existence of two distinct populations was clearly perceived in all the membrane preparations wherein such coupling could take place, and the fraction of coupling-prone receptors (25–70%) was closely linked to the tissue/cellular origin of those membranes (Vauquelin and Maguire 1980; Severne et al. 1986; Vauquelin et al. 1988; Vauquelin and von Mentzer 2007). These observations led to the proposal that coupling-prone and coupling-refractory GPCRs may reside in distinct membrane microenvironments (Severne et al. 1984; Ransnäs and Insel 1988; Neubig et al. 1988; Graeser and Neubig 1993). Experimental support for this hypothesis was provided later on by the discovery of specialized membrane compartments with limited receptor mobility such as caveolae and lipid rafts (Ostrom et al. 2000; Ostrom 2002; Pike 2003; Allen et al. 2007; Charalambous et al. 2008). For D2 receptors, recent findings also suggest that their presence in lipid rafts constitutes a necessary condition for their signaling to take place (Sibley et al. 2011).
Alterations in the D High2 /D low2 ratio echo important patho-physiological manifestations in vivo. Indeed, an important aspect of schizophrenia is that the majority of patients show behavioral super-sensitivity to dopamine-like drugs like amphetamine, methamphetamine, cocaine, apomorphine, or methylphenidate, and this is unrelated to whether or not they are taking antipsychotic medication (Lieberman et al. 1987; Curran et al. 2004). In this respect, substances like amphetamine, phencyclidine, steroids, and ethanol, as well as some brain lesions have also been found to produce psychosis and dopamine super-sensitivity in humans. In this respect, it is of special interest that dopamine super-sensitivity can also be induced in rats and that this goes along with a two- to fourfold increase in the proportion of D High2 receptors in their striatal membranes (Seeman et al. 2006; Seeman 2009, 2011). On the other hand, the total D2 receptor concentration seems to be much less affected both in manipulated rodents and in schizophrenic patients (Farde et al. 1990; Seeman et al. 2002). These findings led to the proposal that primary neuronal defects as well as drug insults that elevate the D High2 state in brain regions may elicit psychosis (Seeman et al. 2006; Seeman 2011). Since a redistribution of D2 receptors between membrane compartments with different susceptibility for G protein coupling could be at stake, it should be of interest to find out whether or not dopamine super-sensitivity is associated with an increased occurrence of D2 receptors in specialized membrane structures like caveolae and lipid rafts. In this respect, it has also been alluded that the altered expression of D2 receptor interacting proteins could promote modifications to the trafficking and/or the signaling profile of those receptors (Kabbani et al. 2005; Kabbani and Levenson 2006).
References
Abi-Dargham A, Laruelle M (2005) Mechanisms of action of second generation antipsychotic drugs in schizophrenia: insights from brain imaging studies. Eur Psychiat 20:15–27
Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, Weiss R, Cooper TB, Mann JJ, Van Heertum RL, Gorman JM, Laruelle M (2000) Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci U S A 97:8104–8109
Agid O, Mamo D, Ginovart N, Vitcu I, Wilson AA, Zipursky RB, Kapur S (2007) Striatal vs extrastriatal dopamine D2 receptors in antipsychotic response: a double-blind PET study in schizophrenia. Neuropsychopharmacol 32:1209–1215
Agnati LF, Guidolin D, Guescini M, Genedani S, Fuxe K (2010) Understanding wiring and volume transmission. Brain Res Rev 64:137–159
Agneter E, Drobny H, Singer EA (1991) Estimation of agonist dissociation constants at central presynaptic alpha2 autoreceptors in the absence of autoinhibition. J Pharmacol Exp Ther 257:19–25
Aizman O, Brismar H, Uhlen P, Zettergren E, Levey AI, Forssberg H, Greengard P, Aperia A (2000) Anatomical and physiological evidence for D1 and D2 dopamine receptor colocalization in neostriatal neurons. Nat Neurosci 3:226–230
Akam E, Strange PG (2004) Inverse agonist properties of atypical antipsychotic drugs. Biochem Pharmacol 67:2039–2045
Albin RL, Young AB, Penney JB (1989) The functional anatomy of basal ganglia disorders. Trends Neurosci 12:366–375
Allen JA, Halverson-Tamboli RA, Rasenick MM (2007) Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci 8:128–140
Alphs LD, Lee HS (1991) Comparison of withdrawal of typical and atypical antipsychotic drugs: a case study. J Clin Psychiat 52:346–348
Amalric M, Koob GF (1993) Functionally selective neurochemical afferents and efferents of the mesocorticolimbidopamine system. Prog Brain Res 99:209–226
Anderson GP (1993) Formoterol: pharmacology, molecular basis of agonism, and mechanism of long duration of a highly potent and selective beta2-adrenoceptor agonist bronchodilator. Life Sci 52:2145–2160
Anderson GP, Lindén A, Rabe KF (1994) Why are long-acting beta-adrenoceptor agonists long-acting? Eur Respir J 7:569–578
Angrist B, Van Kammen DP (1984) CNS stimulants as a tool in the study of schizophrenia. Trends Neurosci 7:388–390
Armstrong D, Strange PG (2001) Dopamine D2 receptor dimer formation: evidence from ligand binding. J Biol Chem 276:22621–22629
Arnt J, Skarsfelt T (1998) Do novel antipsychotics have similar pharmacological characteristics? A review of evidence. Neuropsychopharmacol 18:63–101
Arunlakshana O, Schild HO (1959) Some quantitative uses of drug antagonists. Brit J Pharmacol 14:48–58
Baldessarini RJ, Frankenburg FR (1991) Clozapine. A novel antipsychotic agent. N Engl J Med 324:74–54
Baron JC, Martinot JL, Cambon H, Boulenger JP, Poirier MF, Caillard V, Blin J, Huret JD, Loch C, Maziere B (1989) Striatal dopamine receptor occupancy during and following withdrawal from neuroleptic treatment: correlative evaluation by positron emission tomography and plasma prolactin levels. Psychopharmacology 99:463–472
Barton AC, Black LE, Sibley DR (1991) Agonist-induced desensitization of D2 dopamine receptors in human Y-79 retinoblastoma cells. Mol Pharmacol 39:650–658
Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG (2005) An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122:261–273
Bédard P, Larochelle L, Parent A, Poirier LJ (1969) Dopamine and serotonin in the striatum of the cat. Effect of lesions of the intralaminar nuclei. Arch Neurol 20:239–242
Ben-Jonathan N, Hnasko R (2001) Dopamine as a prolactin (PRL) inhibitor. Endocrine Rev 22:724–763
Björklund A, Falck B, Hromek F, Owman C, West KA (1970) Identification and terminal distribution of the tubero-hypophyseal monoamine fibre systems in the rat by means of stereotaxic and microspectrofluorimetric techniques. Brain Res 17:1–23
Black JW, Leff P (1983) Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci 220:141–162
Brink CB, Wade SM, Neubig RR (2000) Agonist-directed trafficking of porcine alpha2A-adrenergic receptor signaling in Chinese hamster ovary cells: l-isoproterenol selectively activates Gs. J Pharmacol Exp Ther 294:539–547
Burris KD, Molski TF, Xu C, Ryan E, Tottori K, Kikuchi T, Yocca FD, Molinoff PB (2002) Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 302:381–389
Burstein ES, Ma J, Wong S, Gao Y, Pham E, Knapp AE, Nash NR, Olsson R, Davis RE, Hacksell U, Weiner DM, Brann MR (2005) Intrinsic efficacy of antipsychotics at human D2, D3, and D4 dopamine receptors: identification of the clozapine metabolite N-desmethylclozapine as a D2/D3 partial agonist. J Pharmacol Exp Ther 315:1278–1287
Burt DR, Creese I, Snyder SH (1977) Antischizophrenic drugs: chronic treatment elevates dopamine receptor binding in brain. Science 196:326–328
Carlsson A, Carlsson ML (2006) A dopaminergic deficit hypothesis of schizophrenia: the path to discovery. Dialog Clin Neurosci 8:137–142
Caron MG, Beaulieu M, Raymond V, Gagne B, Drouin J, Lefkowitz RJ, Labrie F (1978) Dopaminergic receptors in the anterior pituitary gland: correlation of [3H]dihydroergocryptine binding with the dopaminergic control of prolactin release. J Biol Chem 253:2244–2253
Centonze D, Usiello A, Gubellini P, Pisani A, Borrelli E, Bernardi G, Calabresi P (2002) Dopamine D2 receptor- mediated inhibition of dopaminergic neurons in mice lacking D2L receptors. Neuropsychopharmacol 27:723–726
Charalambous C, Gsandtner I, Keuerleber S, Milan-Lobo L, Kudlacek O, Freissmuth M, Zezula J (2008) Restricted collision coupling of the A2A receptor revisited: evidence for physical separation of two signaling cascades. J Biol Chem 283:9276–9288
Charlton S, Vauquelin G (2010) Elusive equilibrium: the challenge of interpreting receptor pharmacology using calcium assays. Brit J Pharmacol 161:1250–1265
Chio CL, Lajiness ME, Huff RM (1994) Activation of heterologeously expressed D3 dopamine receptors: comparison with D2 dopamine receptors. Mol Pharmacol 45:51–60
Christopoulos A, Parsons AM, Lew MJ, El-Fakahany EE (1999) The assessment of antagonist potency under conditions of transient response kinetics. Eur J Pharmacol 382:217–227
Coldwell MC, Boyfield I, Brown AM, Stemp G, Middlemiss DN (1999) Pharmacological characterization of extracellular acidification rate responses in human D2(long), D3 and D4.4 receptors expressed in Chinese hamster ovary cells. Brit J Pharmacol 127:1135–1144
Copeland RA (2010) The dynamics of drug-target interactions: drug-target residence time and its impact on efficacy and safety. Expert Opin Drug Discov 5:305–310
Copeland RA, Pompliano DL, Meek TD (2006) Drug-target residence time and its implications for lead optimization. Nat Rev Drug Disc 5:730–739
Cragg SJ, Rice ME (2004) DAncing past the DAT at a DA synapse. Trends Neurosci 27:270–277
Creese I, Burt DR, Snyder SH (1976) Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 192:481–483
Csernansky JG, Murphy GM, Faustman WO (1991) Limbic/mesolimbic connections and the pathogenesis of schizophrenia. Biol Psychiat 30:383–400
Curran C, Byrappa N, McBride A (2004) Stimulant psychosis: systematic review. Brit J Psychiat 185:196–204
Dal Toso R, Sommer B, Ewert M, Herb A, Pritchett DB, Bach A, Shivers BD, Seeburg PH (1989) The dopamine D2 receptor: two molecular forms generated by alternative splicing. EMBO J 8:4025–4034
Davis KL, Kahn RS, Ko G, Davidson M (1991) Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiat 148:1474–1486
De Lean A, Stadel JM, Lefkowitz RJ (1980) A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem 255:7108–7117
Demaegdt H, Smitz L, De Backer J-P, Le MT, Bauwens M, Michotte Y, Vanderheyden P, Vauquelin G (2008) Translocation of the insulin regulated aminopeptidase to the cell surface: detection by radioligand binding. Brit J Pharmacol 154:872–881
Demaegdt H, Gard P, De Backer J-P, Lukaszuk A, Szemenyei E, Tóth G, Tourwé D, Vauquelin G (2011) Binding of “AT4 receptor” ligands to the insulin regulated aminopeptidase (IRAP) in intact Chinese hamster ovary cells. Mol Cell Endocrinol 339:34–44
Dowling MR, Charlton SJ (2006) Quantifying the association and dissociation rates of unlabelled antagonists at the muscarinic M3 receptor. Brit J Pharmacol 148:1134–1142
Dreyer JK, Herrik KF, Berg RW, Hounsgaard JD (2010) Influence of phasic and tonic dopamine release on receptor activation. J Neurosci 30:14273–14283
Dyhring T, Nielsen EØ, Sonesson C, Pettersson F, Karlsson J, Svensson P, Christophersen P, Waters N (2010) The dopaminergic stabilizers pridopidine (ACR16) and (-)-OSU6162 display dopamine D2 receptor antagonism and fast receptor dissociation properties. Eur J Pharmacol 628:19–26
Einhorn LC, Oxford GS (1993) Guanine nucleotide binding proteins mediate D2 dopamine receptor activation of a potassium channel in rat lactotrophs. J Physiol 462:563–578
Einhorn LC, Gregerson KA, Oxford GS (1991) D2 dopamine receptor activation of potassium channels in identified rat lactotrophs: whole-cell and single-channel recording. J Neurosci 11:3727–3737
Eklind-Cervenka M, Benson L, Dahlström U, Edner M, Rosenqvist M, Lund LH (2011) Association of candesartan vs losartan with all-cause mortality in patients with heart failure. J Amer Med Assoc 305:175–182
Farah AJ (2005) Atypicality of atypical antipsychotics. Clin Psychiatry 2005(7):268–274
Farde L (1996) The advantage of using positron emission tomography in drug research. Trends Neurol Sci 19:211–214
Farde L, Nordström AL (1993) PET examination of central D2 dopamine receptor occupancy in relation to extrapyramidal syndromes in patients being treated with neuroleptic drugs. Psychoph S 10:94–100
Farde L, Wiesel FA, Stone-Elander S, Halldin C, Nordström AL, Hall H, Sedvall G (1990) D2 dopamine receptors in neuroleptic-naive schizophrenic patients. A positron emission tomography study with [11 C]raclopride. Arch Gen Psychiat 47:213–219
Farde L, Nordström AL, Wiesel FA, Pauli S, Halldin C, Sedvall G (1992) Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine: relation to extrapyramidal side effects. Arch Gen Psychiat 49:538–544
Faron-Górecka A, Górecki A, Kuśmider M, Wasylewski Z, Dziedzicka-Wasylewska M (2008) The role of D1-D2 receptor hetero-dimerization in the mechanism of action of clozapine. Eur Neuropsychopharm 18:682–691
Fierens FLP, Vanderheyden PML, De Backer J-P, Vauquelin G (1999) Insurmountable angiotensin II AT1 receptor antagonists: the role of tight antagonist binding. Eur J Pharmacol 372:199–206
Fierens F, Vanderheyden PML, Roggeman C, Vande Gucht P, De Backer J-P, Vauquelin G (2002) Distinct binding properties of the AT1 receptor antagonist [3H]candesartan to intact cells and membrane preparations. Biochem Pharmacol 63:1273–1279
Fluxe K, Hökfelt T, Johansson O, Jonsson G, Lidbrink P, Ljungdahl A (1974) The origin of the dopamine nerve terminals in limbic and frontal cortex. Evidence for meso-cortico dopamine neurons. Brain Res 82:349–355
Frankle WG, Gil R, Hackett E, Mawlawi O, Zea-Ponce Y, Zhu Z, Kochan LD, Cangiano C, Slifstein M, Gorman JM, Laruelle M, Abi-Dargham A (2004) Occupancy of dopamine D2 receptors by the atypical antipsychotic drugs risperidone and olanzapine: theoretical implications. Psychopharmacology 175:473–480
Furchgott RF (1966) The use of β-haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of receptor-agonist complexes. Adv Drug Res 3:21–55
Furchgott RF, Bursztyn P (1967) Comparison of dissociation constants and of relative efficacy of selected agonists acting on parasympathetic receptors. Ann N Y Acad Sci 144:882–898
Gaddum JH, Hameed KA, Hathaway DE, Stephens FF (1955) Quantitative studies of antagonists for 5-hydroxytryptamine. Q J Exp Physiol 40:49–74
Ganz MB, Pachter JA, Barber DL (1990) Multiple receptors coupled to adenylate cyclase regulate Na-H exchange independent of cAMP. J Biol Chem 265:8989–8992
Gardner B, Hall DA, Strange PG (1996) Pharmacological analysis of dopamine stimulation of [35S]-GTPγS binding via human D2short and D2long dopamine receptors expressed in recombinant cells. Brit J Pharmacol 118:1544–1550
Garris PA, Ciolkowski EL, Pastore P, Wightman RM (1994) Efflux of dopamine from the synaptic cleft in the nucleus accumbens of the rat brain. J Neurosci 14:6084–6093
Gazi L, Nickolls SA, Strange PG (2003a) Functional coupling of the human dopamine D2 receptor with Gαi1, Gαi2, Gαi3 and Gαo G proteins: evidence for agonist regulation of G protein selectivity Brit. J Pharmacol 138:775–786
Gazi L, Wurch T, Lopéz-Giménez JF, Pauwels PJ, Strange PG (2003b) Pharmacological analysis of a dopamine D2Short: Gαo fusion protein expressed in Sf9 cells. FEBS Lett 545:155–160
Gefvert O, Bergstrom M, Langstrom B, Lundberg T, Lindstrom L, Yates R (1998) Time course of central nervous dopamine-D2 and 5-HT2 receptor blockade and plasma drug concentrations after discontinuation of quetiapine (Seroquel) in patients with schizophrenia. Psychopharmacology 135:119–126
Gejman PV, Ram A, Gelernter J, Friedman E, Cao Q, Pickar D, Blum K, Noble EP, Kranzler HR, O’malley S, Hamer DH, Whitsitt F, Rao P, Delisi LE, Virkkunen M, Linnoila M, Goldman D, Gershon ES (1994) No structural mutation in the dopamine-D2 receptor gene in alcoholism or schizophrenia—analysis using denaturing gradient gel-electrophoresis. J Amer Med Assoc 271:204–208
Gerfen CR (1992) The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci 15:285–320
Ghahremani MH, Cheng P, Lembo PM, Albert PR (1999) Distinct roles for Gαi2, Gαi3, and Gβγ in modulation of forskolin- or Gs-mediated cAMP accumulation and calcium mobilization by dopamine D2S receptors. J Biol Chem 274:9238–9245
Ghahremani MH, Forget C, Albert PR (2000) Distinct roles for Gαi2 and Gβγ in signaling to DNA synthesis and Gαi3 in cllular transformation by dopamine D2S receptor activation in BALB/c 3T3 cells. Mol Cell Biol 20:1497–1506
Gilliland SL, Alper RH (2000) Characterization of dopaminergic compounds at hD2short, hD4.2 and hD4.7 receptors in agonist-stimulated [35S]GTPγS binding assays. N-S Arch Pharmacol 361:498–504
Ginovart N, Farde L, Halldin C, Swahn C-G (1997) Effect of reserpine-induced depletion of synaptic dopamine on [11C]raclopride binding to D2-dopamine receptors in the monkey brain. Synapse 25:321–325
Ginovart N, Galineau L, Willeit M, Mizrahi R, Bloomfield PM, Seeman P, Houle S, Kapur S, Wilson AA (2006) Binding characteristics and sensitivity to endogenous dopamine of [11C]-(+)-PHNO, a new agonist radiotracer for imaging the high-affinity state of D2 receptors in vivo using positron emission tomography. J Neurochem 97:1089–1103
Graeser D, Neubig RR (1993) Compartmentation of receptors and guanine nucleotide-binding proteins in NG108-15 cells: lack of cross-talk in agonist binding among the alpha2-adrenergic, muscarinic and opiate receptors. Mol Pharmacol 43:434–443
Grießner M, Bröker P, Lehmann A, Ehrentreich-Förster E, Bier FF (2009) Detection of angiotensin II type 1 receptor ligands by a cell-based assay. Anal Bioanal Chem 395:1937–1940
Grunder G, Landvogt C, Vernaleken I, Buchholz HG, Ondracek J, Siessmeier T, Härtter S, Schreckenberger M, Stoeter P, Hiemke C, Rösch F, Wong DF, Bartenstein P (2006) The striatal and extrastriatal D2/D3 receptor-binding profile of clozapine in patients with schizophrenia. Neuropsychopharmacol 31:1027–1035
Guillin O, Abi-Dargham A, Laruelle M (2007) Neurobiology of dopamine in schizophrenia. Int Rev Neurobiol 78:1–39
Guo N, Guo W, Kralikova M, Jiang M, Schieren I, Narendran R, Slifstein M, Abi-Dargham A, Laruelle M, Javitch JA, Rayport S (2010) Impact of D2 receptor internalization on binding affinity of neuroimaging radiotracers. Neuropsychopharmacol 35:806–817
Hagberg G, Gervert O, Bergström M, Wieselgren IM, Lindström L, Wiesel FA, Langström B (1998) N-[11C]methylspiperone PET, in contrast to [11C]raclopride, fails to detect D2 receptor occupancy by an atypical neuroleptic. Psychiatry Res 82:147–160
Hagger C, Buckley P, Kenny JT, Friedman L, Ubogy D, Meltzer HY (1993) Improvement in cognitive functions and psychiatric symptoms in treatment-refractory schizophrenic patients receiving clozapine. Biol Psychiat 34:702–712
Hall DA, Strange PG (1997) Evidence that antipsychotic drugs are inverse agonists at D2 dopamine receptors. Brit J Pharmacol 121:731–736
Hamblin MW, Creese I (1983) Behavioral and radioligand binding evidence for irreversible dopamine receptor blockade by N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline. Life Sci 32:2247–2255
Hara M, Tozawa F, Itazaki K, Mihara S, Fujimoto M (1998) Endothelin ETB receptors show different binding profiles in intact cells and cell membrane preparations. Eur J Pharmacol 345:339–342
Hartvig P, Eckernäs SA, Lindström L, Ekblom B, Bondesson U, Lundqvist H, Halldin C, Någren K, Långström B (1986) Receptor binding of N-(methyl-11C) clozapine in the brain of rhesus monkey studied by positron emission tomography (PET). Psychopharmacology 89:248–252
Hauber W (1998) Involvement of basal ganglia transmitter systems in movement initiation. Prog Neurobiol 56:507–540
Heise CE, Sullivan S, Crowe PD (2007) Scintillation proximity assay as high-throughput method to identify slowly dissociating nonpeptide ligand binding to the GnRH receptor. J Biomol Screen 12:235–239
Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, Surmeier DJ (2000) D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLCβ1-IP3-calcineurin-signaling cascade. J Neurosci 20:8987–8995
Hersch SM, Ciliax BJ, Gutekunst CA, Flees HD, Heilman CJ, Yung KKL, Paul Bolam J, Ince E, Yi H, Levey AI (1995) Electron microscopic analysis of Dl and D2 dopamine receptor proteins in the dorsal striatum and their synaptic relationships with motor corticostriatal afferents. J Neurosci 75:5222–5237
Hippius H (1989) The history of clozapine. Psychopharmacology 99:S3–S5
Hippius H (1999) A historical perspective of clozapine. J Clin Psychiat 60:S22–S23
Hrabctová S, Nicholson C (2004) Contribution of dead-space microdomains to tortuosity of brain extracellular space. Neurochem Int 45:467–477
Huff RM (1996) Signal transduction pathways modulated by the D2 subfamily of dopamine receptors. Cell Signal 8:453–459
Ichise M, Meyer JH, Yonekura Y (2001) An introduction to PET and SPECT neuroreceptor quantification models. J Nuclear Med 42:755–763
Idanpaan-Heikkila J, Alhava E, Olkinuora M, Palva IP (1977) Agranulocytosis during treatment with clozapine. Eur J Clin Pharmacol 11:193–198
Itokawa M, Toru M, Ito K, Tsuga H, Kameyama K, Haga T, Arinami T, Hamaguchi H (1996) Sequestration of the short and long isoforms of dopamine D2 receptors expressed in Chinese hamster ovary cells. Mol Pharmacol 49:560–566
Jardemark K, Wadenberg ML, Grillner P, Svensson TH (2002) Dopamine D3 and D4 receptor antagonists in the treatment of schizophrenia. Curr Opin Invest Dr 3:101–105
Jiang M, Spicher K, Boulay G, Wang Y, Birnbaumer L (2001) Most central nervous system D2 dopamine receptors are coupled to their effectors by Go. Proc Natl Acad Sci USA 98:3577–3582
Jomphe C, Tiberi M, Trudeau L-E (2006) Expression of D2 receptor isoforms in cultured neurons reveals equipotent autoreceptor function. Neuropharmacology 50:595–605
Jones C, Kapur S, Remington G, Zipursky RB (2000) Transient dopamine D2 occupancy in low EPS-incidence drugs: PET evidence (abstract). Biol Psychiat 47(8 suppl):112
Jordan S, Johnson JL, Regardie K, Chen R, Koprivica V, Tadori Y, Kambayashi J, Kitagawa H, Kikuchi T (2007a) Dopamine D2 receptor partial agonists display differential or contrasting characteristics in membrane and cell-based assays of dopamine D2 receptor signaling. Prog Neuro-Psychoph 31:348–356
Jordan S, Regardie K, Johnson JL, Chen R, Kambayashi J, McQuade R, Kitagawa H, Tadori Y, Kikuchi T (2007b) In vitro functional characteristics of dopamine D2 receptor partial agonists in second and third messenger-based assays of cloned human dopamine D2Long receptor signalling. J Psychopharmacol 21:620–627
Kabbani N, Levenson R (2006) Antipsychotic-induced alterations in D2 dopamine receptor interacting proteins within the cortex. Mol Neurosci 17:299–301
Kabbani N, Hannan M, Levenson R (2005) Unravelling the dopamine receptor signalplex by DRIPs and DRAPS. Curr Proteom 2:209–224
Kalani MY, Vaidehi N, Hall SE, Trabanino RJ, Freddolino PL, Kalani MA, Floriano WB, Kam VW, Goddard WA 3rd (2004) The predicted 3D structure of the human D2 dopamine receptor and the binding site and binding affinities for agonists and antagonists. Proc Natl Acad Sci USA 101:3815–3820
Kane J, Honigfeld G, Singer J, Meltzer H (1988a) Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiat 45:789–796
Kane JM, Honigfeld G, Singer J, Meltzer H (1988b) Clozapine in treatment-resistant schizophrenics. Psychopharmacol Bull 24:62–67
Kanterman RY, Mahan LC, Briley EM, Monsma FJ Jr, Sibley DR, Axelrod J, Felder CC (1991) Transfected D2 dopamine receptors mediate the potentiation of arachidonic acid release in Chinese hamster ovary cells. Mol Pharmacol 39:364–369
Kapur S, Mamo D (2003) Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog Neuropsychopharmacol Biol Psychiatr 27:1081–1090
Kapur S, Seeman P (2000) Antipsychotic agents differ in how fast they come off the dopamine D2 receptors. Implications for atypical antipsychotic action. J Psychiatr Neurosci 25:161–166
Kapur S, Seeman P (2001) Does fast dissociation from the dopamine D2 receptor explain the action of atypical antipsychotics?: a new hypothesis. Am J Psychiat 158:360–369
Kapur S, Zipursky RB, Remington G (1999) Clinical and theoretical implications of 5-HT2 and D2 receptor occupancy of clozapine, risperidone, and olanzapine in schizophrenia. Am J Psychiat 156:286–293
Kapur S, Zipursky R, Jones C, Shammi CS, Remington G, Seeman P (2000) A positron emission tomography study of quetiapine in schizophrenia: a preliminary finding of an antipsychotic effect with only transiently high dopamine D2 receptor occupancy. Arch Gen Psychiat 57:553–559
Karbe H, Wienhard K, Haniacher K, Huber M, Lerholz K, Coenen HH, Stocklin G, Lovenich A, Heiss WD (1991) Positron emission tomography with [18F]methylspipcronc demonstrates D2 dopamine receptor binding differences of clozapine and haloperidol. J Neural Transm 86:163–173
Kawagoe KT, Garris PA, Wiedemann DJ, Wightman RM (1992) Regulation of transient dopamine concentration gradients in the microenvironment surrounding nerve terminals in the rat striatum. Neuroscience 51:55–64
Kebabian JW, Calne DB (1979) Multiple receptors for dopamine. Nature 277:93–96
Kegeles LS, Slifstein M, Frankle WG, Xu X, Hackett E, Bae SA, Gonzales R, Kim JH, Alvarez B, Gil R, Laruelle M, Abi-Dargham A (2008) Dose-occupancy study of striatal and extrastriatal dopamine D2 receptors by aripiprazole in schizophrenia with PET and [18 F]fallypride. Neuropsychopharmacol 33:3111–3125
Kenakin TP (1993) Stimulus–response mechanisms. In: Kenakin TP (ed) Pharmacologic analysis of drug–receptor interactions. Raven, New York, pp 39–68
Kenakin T (1996) The classification of seven transmembrane receptors in recombinant expression systems. Pharmacol Rev 48:413–463
Kenakin T (2003) Ligand-selective receptor conformations revisited: the promise and the problem. Trends Pharmacol Sci 24:346–354
Kenakin T, Jenkinson S, Watson C (2006) Determining the potency and molecular mechanism of action of insurmountable antagonists. J Pharmacol Exp Ther 319:710–723
Kessler RM, Whetsell WO, Ansari MS, Votaw JR, de Paulis T, Clanton JA, Schmidt DE, Mason NS, Manning RG (1993) Identification of extrastriatal dopamine D2 receptors in post mortem human brain with [123I]epidepride. Brain Res 609:237–243
Kessler RM, Ansari MS, Riccardi P, Li R, Jayathilake K, Dawant B et al (2006) Occupancy of striatal and extrastriatal dopamine D2 receptors by clozapine and quetiapine. Neuropsychopharmacol 3:1991–2001
Kim KM, Valenzano KJ, Robinson SR, Yao WD, Barak LS, Caron MG (2001) Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled kinases and beta-arrestins. J Biol Chem 276:37409–37414
Kinon BJ, Lieberman JA (1996) Mechanisms of action of atypical antipsychotic drugs: a critical analysis. Psychopharmacology 124:2–34
Kjeldsen SE, Stålhammar J, Hasvold P, Bodegard J, Olsson U, Russell D (2010) Effects of losartan vs candesartan in reducing cardiovascular events in the primary treatment of hypertension. J Hum Hypertens 24:263–273
Koepp MJ, Gunn RN, Lawrence AD, Cunningham VJ, Dagher A, Jones T, Brooks DJ, Bench CJ, Grasby PM (1998) Evidence for striatal dopamine release during a video game. Nature 393:266–268
Korner M, Gilon C, Schramm M (1982) Locking of hormone in the beta-adrenergic receptor by attack on a sulfhydryl in an associated component. J Biol Chem 257(7):3389–3396
Kortekaas R, Maguire RP, Cremers TI, Dijkstra D, van Waarde A, Leenders KL (2004) In vivo binding behavior of dopamine receptor agonist (+)-PD 128907 and implications for the “ceiling effect” in endogenous competition studies with [11C]raclopride—a positron emission tomography study in Macaca mulatta. J Cerebr Blood F Met 24:531–535
Kramer MS, Last B, Getson A, Reines SA (1997) The effects of a selective D4 dopamine receptor antagonist (L-745,870) in acutely psychotic inpatients with schizophrenia. Arch Gen Psychiat 4:567–572
Langlois X, Megens A, Lavreysen H, Wouters R, Peeters L, te Riele P, Hendrickx H, Mahieu M, de Bruyn M, Macdonald G (2010) Pharmacology of JNJ-37822681, a specific and fast-dissociating D2 antagonist for the treatment of schizophrenia. Poster, presented at the 23nd Congress of European College of Neuropsychopharmacology; Amsterdam, 9/2010. Eur Neuropsychopharmacol 20:S502
Laruelle M (2000) Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cerebr Blood F Met 20:423–451
Laruelle M (2003) Dopamine transmission in the schizophrenic brain. In: Weinberger DR, Hirsch S (eds) Schizophrenia, 2nd edn. Blackwell Publishing, Oxford, pp 365–387
Laruelle M, D’Souza CD, Baldwin RM, Abi-Dargham A, Kanes SJ, Fingado CL, Seibyl JP, Zoghbi SS, Bowers MB, Jatlow P, Charney DS, Innis RB (1997a) Imaging D2 receptor occupancy by endogenous dopamine in humans. Neuropsychopharmacol 17:162–174
Laruelle M, Iyer RN, Al-Tikriti MS, Zea-Ponce Y, Malison R, Zoghbi SS, Baldwin RM, Kung HF, Charney DS, Hoffer PB, Innis RB, Bradberry CW (1997b) Microdialysis and SPECT measurements of amphetamine-induced dopamine release in nonhuman primates. Synapse 25:1–14
Lawler CP, Prioleau C, Lewis MM, Mak C, Jiang D, Schetz JA, Gonzalez AM, Sibley DR, Mailman RB (1999) Interactions of the novel antipsychotic aripiprazole (OPC-14597) with dopamine and serotonin receptor subtypes. Neuropsychopharmacol 20:612–627
Lee SP, So CH, Rashid AJ, Varghese G, Cheng R, Lanca AJ, O’Dowd BF, George SR (2004) Dopamine D1 and D2 receptor co-activation generates a novel phospholipase C-mediated calcium signal. J Biol Chem 279:35671–35678
Leff P, Martin GR (1986) Peripheral 5-HT2-like receptors. Can they be classified with the available antagonists? Brit J Pharmacol 88:585–593
Leysen JE, Gommeren WJ (1984) The dissociation of unlabelled dopamine antagonists and agonists from the dopamine D2-receptor. Implication of an original filter method. J Receptor Res 4:817–845
Lidow MS, Goldman-Rakic PS (1994) A common action of clozapine, haloperidol, and remoxipride on D1- and D2- dopaminergic receptors in the primate cerebral cortex. Proc Natl Acad Sci USA 91:4353–4356
Lidow MS, Goldman-Rakic PS (1997) Differential regulation of D2 and D4 dopamine receptor mRNAs in the primate cerebral cortex vs neostriatum: effects of chronic treatment with typical and atypical antipsychotic drugs. J Pharmacol Exp Ther 283:939–946
Lieberman JA, Kane JM, Alvir J (1987) Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology 91:415–433
Lindgren N, Usiello A, Goiny M, Haycock J, Erbs E, Greengard P, Hokfelt T, Borrelli E, Fisone G (2003) Distinct roles of dopamine D2L and D2S receptor isoforms in the regulation of protein phosphorylation at presynaptic and postsynaptic sites. Proc Natl Acad Sci USA 100:4305–4309
Lipworth BJ (2002) Antagonism of long-acting beta2-adrenoceptor agonism. Brit J Clin Pharmacol 54:231–245
Liu YF, Civelli O, Grandy DK, Albert PR (1992) Differential sensitivity of the short and long human dopamine D2 receptor subtypes to protein kinase C. J Neurochem 59:2311–2317
Luttrell LM, Lefkowitz RJ (2002) The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci 115:455–465
Mager DE, Wyska E, Jusko WJ (2003) Diversity of mechanism-based pharmacodynamic models. Drug Metab Dispos 31:510–519
Malany S, Hernandez LM, Smith WF, Crowe PD, Hoare SRJ (2009) Analytical method for simultaneously measuring ex vivo drug receptor occupancy and dissociation rate: application to (R)-dimethindene occupancy of central histamine H1 receptors. J Recept Sig Transd Res 29:84–93
Malmberg A, Mohell N (1995) Characterization of [3H]quinpirole binding to human dopamine D2A and D3 receptors: effects of ions and guanine nucleotides. J Pharmacol Exp Ther 274:790–797
Malmberg A, Mohell N, Backlund Höök B, Johansson AM, Hacksell U, Nordvall G (1998) Interactions of ligands with active and inactive conformations of the dopamine D2 receptor. Eur J Pharmacol 346:299–307
Marchese A, Paing MM, Temple BR, Trejo J (2008) G protein coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol 48:601–629
Masri B, Salahpour A, Didriksen M, Ghisi V, Beaulieu JM, Gainetdinov RR, Caron MG (2008) Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc Natl Acad Sci USA 105:13656–13661
Meibohm B, Derendorf H (1997) Basic concepts of pharmacokinetic/pharmacodynamic (PK/PD) modelling. Int J Clin Pharm Th 35:401–413
Meller E, Bohmaker K, Namba Y, Friedhoff AJ, Goldstein M (1987) Relationship between receptor occupancy and response at striatal dopamine autoreceptors. Mol Pharmacol 31:592–598
Meller E, Enz A, Goldstein M (1988) Absence of receptor reserve at striatal dopamine receptors regulating cholinergic neuronal activity. Eur J Pharmacol 155:151–154
Meltzer HY (1999) The role of serotonin in antipsychotic drug action. Neuropsychopharmacol 21:106S–115S
Meltzer HY (2002) Mechanism of action of atypical antipsychotic drugs. In: Davis KL, Charney D, Coyle JT, Nemeroff C (eds) Neuropsychopharmacology: the fifth generation of progress. Lippincott Williams & Wilkins, Philadelphia, pp 819–831
Miyake N, Thompson J, Skinbjerg M, Abi-Dargham A (2010) Presynaptic dopamine in schizophrenia. CNS Neurosci Ther 17:104–109
Miyamoto S, Duncan GE, Marx CE, Lieberman JA (2005) Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiat 10:79–104
Mogenson GJ, Jones DL, Yim CY (1980) From motivation to action: functional interface between the limbic system and the motor system. Prog Neurobiol 14:69–97
Mogenson GJ, Yang CR, Yim CY (1988) Influence of dopamine on limbic inputs to the nucleus accumbens. Ann N Y Acad Sci 537:86–100
Moreland RB, Nakane M, Donnelly-Roberts DL, Miller LN, Chang R, Uchic ME, Terranova MA, Gubbins EJ, Helfrich RJ, Namovic MT, El-Kouhen OF, Masters JN, Brioni JD (2004) Comparative pharmacology of human dopamine D2-like receptor stable cell lines coupled to calcium flux through Gaqo5. Biochem Pharmacol 68:761–772
Morsing P, Adler G, Brandt-Eliasson U, Karp L, Ohlson K, Renberg L, Sjöquist PO, Abrahamsson T (1999) Mechanistic differences of various AT1-receptor blockers in isolated vessels of different origin. Hypertension 33:1406–1413
Motulsky HJ, Mahan LC (1984) The kinetics of competitive radioligand binding predicted by the law of mass action. Mol Pharmacol 25:1–9
Mukherjee J, Christian BT, Narayanan TK, Shi S, Mantil J (2001) Evaluation of dopamine D2 receptor occupancy by clozapine, risperidone, and haloperidol in vivo in the rodent and nonhuman primate brain using [18F]-fallypride. Neuropsychopharmacol 25:476–488
Narendran R, Hwang DR, Slifstein M, Talbot PS, Erritzoe D, Huang Y, Cooper TB, Martinez D, Kegeles LS, Abi-Dargham A, Laruelle M (2004) In vivo vulnerability to competition by endogenous dopamine: comparison of the D2 receptor agonist radiotracer (-)-N-[11 C]propyl-norapomorphine ([11 C]NPA) with the D2 receptor antagonist radiotracer [11 C]-raclopride. Synapse 52:188–208
Narendran R, Hwang DR, Slifstein M, Hwang Y, Huang Y, Ekelund J, Guillin O, Scher E, Martinez D, Laruelle M (2005) Measurement of the proportion of D2 receptors configured in state of high affinity for agonists in vivo: a positron emission tomography study using [11 C]N-propyl-norapomorphine and [11 C]raclopride in baboons. J Pharmacol Exp Ther 315:80–90
Neubig RR, Gantzos RD, Brasier RS (1985) Agonist and antagonist binding to alpha2-adrenergic receptors in purified membranes from human platelets. Implications of receptor-inhibitory nucleotide-binding protein stoichiometry. Mol Pharmacol 28:475–486
Neubig RR, Gantzos RD, Thomsen WJ (1988) Mechanism of agonist and antagonist binding to alpha2 adrenergic receptors: evidence for a precoupled receptor.guanine nucleotide protein complex. Biochemistry 27:2374–2384
Neve KA, Kozlowski MR, Rosserg MP (1992) Dopamine D2 receptor stimulation of Na+/H+ exchange assessed by quantification of extracellular acidification. J Biol Chem 267:25748–25753
Ng GY, O’Dowd BF, Lee SP, Chung H, Brann MR, Seeman P, George SR (1996) Dopamine D2 receptor dimers and receptor-blocking peptides. Biochem Biophys Res Commun 227:200–204
Nickolls SA, Strange PG (2004) The influence of G protein subtype on agonist action at D2 dopamine receptors. Neuropharmacology 47:860–872
Nilsson CL, Eriksson E (1993) Haloperidol increases prolactin release and cyclic AMP formation in vitro: inverse agonism at dopamine D2 receptors? J Neural Transm 92:213–220
Nordström A-L, Farde L, Halldin C (1992) Time course of D2-dopamine receptor occupancy examined by PET after single oral doses of haloperidol. Psychopharmacology 106:433–438
Ojima M, Inada Y, Shibouta Y, Wada T, Sanada T, Kubo K, Nishikawa K (1997) Candesartan (CV-11974) dissociates slowly from the angiotensin AT1 receptor. Eur J Pharmacol 319:137–146
Oldham WM, Hamm HE (2008) Heterotrimeric G-protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol 9:60–71
Olsen CK, Brennum LT, Kreilgaard M (2008) Using pharmacokinetic-pharmacodynamic modelling as a tool for prediction of therapeutic effective plasma levels of antipsychotics. Eur J Pharmacol 584:318–327
Olson T (2005) Does clozapine work by blocking spikes and sparing bursts? Med Hypotheses 65:68–78
Olsson H, Farde L (2001) Potentials and pitfalls using high affinity radioligands in PET and SPET determinations on regional drug induced D2 receptor occupancy—a simulation study based on experimental data. NeuroImage 14:936–945
Ostrom RS (2002) New determinants of receptor-effector coupling: trafficking and compartmentation in membrane microdomains. Mol Pharmacol 61:473–476
Ostrom RS, Post SR, Insel PA (2000) Stoichiometry and compartmentation in G protein-coupled receptor signaling: implications for therapeutic interventions involving Gs. J Pharmacol Exp Ther 294:407–412
Packeu A, De Backer J-P, Van Liefde I, Vanderheyden P, Vauquelin G (2008) Antagonist–dopamine D2L-receptor interactions in intact cells. Biochem Pharmacol 75:2192–2203
Packeu A, Béghin T, De Backer J-P, Vauquelin G (2010a) Antagonist-D2S-dopamine receptor interactions in intact recombinant Chinese hamster ovary cells. Fund Clin Pharmacol 24:293–303, 513 (title correction)
Packeu A, De Backer J-P, Vauquelin G (2010b) Non-competitive interaction between raclopride and spiperone on human D2L-receptors in intact recombinant Chinese hamster ovary cells. Fund Clin Pharmacol 24:283–291
Packeu A, Wennerberg M, Ballendran A, Vauquelin G (2010c) Estimation of the dissociation rate of unlabelled ligand-receptor complexes by a “two-step” competition binding approach. Brit J Pharmacol 161:1311–1328
Paspalas CD, Rakic P, Goldman-Rakic PS (2006) Internalization of D2 dopamine receptors is clathrin-dependent and select to dendro-axonic appositions in primate prefrontal cortex. Eur J Neurosci 24:1395–1403
Pauwels PJ, Tardif S, Wurch T, Colpaert FC (2001a) Real-time analysis of dopamine: antagonist interactions at recombinant human D2long receptor upon modulation of its activation state. Brit J Pharmacol 134:88–97
Pauwels PJ, Finana F, Tardif S, Wurch T, Colpaert FC (2001b) Dynamic dopamine-antagonist interactions at recombinant human dopamine D2short receptor: dopamine-bound versus antagonist-bound receptor states. J Pharmacol Exp Ther 297:133–140
Pettersson F, Pontén H, Waters N, Waters S, Sonesson C (2010) Synthesis and evaluation of a set of 4-phenylpiperidines and 4-phenylpiperazines as D2 receptor ligands and the discovery of the dopaminergic stabilizer 4-[3-(methylsulfonyl)phenyl]-1-propylpiperidine (huntexil, pridopidine, ACR16). J Med Chem 53:2510–2520
Petty RG (1999) Prolactin and antipsychotic medications: mechanism of action. Schizophr Res 35:S67–S73
Pierce KL, Lefkowitz RJ (2001) Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci 2:727–733
Pike LJ (2003) Lipid rafts: bringing order to chaos. J Lipid Res 44:655–667
Pilowsky LS, Mulligan RS, Acton PD, Ell PJ, Costa DC, Kerwin RW (1997) Limbic selectivity of clozapine. Lancet 350:490–491
Ploeger BA, van der Graaf PH, Danhof M (2009) Incorporating receptor theory in mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling. Drug Metab Pharmacok 24:3–15
Raleigh F (1996) Use of novel antipsychotic drugs. Pharmacotherapy 16:160S–165S
Ransnäs LA, Insel PA (1988) Quantitation of the guanine nucleotide binding regulatory protein Gs in S49 cell membranes using antipeptide antibodies to αs. J Biol Chem 263:9482–9485
Rayport S, Sulzer D (1995) Visualization of antipsychotic drug binding to living mesolimbic neurons reveals D2 receptor, acidotropic, and lipophilic components. J Neurochem 65:691–703
Remington G, Kapur S (2000) Atypical antipsychotics: are some more atypical than others? Psychopharmacology 148:3–15
Rhodes DG, Sarmiento JG, Herbette LG (1985) Kinetics of binding of membrane-active drugs to receptor sites. Diffusion-limited rates for a membrane bilayer approach of 1,4-dihydropyridine calcium channel antagonists to their active site. Mol Pharmacol 27:612–623
Rice ME, Cragg SJ (2008) Dopamine spillover after quantal release: rethinking dopamine transmission in the nigrostriatal pathway. Brain Res Rev 58:303–313
Robert V, Pinton P, Tosello V, Rizzuto R, Pozzan T (2000) Recombinant aequorin as tool for monitoring calcium concentration in subcellular compartments. Meth Enzymol 327:440–456
Roberts DJ, Strange PG (2005) Mechanisms of inverse agonist action at D2 dopamine receptors. Brit J Pharmacol 145:34–42
Roberts DJ, Lin H, Strange PG (2004a) Investigation of the mechanism of agonist and inverse agonist action at D2 dopamine receptors. Biochem Pharmacol 67:1657–1665
Roberts DJ, Lin H, Strange PG (2004b) Mechanisms of agonist action at D2 dopamine receptors. Mol Pharmacol 66:1573–1579
Salimpoor VN, Benovoy M, Larcher K, Dagher Aand Zatorre RJ (2010) Anatomically distinct dopamine release during anticipation and experience of peak emotion to music. Nat Neurosci 14:257–262
Samama P, Cotecchia S, Costa Tand Lefkowitz RJ (1993) A mutation-induced activated state of the beta2-adrenergic receptor. Extending the ternary complex model. J Biol Chem 268:4625–4636
Sanger DJ (2004) The search for novel antipsychotics: pharmacological and molecular targets. Expt Opin Ther Tar 8:631–641
Schmidt M, Jansens L, Kent J, Anghelecu I, Husken G, Sinha V, Mannaert E (2010) Efficay and safety of JNJ-37822681, a fast-dissociating D2 antagonist in the treatment of schizophrenia. Eur Neuropsychopharmacol 20:S484–S485
Schotte A, Janssen PF, Gommeren W, Luyten WH, Van Gompel P, Lesage AS, De Loore K, Leysen JE (1996) Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology 124:57–73
Schultz W (2007) Multiple dopamine functions at different time courses. Annu Rev Neurosci 30:259–288
Seeman P (1992) Dopamine receptor sequences. Therapeutic levels of neuroleptics occupy D2 receptors, clozapine occupies D4. Neuropsychopharmacol 7:261–284
Seeman P (1995) Therapeutic receptor-blocking concentrations of neuroleptics. Int Clin Psychopharmacol 10(Suppl 3):5–13
Seeman P (2002) Atypical antipsychotics: mechanism of action. Can J Psychiat 47:27–38
Seeman P (2005) An update of fast-off dopamine D2 atypical antipsychotics. Am J Psychiat 162:1984–1985
Seeman P (2008) Dopamine D2High receptors on intact cells. Synapse 62:314–318
Seeman P (2009) Dopamine D High2 receptors measured ex vivo are elevated in amphetamine-sensitized animals. Synapse 63:186–192
Seeman P (2011) All roads to schizophrenia lead to dopamine supersensitivity and elevated dopamine D High2 receptors. CNS Neurosci Ther 17:118–132
Seeman P, Kapur S (1997) Clozapine occupies high levels of dopamine D2 receptors. Life Sci 160:PL 207–PL 216
Seeman P, Tallerico T (1998) Antipsychotic drugs which elicit little or no parkinsonism bind more loosely than dopamine to brain D2 receptors, yet occupy high levels of these receptors. Mol Psychiat 3:123–134
Seeman P, Tallerico T (1999) Rapid release of antipsychotic drugs from dopamine D2 receptors: an explanation for low receptor occupancy and early clinical relapse upon withdrawal of clozapine or quetiapine. Am J Psychiat 156:876–884
Seeman P, Van Tol HH (1995) Deriving the therapeutic concentrations for clozapine and haloperidol: the apparent dissociation constant of a neuroleptic at the dopamine D2 or D4 receptor varies with the affinity of the competing radioligand. Eur J Pharmacol 291:59–66
Seeman P, Chau-Wong M, Tedesco J, Wong K (1975) Brain receptors for antipsychotic drugs and dopamine: direct binding assays. Proc Nat Acad Sci USA 72:4376–4380
Seeman P, Lee T, Chau-Wong M, Wong K (1976) Antipsychotic drug doses and neurolepticrdopamine receptors. Nature 261:717–719
Seeman P, Guan HC, Civelli O, Van Tol HHM, Sunahara RK, Niznik HB (1992) The cloned dopamine D2 receptor reveals different densities for dopamine receptor antagonist ligands. Implications for human brain positron emission tomography. Eur J Pharmacol 227:139–146
Seeman P, Corbett R, Nam D, Van Tol HHM (1996) Dopamine and serotonin receptors: amino acid sequences, and clinicalroleinneu-roleptic parkinsonism. Jpn J Pharmacol 71:187–204
Seeman P, Corbett R, Van Tol HHM (1997) Atypical neuroleptics have low affinity for dopamine D2 receptors or are selective for D4 receptors. Neuropsychopharmacol 16:93–110, discussion 111-135
Seeman P, Nam D, Ulpian C, Liu IS, Tallerico T (2000) New dopamine receptor, D2(Longer), with unique TG splice site, in human brain. Brain Res Mol Brain Res 76:132–141
Seeman P, Tallerico T, Ko F, Tenn C, Kapur S (2002) Amphetamine- sensitized animals show a marked increase in dopamine D high2 receptors occupied by endogenous dopamine, even in the absence of acute challenges. Synapse 46:235–239
Seeman P, Tallerico T, Ko F (2003) Dopamine displaces [3H]domperidone from high-affinity sites of the dopamine D2 receptor, but not [3H]raclopride or [3H]spiperone in isotonic medium: implications for human positron emission tomography. Synapse 49:209–215
Seeman P, Schwarz J, Chen JF, Szechtman H, Perreault M, McKnight GS, Roder JC, Quirion R, Boksa P, Srivastava LK, Yanai K, Weinshenker D, Sumiyoshi T (2006) Psychosis pathways converge via D high2 dopamine receptors. Synapse 60:319–346
Seneca N, Finnema SJ, Farde L, Gulyás B, Wikström HV, Halldin C, Innis RB (2006) Effect of amphetamine on dopamine D2 receptor binding in nonhuman primate brain: a comparison of the agonist radioligand [11C]MNPA and antagonist [11C]raclopride. Synapse 59:260–269
Sesack SR, Aoki C, Pickel VM (1994) Ultrastructural localization of D2 receptor-like immunoreactivity in midbrain dopamine neurons and their striatal targets. J Neurosci 14:88–106
Sesack SR, Carr DB, Omelchenko N, Pinto A (2003) Anatomical substrates for glutamate-dopamine interactions: evidence for specificity of connections and extrasynaptic actions. Ann N Y Acad Sci 1003:36–52
Severne Y, Coppens D, Bottari S, Riviere M, Kram R, Vauquelin G (1984) The influence of the beta-adrenergic receptor concentration on their functional coupling to the adenylate cyclase system. Proc Natl Acad Sci USA 81:4637–4641
Severne Y, IJzerman A, Nerme V, Zimmerman H, Vauquelin G (1986) Shallow agonist competition binding curves for beta-adrenergic receptors: the role for tight agonist binding. Mol Pharmacol 31:69–73
Sibley DR (1995) Molecular biology of dopamine receptors. In: Ariano MA, Surmeier DJ (eds) Molecular and cellular mechanisms of neostriatal function. Landes RG, Austin, pp 255–272
Sibley DR, Leff SE, Creese I (1982) Interactions of novel dopaminergic ligands with D-1 and D-2 dopamine receptors. Life Sci 31:637–645
Sibley DR, Mahan LC, Creese I (1983) Dopamine receptor binding on intact cells. Absence of a high-affinity agonist-receptor binding state. Mol Pharmacol 23:295–302
Sibley D, Hazelwood L, Roof R, Free RB, Han Y, Javitch J (2011) Membrane lipid rafts are required for D2 dopamine receptor signaling. Poster, presented at the19th Congress of the European Congress of Psychiatry; Prague, 3/2011. Eur Psychiat 26(suppl 1):910
Skinbjerg M, Liow J-S, Seneca N, Hong J, Lu S, Thorsell A, Heilig M, Pike VW, Halldin C, Sibley DR, Innis RB (2010) D2 dopamine receptor internalization prolongs the decrease of radioligand binding after amphetamine: a PET study in a receptor internalization-deficient mouse model. NeuroImage 50:1402–1407
Skinbjerg M, Namkung Y, Halldin C, Innis RB, Sibley DR (2009) Pharmacological characterization of 2-methoxy-N-propylnorapomorphine’s interactions with D2 and D3 dopamine receptors. Synapse 63:462–475
Smith C, Rahman T, Toohey N, Mazurkiewicz J, Herrick-Davis K, Teitler M (2006) Risperidone irreversibly binds to and inactivates the h5-HT7 serotonin receptor. Mol Pharmacol 70:1264–1270
Spivak CE, Oz M, Beglan CL, Shrager RI (2006) Diffusion delays and unstirred layer effects at monolayer cultures of Chinese hamster ovary cells: radioligand binding, confocal microscopy, and mathematical simulations. Cell Biochem Biophys 45:43–58
Sternweis PC, Robishaw JD (1984) Isolation of two proteins with high affinity for guanine nucleotides from membranes of bovine brain. J Biol Chem 259:13806–13813
Suhara T, Okauchi T, Sudo Y, Takano A, Kawabe K, Maeda J, Kapur S (2002) Clozapine can induce high dopamine D2 receptor occupancy in vivo. Psychopharmacology 160:107–112
Surmeier DJ, Song WJ, Yan Z (1996) Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci 16:6579–6591
Swinney DC (2004) Biochemical mechanisms of drug action: what does it take for success? Nat Rev Drug Discov 3:801–808
Swinney DC (2006a) Biochemical mechanisms of new molecular entities (NMEs) approved by United States FDA during 2001-2004: mechanisms leading to optimal efficacy and safety. Curr Top Med Chem 6:461–478
Swinney DC (2006b) Can binding kinetics translate to a clinically differentiated drug? From theory to practice. Lett Drug Des Discov 3:569–574
Swinney DC (2008) Applications of binding kinetics to drug discovery: translation of binding mechanisms to clinically differentiated therapeutic responses. Int J Pharm Med 22:23–34
Swinney DC (2009) The role of binding kinetics in therapeutically useful drug action. Curr Opin Drug Disc Dev 12:31–39
Szczuka A, Packeu A, Wennerberg M, Vauquelin G (2009) Molecular mechanism of the persistent bronchodilatory effect of the partial beta2-adrenoceptor agonist salmeterol. Brit J Pharmacol 158:183–194
Tadori Y, Forbes RA, McQuade RD, Kikuchi T (2009) Receptor reserve-dependent properties of antipsychotics at human dopamine D2 receptors. Eur J Pharmacol 607:35–40
Talvik M, Nordström AL, Nyberg S, Olsson H, Halldin C, Farde L (2001) No support for regional selectivity in clozapine-treated patients: a PET study with [11 C]raclopride and [11 C]FLB 457. Am J Psychiat 58:926–930
Tauscher J, Jones C, Remington G, Zipursky RB, Kapur S (2002) Significant dissociation of brain and plasma kinetics with antipsychotics. Mol Psychiatr 7:317–321
Tresadern G, Bartolome JM, Macdonald GJ, Langlois X (2011) Molecular properties affecting fast dissociation from the D2 receptor. Bioorg Med Chem 19:2231–2241
Tummino PJ, Copeland RA (2008) Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry 47:5481–5492
Turrone P, Kapur S, Seeman MV, Flint AJ (2002) Elevation of prolactin levels by atypical antipsychotics. Am J Psychiat 159:133–135
Turu G, Várnai P, Gyombolai P, Szidonya L, Offertaler L, Bagdy G, Kunos G, Hunyady L (2009) Paracrine transactivation of the CB1 cannabinoid receptor by AT1 angiotensin and other Gq/11 protein-coupled receptors. J Biol Chem 284:16914–16921
Usiello A, Baik JH, Rouge-Pont F, Picetti R, Dierich A, LeMeur M, Piazza PV, Borrelli E (2000) Distinct functions of the two isoforms of dopamine D2 receptors. Nature 408:199–203
Vallar L, Muca C, Magni M, Albert P, Bunzow J, Meldolesi J, Civelli O (1990) Differential coupling of dopaminergic D2 receptors expressed in different cell types. Stimulation of phosphatidylinositol 4,5-bisphosphate hydrolysis in Ltk- fibroblasts, hyperpolarization, and cytosolic-free Ca2+ concentration decrease in GH4C1 cells. J Biol Chem 265:10320–10326
van Os J, Kapur S (2009) Schizophrenia. Lancet 374:635–645
Van Rossum JM (1967) The significance of dopamine-receptor blockade for the action of neuroleptic drugs. In: Brill H, Cole JO, Deniker P, Hippius H, Bradley PB (eds) Neuro-psycho-pharmacology, Proceedings of the 5th Int. Congress of the Collegium Internationale Neuro-Psycho-pharmacologicum, March 1966. Excerpta Medica Foundation, Amsterdam, pp 321–329
Vanhauwe JF, Fraeyman N, Francken BJ, Luyten WH, Leysen JE (1999) Comparison of the ligand binding and signaling properties of human dopamine D2 and D3 receptors in Chinese hamster ovary cells. J Pharmacol Exp Ther 290:908–916
Vauquelin G (2010) Rebinding: or why drugs may act longer in vivo than expected from their in vitro target residence time. Expert Opin Drug Discov 5:927–941
Vauquelin G, Charlton S (2010) Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Brit J Pharmacol 161:488–508
Vauquelin G, Maguire ME (1980) Inactivation of beta-adrenergic receptors by N-ethylmaleimide in S49 lymphoma cells: receptor heterogeneity in relation to coupling to adenylate cyclase. Mol Pharmacol 18:362–369
Vauquelin G, Packeu A (2009) Ligands, their receptors and … plasma membranes. Mol Cell Endocrinol 311:1–10
Vauquelin G, Szczuka A (2007) Kinetic versus allosteric mechanisms to explain insurmountable antagonism and delayed ligand dissociation. Neurochem Int 51:254–260
Vauquelin G, Van Liefde I (2006) Slow antagonist dissociation and long-lasting in vivo receptor protection. Trends Pharmacol Sci 27:356–359
Vauquelin G, von Mentzer P (2007) In: Vauquelin G, von Mentzer B (eds) G-protein coupled receptors: Molecular pharmacology. John Wiley and Sons Ltd, Chichester, pp 53–64, 163-175 and 185-191
Vauquelin G, Severne Y, Convents A, Nerme V, Abrahamsson T (1988) Agonist-mediated activation of adrenergic receptors. In: Melchiorre C, Giannella M (eds) Recent advances in receptor chemistry. Elsevier Science Publishers, Amsterdam, pp 43–61
Vauquelin G, Morsing P, Fierens FLP, De Backer JP, Vanderheyden PML (2001) A two-state receptor model for the interaction between angiotensin II AT1 receptors and their non-peptide antagonists. Biochem Pharmacol 61:277–284
Vauquelin G, Van Liefde I, Vanderheyden P (2002a) Models and methods for studying insurmountable antagonism. Trends Pharmacol Sci 23:514–518
Vauquelin G, Van Liefde I, Birzbier BB, Vanderheyden PML (2002b) New insights in insurmountable antagonism. Fund Clin Pharmacol 16:263–272
Vauquelin G, Fierens F, Van Liefde I (2006) Long-lasting AT1 receptor binding and protection by candesartan: comparison to other biphenyl-tetrazole sartans. J Hypertens 24:S23–S30
Verheijen I, Tourlousse D, Vanderheyden PML, De Backer JP, Vauquelin G (2004) Effect of saponin and filipin on antagonist binding to AT1 receptors in intact cells. Biochem Pharmacol 67:601–606
Wang Y, Xu R, Sasaoka T, Tonegawa S, Kung MP, Sankoorikal EB (2000) Dopamine D2long receptor-deficient mice display alterations in striatum-dependent functions. J Neurosci 20:8305–8314
Weinberger DR, Laruelle M (2001) Neurochemical and neuropharmacological imaging in schizophrenia. In: Davis KL, Charney D, Coyle JT, Nemeroff C (eds) Neuropsychopharmacology: the fifth generation of progress. Lippincott Williams & Wilkins, Philadelphia, pp 833–855
Weiner DM, Burstein ES, Nash N, Croston GE, Currier EA, Vanover KE, Harvey SC, Donohue E, Hansen HC, Andersson CM, Spalding TA, Gibson DF, Krebs-Thomson K, Powell SB, Geyer MA, Hacksell U, Brann MR (2001) 5-Hydroxytryptamine2A receptor inverse agonists as antipsychotics. J Pharmacol Exp Ther 299:268–276
Wilson JM, Sanyal S, Van Tol HHM (1998) Dopamine D2 and D4 receptor ligands: relation to antipsychotic action. Eur J Pharmacol 351:273–286
Wilson J, Lin H, Fu D, Javitch JA, Strange PG (2001) Mechanisms of inverse agonism of antipsychotic drugs at the D2 dopamine receptor: use of a mutant D2 dopamine receptor that adopts the activated conformation. J Neurochem 77:493–504
Xiberas X, Martinot JL, Mallet L, Artiges E, Loc’H C, Mazière B, Paillère-Martinot ML (2001) Extrastriatal and striatal D2 dopamine receptor blockade with haloperidol or new antipsychotic drugs in patients with schizophrenia. Brit J Psychiat 179:503–508
Yung KKL, Bolam JP, Smith AD, Hersch SM, Ciliax BJ, Levey AI (1995) Immunocytochemical localization of D1 and D2 dopamine receptors in the basal ganglia of the rat: light and electron microscopy. Neuroscience 65:709–730
Zhang R, Monsma F (2009) The importance of drug-target residence time. Curr Opin Drug Discov Devel 12:488–496
Zhang R, Monsma F (2010) Binding kinetics and mechanism of action: toward the discovery and development of better and best in class drugs. Expert Opin Drug Discov 5:1023–1029
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vauquelin, G., Bostoen, S., Vanderheyden, P. et al. Clozapine, atypical antipsychotics, and the benefits of fast-off D2 dopamine receptor antagonism. Naunyn-Schmiedeberg's Arch Pharmacol 385, 337–372 (2012). https://doi.org/10.1007/s00210-012-0734-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-012-0734-2