
Abstract
Activation of 5-HT4 receptors in failing ventricles elicits a cAMP-dependent positive inotropic response which is mainly limited by the cGMP-inhibitable phosphodiesterase (PDE) 3. However, PDE4 plays an additional role which is demasked by PDE3 inhibition. The objective of this study was to evaluate the effect of cGMP generated by particulate and soluble guanylyl cyclase (GC) on the 5-HT4-mediated inotropic response. Extensive myocardial infarctions were induced by coronary artery ligation in Wistar rats, exhibiting heart failure 6 weeks after surgery. Contractility was measured in left ventricular preparations. Cyclic GMP was measured by EIA. In ventricular preparations, ANP or BNP displayed no impact on 5-HT4-mediated inotropic response. However, CNP increased the 5-HT4-mediated inotropic response as well as the β1-adrenoceptor (β1-AR)-mediated response to a similar extent as PDE3 inhibition by cilostamide. Pretreatment with cilostamide eliminated the effect of CNP. Inhibition of nitric oxide (NO) synthase and soluble GC by l-NAME and ODQ, respectively, attenuated the 5-HT4-mediated inotropic response, whereas the NO donor Sin-1 increased this response. The effects were absent during PDE3 inhibition, suggesting cGMP-dependent inhibition of PDE3. However, in contrast to the effects on the 5-HT4 response, Sin-1 inhibited whereas l-NAME and ODQ enhanced the β1-AR-mediated inotropic response. cGMP generated both by particulate (NPR-B) and soluble GC increases the 5-HT4-mediated inotropic response in failing hearts, probably through inhibition of PDE3. β1-AR and 5-HT4 receptor signalling are subject to opposite regulatory control by cGMP generated by soluble GC in failing hearts. Thus, cGMP from different sources is functionally compartmented, giving differential regulation of different Gs-coupled receptors.
Similar content being viewed by others

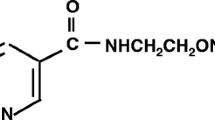
Avoid common mistakes on your manuscript.
Introduction
The failing human and rat cardiac ventricles, regardless of heart failure aetiology, become sensitive to serotonin (5-HT) which elicits a positive inotropic response through stimulation of 5-HT4 receptors (Brattelid et al. 2004, 2007; Qvigstad et al. 2005). 5-HT4 receptors are Gs-coupled, and their positive inotropic and lusitropic effects are assumed to be mediated by cAMP both in rat and human myocardium (Kaumann and Levy 2006; Afzal et al. 2008; Qvigstad et al. 2005; Gergs et al. 2009) in similarity with β-adrenoceptors (β-ARs) (Brodde and Michel 1999). Stimulation of β-ARs is increased in heart failure, and chronic stimulation is considered deleterious for the failing heart due to elevated levels of cAMP that activates processes leading to increased energy expenditure and calcium overload (Lohse et al. 2003). Accordingly, in line with treatment with β-AR antagonists (β-blockers), treatment with a 5-HT4 receptor antagonist has shown beneficial effects in rats with heart failure (Birkeland et al. 2007). Moreover, treatment with piboserod, a 5-HT4 receptor antagonist, has been shown to increase left ventricular ejection fraction in patients with heart failure (Kjekshus et al. 2009).
Cyclic GMP (cGMP) is a cyclic nucleotide synthesized by guanylyl cyclase (GC) and activates cGMP-dependent protein kinase (PKG), which mediates a series of cardiac effects such as metabolic responses and modulation of cardiac contractility (Shah and MacCarthy 2000). Two types of GC exist, differing in cellular localization and activation by specific ligands. Particulate GC (pGC) is located in the plasma membrane as an integral part of the A and B type natriuretic peptide receptors (NPRs) and is activated by these peptides (Kuhn 2003), whereas soluble GC (sGC) is present in cytosol and is activated by nitric oxide (NO) (Kuhn 2003). Atrial (ANP) and brain (BNP) natriuretic peptides mediate their effects by binding to and activation of the cell surface GC natriuretic peptide receptor A (NPR-A), whereas CNP activates the B-type natriuretic peptide receptor (NPR-B) (Kuhn 2003). Plasma levels of ANP, BNP and CNP produced by the myocardium are increased in heart failure (Kalra et al. 2003; Mukoyama et al. 1991). It is well known that NO, which is traditionally known as a vasodilator, also affects myocardial function and modulates cardiac excitation–contraction coupling in multifaceted ways (Massion et al. 2003). The NO-dependent signalling attenuates β-adrenergic responses, and the activity of myocardial NO synthase (NOS) is reported to be enhanced in heart failure (Bendall et al. 2004; Damy et al. 2003, 2004; Layland et al. 2002). As both natriuretic peptides and NO are increased in heart failure, their increased local effects may increase cGMP levels within the cardiac tissue. We recently found that both BNP and CNP increased cGMP in isolated cardiomyocytes from rat failing hearts (Qvigstad et al. 2010). Thus, both NPR-A and NPR-B receptors are operative in failing cardiomyocytes.
Cyclic nucleotide phosphodiesterases (PDEs) are the only enzymes that degrade cyclic nucleotides (cAMP or cGMP) and thus play crucial roles in regulating the amplitude, duration and localization of cyclic nucleotide signalling (Fischmeister et al. 2006). Cyclic GMP-dependent signalling may cross-talk with cAMP-dependent signalling, as varying concentrations of cGMP can regulate the activity of PDEs hydrolyzing cAMP and thus alter the concentration of cAMP and hence the downstream signalling (Zaccolo and Movsesian 2007). We have previously reported that, in failing cardiac ventricles, PDE3 is the major PDE isoform regulating the inotropic responses to β-ARs (Afzal et al. 2011) and 5-HT4 receptors (Afzal et al. 2008). However, a role of PDE4 is demasked when PDE3 is already inhibited. PDE3 can degrade both cAMP and cGMP but has a lower enzymatic activity towards cGMP, and increasing concentrations of cGMP effectively inhibit the degradation of cAMP (Degerman et al. 1997). We thus investigated whether agents increasing cGMP would amplify the responses to 5-HT4 receptor stimulation in failing myocardium.
Accordingly, the aims of this study were to evaluate the regulatory effects of increased cGMP levels generated by pGC and sGC, respectively, to elucidate whether separate functional cGMP pools might exist. Thus, we studied the functional influences of natriuretic peptides through pGC and NO-dependent signalling through sGC on the 5-HT4-elicited positive inotropic response in failing rat ventricle. We show that CNP, but not ANP or BNP, increases the 5-HT4-elicited positive inotropic response through inhibition of PDE3, probably exerted by a localized pool of cGMP. Moreover, cGMP generated by NO/sGC increases the 5-HT4-mediated inotropic response, most likely by inhibiting PDE3. This was in striking contrast to the β1-AR-mediated inotropic response, which was attenuated by stimulation of sGC.
Methods
Animal model
Animal care was according to the Norwegian Animal Welfare Act, which conforms to the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health, and was approved by the Norwegian Animal Research Authority. As described, an extensive myocardial infarction was induced in 320-g male Wistar rats by coronary artery ligation for 6 weeks. Left ventricular end diastolic pressures (LVEDP) were measured by catheterization as described earlier (Sjaastad et al. 2003). In order to obtain well-characterized heart failure, rats were only included in the study when the infarct size was larger than 30% of the left ventricular inner surface and the LVEDP was 15 mmHg or higher after 6 weeks (HF) (Sjaastad et al. 2000).
Papillary muscles/ventricular strips
Left ventricular papillary muscles and ventricular strips were prepared and mounted in organ baths (31°C) with a physiological salt solution with a Ca2+ concentration of 1.8 mM and equilibrated and field-stimulated at 1 Hz (Sjaastad et al. 2003). The contraction–relaxation cycles (CRC) were recorded and analyzed as described previously (Sjaastad et al. 2003) with respect to maximal developed force (F max, millinewton) and maximal development of force ((dF/dt)max). The (dF/dt)max was used as an index of contractility, and inotropic response to agonists was expressed as increases in (dF/dt)max in percentage of control levels. Blockers of α1-adrenoceptors (prazosin 1 μM), β-adrenoceptors (timolol 1 μM, except the experiments with (−)-noradrenaline), muscarinic cholinergic receptors (atropine 1 μM) and 5-HT2A receptors (ketanserin 0.1 μM) were added 90 min before the agonists in order to obtain selective receptor stimulation and to avoid possible indirect effects by release of endogenous substances. PDE3 inhibitor (cilostamide 1 μM), PDE4 inhibitor (rolipram 10 μM), NO-synthase inhibitor (l-NAME 100 μM) and sGC inhibitor (ODQ 10 μM) were added as specified 45 min before the agonists. The NO donor (Sin-1 300 μM), ANP (1 μM), BNP (1 μM) and CNP (300 nM) were added 20 min before the agonist as specified. These drugs did not influence the basal CRC characteristics or electrical stimulation threshold (data not shown), except 300 nM CNP, which reduced the basal (dF/dt)max by 12.5 ± 1.3% (n = 19). (−)-Noradrenaline in the presence of 50 nM ICI118551 (and 1 μM prazosin) was used to selectively stimulate β1-ARs. 5-HT or (−)-noradrenaline was added to the organ bath cumulatively (concentration–response relationship). Concentration–response curves were constructed by estimating centiles (EC10 to EC100) and calculating the corresponding means and the horizontal positioning expressed as –logEC50 (Sjaastad et al. 2003). Compared to stimulation of β1-ARs, stimulation of 5-HT4 receptors in failing myocardium with 5-HT gives a lower inotropic response. Therefore, comparable to the behaviour of a partial agonist, any enhancement of the response increases the maximal response (efficacy) and not necessarily the potency (Afzal et al. 2008). Thus, in order to visualize the effect of any intervention, the concentration–response curves of 5-HT are plotted as percent above basal. In contrast, the concentration–response curves of the “full agonist” (−)-noradrenaline are normalized to the individual maxima, as any enhancement of the response increases the sensitivity (lowers EC50) of the response without any significant change in the efficacy. However, the individual maximal values of inotropic responses to both 5-HT and (−)-noradrenaline are presented in Tables 2 and 3. Since the inotropic response to 5-HT4 stimulation induced during heart failure is remarkably reproducible in papillary muscles, these muscles were reserved for these studies. The inotropic response to noradrenaline, however, always appears as full agonism, and as changes in EC50 will not be influenced by random variation in maxima, ventricular strips could accordingly be used for these studies. Therefore, left ventricular papillary muscles were used in experiments with 5-HT, and left ventricular strips were used in experiments with (−)-noradrenaline.
cGMP content in left ventricular strips
Left ventricular strips (described above) were equilibrated for 90 min and freeze-clamped 45 min after addition of ODQ (inhibitor of sGC) or l-NAME (NO synthase inhibitor) and 20 min after Sin-1 (NO donor). Frozen tissue was homogenized at 4°C in 1 ml 5% trichloroacetic acid by a Retch MM301 mechanical mill. Total cGMP levels were measured by cGMP EIA kit (Cayman Chemicals) according to the manufacturer's protocol.
Materials
5-HT hydrochloride, (−)-Arterenol ([−]-noradrenaline) bitartrate (hydrate), timolol maleate, prazosine hydrochloride and atropine sulphate were purchased from Sigma-Aldrich (St. Louis, MO, USA). Rolipram (4-(3-(cyclopentyloxy)-4-methoxyphenyl)pyrrolidin-2-one), cilostamide (N-cyclohexyl-N-methyl-4-(1,2-dihydro-2-oxo-6-quinolyloxy)butyramide), Sin-1 (amino-3-morpholinyl-1,2,3-oxadiazolium) chloride, ICI118551 ((±)-erythro-(S*,S*)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol) hydrochloride, ODQ (1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one), l-NAME (NG-Nitro-l-arginine methyl ester) hydrochloride and ketanserin (3-[2-[4-(4-fluorobenzoyl)-1-piperidinyl]ethyl]-2,4[1H,3 H]-quinazolinedione) tartrate were from Tocris Bioscience (Bristol, UK). Isoflurane (Forene) was from Abbot Scandinavia (Solna, Sweden).
Statistics
All results are expressed as mean±SEM unless otherwise indicated, and statistical significance was assessed with unpaired or paired Student's t tests as appropriate. When appropriate, Bonferroni corrections were made. p < 0.05 was regarded to reflect statistically significant differences.
Results
All the rats included in the study had anterolateral infarcts larger than 30% of the left ventricular inner surface, LVEDP >15 mmHg at 6 weeks and signs of HF, including tachypnoea and pulmonary congestion. For animal characteristics and haemodynamic data, see Table 1. For simplification and in order to prompt direct comparison between the different experiments, the data from the concentration–response curves (discussed below) are listed in Tables 2 and 3 in addition to the presentation in the figures.
Effect of natriuretic peptides on the 5-HT4-elicited inotropic response
CNP and not ANP or BNP increases the 5-HT4-elicited inotropic response
We have previously demonstrated that both NPR-A and NPR-B receptors are operative in both cardiomyocytes and ventricular strips from failing rat ventricles, as both BNP and CNP increased cGMP levels (Qvigstad et al. 2010). We determined the effect of ANP, BNP and CNP on 5-HT4-mediated inotropic response in papillary muscles from failing rat left ventricles. Stimulation of NPR-A by 1 μM ANP or 1 μM BNP did not change the responsiveness to 5-HT (Fig. 1). However, stimulation of NPR-B by 300 nM CNP significantly increased the inotropic response to 5-HT compared to control (Figs. 1 and 2a).

Concentration–response curves of inotropic responses to 5-HT4 receptor stimulation in the absence (control) or presence of natriuretic peptides; 1 μM ANP, 1 μM BNP or 300 nM CNP. Natriuretic peptides were added 20 min before 5-HT. Ordinate: inotropic response expressed as increase in (dF/dt)max in percent above basal. Vertical bars represent SEM of maximal inotropic responses. Abscissa: concentration of the agonist. Horizontal bars represent SEM of –logEC50. Double asterisks: p < 0.01 vs. control

a Concentration–response curves of inotropic responses to 5-HT4 receptor stimulation in papillary muscles in the absence (control) or presence of 300 nM CNP or in combination with PDE inhibitors (Cil, 1 μM cilostamide; Rol, 10 μM rolipram). b Concentration–response curves of inotropic responses (normalized) to β1-adrenoceptor stimulation by (−)-noradrenaline (50 nM ICI118551 present) in absence (control) or presence of CNP or in combination with PDE inhibitors (Cil, 1 μM cilostamide; Rol, 10 μM rolipram) in left ventricular strips. CNP and PDE inhibitors were added 20 and 45 min before the agonist, respectively. Ordinate: inotropic response expressed as increase in (dF/dt)max in percent above basal (a) or as percent of maximum (normalized) (b). Vertical bars represent SEM of maximal inotropic responses. Abscissa: concentration of the agonist. Horizontal bars represent SEM of –logEC50. Double asterisks: p < 0.01 CNP vs. control (for maximal response in a, and –logEC50 in b). Single crosses: p < 0.01 CNP/Rol vs. CNP (for maximal response in a, and –logEC50 in b). Double crosses: p < 0.01 –logEC50 CNP/Rol vs. CNP
CNP increases the 5-HT4-elicited inotropic response through inhibition of PDE3
Despite the increase in the responsiveness, there was no change in the sensitivity to 5-HT (EC50) in the presence of CNP compared to control (Fig. 2a). During PDE4 inhibition (10 μM rolipram) in the presence of 300 nM CNP, there was a significant increase in both the responsiveness and the sensitivity to 5-HT compared to the presence of CNP alone (Fig. 2a). Similar patterns were observed during PDE3 inhibition alone (1 μM cilostamide) and during concomitant PDE3/4 inhibition (Fig. 2a), consistent with our previous report that PDE3 inhibition enhances the inotropic response to 5-HT and concomitant PDE3 and PDE4 inhibition further enhances the responsiveness and also enhances the sensitivity to 5-HT compared to PDE3 inhibition alone (Afzal et al. 2008). To evaluate whether CNP enhanced the 5-HT4-mediated inotropic response through inhibition of PDE3, we tested the effect of CNP (300 nM) in the presence of cilostamide (1 μM). Preincubation with both cilostamide and CNP simultaneously did not significantly further increase the inotropic response to 5-HT compared to CNP or cilostamide alone, consistent with a PDE3-dependent mechanism of CNP (Fig. 2a).
CNP also increases the β1-AR-mediated inotropic response through PDE3 inhibition
In order to perform a direct comparison with the modulation of 5-HT4-mediated response, we also tested the effect of CNP on the β1-AR-mediated inotropic response in the present study. In accordance with our previous report (Qvigstad et al. 2010), 300 nM CNP sensitized the β1-AR-mediated inotropic response, with a further increase in sensitivity during concomitant PDE4 inhibition with 10 μM rolipram (Fig. 2b). Moreover, additional presence of 1 μM cilostamide did not further sensitize the β1-mediated inotropic response compared to CNP alone (Fig. 2b), consistent with CNP increasing the β1-AR-mediated inotropic response through PDE3 inhibition (Qvigstad et al. 2010).
Effect of endogenous nitric oxide on the 5-HT4-elicited inotropic response
NO increases the 5-HT4-elicited inotropic response
The NO-dependent signalling is increased in heart failure and thus the cGMP levels. As increase in cGMP levels by pGC enhances the 5-HT4-elicited inotropic response, we determined the effect of cGMP generated by sGC on the inotropic response to 5-HT. Inhibition of NOS (100 μM l-NAME) decreased both the cGMP levels (Fig. 3) and the inotropic response to 5-HT (Fig. 4a). Inhibition of sGC (10 μM ODQ) nominally decreased the cGMP levels without reaching statistical significance but significantly decreased the inotropic response to 5-HT. Thus, both approaches indicate that endogenous NO contributes to increase the 5-HT4-mediated inotropic response in the failing ventricle, as inhibition of the NO pathway decreases the response. In addition, the NO donor Sin-1 (300 μM) increased the cGMP levels (Fig. 3) and also nominally increased the inotropic response to 5-HT (Fig. 4a), although without reaching statistical significance (p = 0.08 after Bonferroni correction).

Cyclic GMP levels in left ventricular strips in absence (control) or presence of 100 μM l-NAME (NOS inhibitor), 10 μM ODQ (sGC inhibitor) or 300 μM Sin-1 (NO donor). l-NAME and ODQ were added 45 min before and Sin-1 20 min before the ventricular strips were snap-frozen. Number of experiments “n” inside the bars; mean±SEM. Asterisk: p < 0.05 vs. control. Number sign: p = 0.16 vs. control (after Bonferroni correction)

a, b Concentration–response curves of inotropic responses to 5-HT4 receptor stimulation in the absence (control) and presence of 100 μM l-NAME (NOS inhibitor), 10 μM ODQ (sGC inhibitor), 300 μM Sin-1 (NO donor) or in combination with PDE inhibitors (Cil, 1 μM cilostamide; Rol, 10 μM rolipram) in papillary muscles. All the inhibitors were added 45 min before 5-HT, whereas Sin-1 was added 20 min before 5-HT. Ordinate: inotropic response expressed as increase in (dF/dt)max in percent above basal. Vertical bars represent SEM of inotropic response. Abscissa: concentration of the agonist. Horizontal bars represent SEM of –logEC50. Asterisk: p < 0.05 vs. control. Number sign: p = 0.08 vs. control (after Bonferroni correction)
NO increases the 5-HT4-elicited inotropic response presumably through PDE3 inhibition
To determine whether cGMP generated by the sGC increased the 5-HT4-mediated inotropic response through inhibition of PDE3, concentration–response experiments were conducted in the presence of 100 μM l-NAME or 300 μM Sin-1 in papillary muscles preincubated with 1 μM cilostamide. During this PDE3 inhibition, intervention with NO signalling by l-NAME or Sin-1 did not exhibit any additional effect on the 5-HT4-mediated inotropic response compared to PDE3 inhibition alone, suggesting that PDE3 is inhibited by NO-generated cGMP (Fig. 4b). Accordingly, during combined PDE3/4 inhibition, there was no difference between the absence or presence of 100 μM l-NAME (Fig. 4b). Additionally, during PDE4 inhibition, 300 μM Sin-1 significantly increased the inotropic response to 5-HT compared to control (Fig. 4a), thus providing a more substantial effect than 300 μM Sin-1 alone, which only nominally increased the inotropic response compared to control. These results suggest that the increased NO signalling here most likely acts through inhibition of PDE3 due to the increased cGMP levels.
NO decreases the β1-AR-mediated inotropic response
For comparison, we determined the effect of increased (Sin-1) and decreased (l-NAME, ODQ) NO signalling on the β1-AR-mediated inotropic response. Sin-1 decreased, whereas l-NAME and ODQ increased the sensitivity of the β1-AR-mediated inotropic response (Fig. 5). These results are contrary to those on the 5-HT4-elicited inotropic response, but in accordance with previous reports that NO signalling decreases β1-adrenergic signalling (Bendall et al. 2004; Damy et al. 2003, 2004).

Concentration–response curves of inotropic responses (normalized) to β1-adrenoceptor stimulation by (−)-noradrenaline (50 nM ICI118551 present) in absence (control) or presence of 100 μM l-NAME (NOS inhibitor), 10 μM ODQ (sGC inhibitor) or 300 μM Sin-1 (NO donor) in left ventricular strips. l-NAME and ODQ were added 45 min before and Sin-1 20 min before the (−)-noradrenaline. Ordinate: inotropic response expressed as increase in (dF/dt)max in percent of maximum (normalized). Abscissa: concentration of the agonist. Horizontal bars represent SEM of –logEC50. Double asterisks: p < 0.01 vs. control
Discussion and conclusions
We demonstrate here that the stimulation of NPR-B by CNP increases the 5-HT4-mediated inotropic response in failing rat heart ventricles. This effect is most likely caused by cGMP-mediated PDE3 inhibition, as the PDE3 inhibitor cilostamide increased the response to a similar extent as CNP without any additive effect. Similar results were obtained for the β1-AR-mediated inotropic response both in this study and previously (Qvigstad et al. 2010). We also presently demonstrate that stimulation of NPR-A by ANP or BNP does not affect the 5-HT4-mediated inotropic response, which is in line with the previously reported lack of effect on the β1-AR-mediated inotropic response (Qvigstad et al. 2010). These differences between stimulation of NPR-B and NPR-A are quite remarkable, as both receptors increase cGMP levels in isolated cardiomyocytes from failing hearts (Qvigstad et al. 2010). Thus, these results suggest the two NPRs generate different cGMP pools, only one of which is available to the PDE3 regulating cAMP-mediated intropic response. We also demonstrate that cyclic GMP generated by sGC enhances the 5-HT4-mediated inotropic response by PDE3 inhibition. This is in contrast to the β1-AR-mediated inotropic response, which is inhibited by increased cGMP levels produced by NO-stimulated sGC. The divergent influence of cGMP generated from sGC on these different Gs-coupled receptor systems suggests both different compartments within this cGMP pool and separate compartments for 5-HT4 receptor- and β1-AR-mediated signalling pathways.
Amplification of 5-HT4-mediated inotropic response by CNP
NPR-B stimulation by CNP increased the 5-HT4-mediated inotropic response to a similar extent as the PDE3 inhibitor cilostamide, and additional PDE3 inhibition did not promote any further increase. This is consistent with inhibition of PDE3 by cGMP generated following stimulation by CNP. Furthermore, this was substantiated by the observation that in the presence of PDE4 inhibition by rolipram, the effect of CNP on the inotropic response was further enhanced to a similar level as by concomitant PDE3/4 inhibition, i.e. an increase in the maximal response accompanied by a sensitization of the inotropic response, as the role of PDE4 is demasked by inhibition of PDE3 (Afzal et al. 2008, 2011). Similarly, we demonstrated that the β1-AR-mediated inotropic response is also enhanced by CNP. This is in accordance with and confirms our previous report (Qvigstad et al. 2010), showing that β1-AR-mediated inotropic response is enhanced by CNP, most likely due to PDE3 inhibition, and that CNP stimulation increased the cAMP levels during β1-AR-activation to similar levels as cilostamide. Wen et al. (2004) found that CNP, by activating pGC, increased cAMP accumulation by inhibiting PDE3 in rabbit atria. In line with this, our functional data support a profound inhibition of PDE3 by cGMP generated in response to NPR-B activation by CNP in failing rat ventricle. Moreover, a recent study has shown that in hypertrophic human ventricle, the balance of effects of activating the cGMP-PKG pathway is shifted from effects of PKG activation towards effects of PDE3 inhibition, due to decreased PKG activity (Nagendran et al. 2007).
Amplification of the 5-HT4-mediated inotropic response by sGC-generated cGMP
The activity of NOS is reported to be increased in experimental and human heart failure (Damy et al. 2003, 2004). We found that inhibitors of NOS (l-NAME) and sGC (ODQ) decreased the cGMP levels in ventricular strips, indicating a certain endogenous NOS activity. This also makes it probable that the observed effects in the presence of these inhibitors can be ascribed to decrease in cGMP levels. Inhibition of NO signalling by l-NAME and ODQ attenuated the 5-HT4-mediated inotropic response, indicating that endogenous NO contributes to increase the 5-HT4-mediated inotropic response in failing hearts. Moreover, the NO donor Sin-1 increased cGMP levels as well as the 5-HT4-mediated inotropic response, suggesting that increasing the cGMP levels by sGC activation increases the 5-HT4-mediated response. During PDE3 inhibition by cilostamide, none of the effects of l-NAME, ODQ and Sin-1 on the 5-HT4-mediated response were observed, and the inotropic responses were similar to those in the presence of PDE3 inhibition alone. This indicates that increased response to cGMP is most likely attributable to inhibition of PDE3, as when PDE3 is already inhibited, no additional inhibition by cGMP is achievable. This was also supported by the observation that during PDE4 inhibition by rolipram, Sin-1-dependent increase of 5-HT4-mediated inotropic response with respect to control was larger compared to that in the absence of PDE4 inhibition (Fig. 4a). These results are in accordance with a previous study showing that NO stimulates Ca2+ channel current in human atrial myocytes by inhibiting PDE3, and the stimulatory effect of Sin-1 on Ca2+ channel current was masked in the presence of the PDE3 inhibitor milrinone (Kirstein et al. 1995). Interestingly, Sin-1 and rolipram in combination only increased the maximal inotropic response without an increase in the sensitivity of the response (Fig. 4a). This is in contrast to concomitant inhibition of PDE3/4 with PDE3 inhibited by either cilostamide or CNP, where there is also an increase in the sensitivity (Fig. 2a), which reflects a full activation of the cAMP-dependent signalling (Afzal et al. 2008). Possible explanations for the lacking increase in the sensitivity of inotropic response in the presence of Sin-1 and rolipram might be that compared to β-AR-mediated inotropic response, the inotropic response to 5-HT4 receptor stimulation is submaximal (Afzal et al. 2008), and Sin-1 is causing a less efficient PDE3 inhibition than cilostamide and CNP. This is supported by a lower maximal inotropic effect of 5-HT in the presence of Sin-1/rolipram than in the presence of cilostamide/rolipram or CNP/rolipram. This amplification of the 5-HT4-mediated inotropic effect by NO signalling is in marked contrast to the inhibitory effect on the β1-AR-mediated inotropic response, as also confirmed in the present study. The proposed mechanism for this attenuation of β-AR-mediated inotropic response is PKG-dependent inhibition of l-type Ca2+ channels and subsequent suppression of β-adrenergic inotropic effect (Wang et al. 2009). Moreover, PKG-dependent phosphorylation of troponin I may also contribute to attenuate the β-adrenergic response (Layland et al. 2002).
Evidence for compartmented cGMP pools
The present study provides some functional evidence for apparently four compartmented cGMP pools in rat failing myocardium by exploring the influence of different agents increasing cGMP on the inotropic effects of the two Gs-coupled receptors 5-HT4 and β1-AR, respectively (Fig. 6): (1) cGMP generated by NPR-B enhancing the response to both Gs-coupled receptors, probably by inhibiting PDE3; (2) cGMP generated by NPR-A with no influence on either of the two Gs-coupled receptors; (3) a pool of cGMP generated by sGC enhancing the response to 5-HT4 only, probably by inhibiting PDE3; and (4) a pool of cGMP generated by sGC attenuating the response to β1-AR. Our results showing that NO inhibits β-adrenergic signalling are in line with previous studies by others (Bendall et al. 2004; Damy et al. 2003; Layland et al. 2002). The opposite influence of the NO system on 5-HT4 and β1-AR-mediated effects illustrates a major qualitative difference between the signalling of these two Gs-coupled receptors, which otherwise possess distinct similarities. The findings suggest a differential compartmented localization of 5-HT4 and β1-AR and indicate a highly selective and intricate regulation of inotropic responses mediated by Gs-coupled receptors. This is further illustrated by similar effects of pGC-generated cGMP on β1-ARs and 5-HT4 receptors in spite of opposing effects on the receptors by sGC-generated cGMP. These observations together provide evidence of an intricate compartmentation of different cGMP pools affecting the downstream signalling of different receptors in similar or opposite ways. Why the cGMP pool generated by NPR-A is unable to influence the response to the Gs-coupled receptors studied remains obscure. A recent study has shown differential effects of cGMP on cAMP levels via different PDEs in spatially distinct subcellular domains (Stangherlin et al. 2011). Other studies have also reported observations indicating compartmented signalling of cGMP, depending on the source of cGMP. ANP-stimulated cGMP displayed no impact on β-AR-stimulated contractility in either the presence or absence of PDE5 inhibitor, whereas NO-stimulated cGMP elevation reduced the β-AR-stimulated contractility, and the effect was enhanced during PDE5 inhibition (Takimoto et al. 2007). Moreover, the same study showed that ANP-stimulated pGC did not activate PKG despite substantial increase in cGMP levels, whereas PDE5 inhibition efficiently activated PKG. A study in mouse cardiomyocytes showed that stimulation of both pGC and sGC exerted similar negative inotropic response by cGMP-dependent pathways, but only pGC activation reduced Ca2+ transients, whereas sGC activation had only marginal effects (Su et al. 2005). Other studies showed that the particulate cGMP pool is controlled by PDE2, whereas the soluble cGMP pool is controlled by PDE5, and the differential cardiac effects of the cGMP were attributable to different spatiotemporal distribution (Castro et al. 2006).

Illustration of the proposed functional compartmentation of 5-HT4 receptor, β1-adrenoceptor and cyclic nucleotides in cardiac myocytes. The inotropic response to 5-HT4 receptor and β1-adrenoceptor stimulation is mediated by cAMP and regulated by PDE3. 1: cGMP generated by NPR-B inhibits PDE3 and increases the 5-HT4 receptor- and β1-adrenoceptor-mediated inotropic response. 2: cGMP generated by NPR-A has no access to the PDE3 regulating inotropic response. 3: A pool of cGMP generated by sGC increases the 5-HT4-mediated inotropic response by inhibiting PDE3. 4: A pool of cGMP generated by sGC has no access to the PDE3 regulating the β1-adrenoceptor-mediated inotropic response, but inhibits it by some other mechanism, possibly through PKG
The cAMP signals generated by different Gs-protein-coupled receptors in cardiomyocytes and controlled by PDEs are known to be compartmented (Rochais et al. 2006; Galindo-Tovar et al. 2009). As the cGMP-dependent pathways are able to cross-talk with cAMP-dependent pathways and as different cross-talk mechanisms are operating, discrepancy between the influence on the function of receptors is not unexpected. To elucidate mechanisms involved, it would be useful to determine the cAMP levels under different experimental conditions and relate them to contractile responses. However, based on our experience (Afzal et al. 2008, 2011), the interpretation of such measurements in ex vivo whole-tissue preparation is complex, i.e. immeasurable cAMP increase may be associated with clear functional amplification, and a marked cAMP increase may appear without functional enhancement. In order to understand these mechanisms, use of selective FRET-based sensors with the ability to measure cAMP or cGMP in distinct compartments would be a more reasonable approach. Compartmentations may rely on different locations of the receptors in the membrane, the involved cyclases, the cyclic nucleotide PDEs and protein kinases with their respective targets. Such phenomena may occur within each cardiomyocyte and/or between cardiomyocytes. Presently, a further elaboration of the concept of compartmentation would require methods that identify the relative location of the components involved in the signalling pathways in the myocytes as well as their mutual and dynamic interaction with each other.
Potentially harmful influence of cGMP on 5-HT4-mediated effects in failing myocardium
We have shown that both BNP and CNP elicited a robust increase in cGMP levels in ventricular tissue and cardiomyocytes from failing hearts (Qvigstad et al. 2010). Moreover, ANP also profoundly increased the cGMP levels in myocardial tissue (Takimoto et al. 2007). Despite the increase in cGMP levels by both NPR-A and NPR-B, only NPR-B-mediated increases in cGMP levels enhance the 5-HT4 receptor and β1-AR-mediated inotropic responses in failing rat ventricles, indicating that NPR-B might play a more prominent role in regulating the cardiac contractility in heart failure. This is supported by another study which reported increased activity of NPR-B compared to NPR-A, and thus a more dominant role of NPR-B than NPR-A receptors in failing mouse hearts compared to Sham hearts (Dickey et al. 2007). Moreover, in the absence of NPR-A in genetic mouse models, the contractile responses to CNP activating NPR-B were enhanced compared to mice expressing NPR-A (Pierkes et al. 2002), which might be similar to the situation in the failing myocardium as described above. In heart failure, CNP plasma levels show a significant increase which parallels clinical severity and correlates negatively with indices of left ventricular function such as ejection fraction and (dP/dt)max (Del Ry et al. 2005, 2008). Activation of 5-HT4 receptors elevates cAMP, and longstanding cAMP-dependent effects are associated with deleterious cardiac effects (Lohse et al. 2003). 5-HT4 receptor stimulation may also promote arrhythmia (Kaumann and Levy 2006). The increased levels of CNP and increased NO signalling in heart failure might contribute to accentuate these potentially harmful effects of 5-HT4 receptor activation in patients with heart failure.
Conclusions
In conclusion, cGMP generated by both pGC, through NPR-B, and sGC increases the 5-HT4-mediated inotropic response. This is in contrast to β1-ARs, where pGC-generated cGMP (through NPR-B) increases the inotropic response, whereas sGC-generated cGMP decreases the inotropic response. These results provide evidence of distinctly different functional cGMP pools regulating the inotropic response to activation of different receptors depending on the source of cGMP (Fig. 6). Compartmented localization of the Gs-coupled receptors may also be involved in this differential regulation of their function.
References
Afzal F, Andressen KW, Mørk HK, Aronsen JM, Sjaastad I, Dahl CP, Skomedal T, Levy FO, Osnes JB, Qvigstad E (2008) 5-HT4-elicited positive inotropic response is mediated by cAMP and regulated by PDE3 in failing rat and human cardiac ventricles. Br J Pharmacol 155:1005–1014
Afzal F, Aronsen JM, Moltzau LR, Sjaastad I, Levy FO, Skomedal T, Osnes JB, Qvigstad E (2011) Differential regulation of β2-adrenoceptor-mediated inotropic and lusitropic response by PDE3 and PDE4 in failing and non-failing rat cardiac ventricle. Br J Pharmacol 162:54–71
Bendall JK, Damy T, Ratajczak P, Loyer X, Monceau V, Marty I, Milliez P, Robidel E, Marotte F, Samuel JL, Heymes C (2004) Role of myocardial neuronal nitric oxide synthase-derived nitric oxide in β-adrenergic hyporesponsiveness after myocardial infarction-induced heart failure in rat. Circulation 110:2368–2375
Birkeland JA, Sjaastad I, Brattelid T, Qvigstad E, Moberg ER, Krobert KA, Bjørnerheim R, Skomedal T, Sejersted OM, Osnes JB, Levy FO (2007) Effects of treatment with a 5-HT4 receptor antagonist in heart failure. Br J Pharmacol 150:143–152
Brattelid T, Qvigstad E, Lynham JA, Molenaar P, Aass H, Geiran O, Skomedal T, Osnes J-B, Levy FO, Kaumann AJ (2004) Functional serotonin 5-HT4 receptors in porcine and human ventricular myocardium with increased 5-HT4 mRNA in heart failure. Naunyn-Schmied Arch Pharmacol 370:157–166
Brattelid T, Qvigstad E, Birkeland JAK, Swift F, Bekkevold SVS, Krobert KA, Sejersted OM, Skomedal T, Osnes JB, Levy FO, Sjaastad I (2007) Serotonin responsiveness through 5-HT2A and 5-HT4 receptors is differentially regulated in hypertrophic and failing rat cardiac ventricle. J Mol Cell Cardiol 43:767–779
Brodde OE, Michel MC (1999) Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev 51:651–690
Castro LR, Verde I, Cooper DM, Fischmeister R (2006) Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation 113:2221–2228
Damy T, Ratajczak P, Robidel E, Bendall JK, Oliviero P, Boczkowski J, Ebrahimian T, Marotte F, Samuel JL, Heymes C (2003) Up-regulation of cardiac nitric oxide synthase 1-derived nitric oxide after myocardial infarction in senescent rats. FASEB J 17:1934–1936
Damy T, Ratajczak P, Shah AM, Camors E, Marty I, Hasenfuss G, Marotte F, Samuel JL, Heymes C (2004) Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet 363:1365–1367
Degerman E, Belfrage P, Manganiello VC (1997) Structure, localization, and regulation of cGMP-inhibited phosphodiesterase (PDE3). J Biol Chem 272:6823–6826
Del Ry S, Passino C, Maltinti M, Emdin M, Giannessi D (2005) C-type natriuretic peptide plasma levels increase in patients with chronic heart failure as a function of clinical severity. Eur J Heart Fail 7:1145–1148
Del Ry S, Maltinti M, Cabiati M, Emdin M, Giannessi D, Morales MA (2008) C-type natriuretic peptide and its relation to non-invasive indices of left ventricular function in patients with chronic heart failure. Peptides 29:79–82
Dickey DM, Flora DR, Bryan PM, Xu X, Chen Y, Potter LR (2007) Differential regulation of membrane guanylyl cyclases in congestive heart failure: natriuretic peptide receptor (NPR)-B, not NPR-A, is the predominant natriuretic peptide receptor in the failing heart. Endocrinology 148:3518–3522
Fischmeister R, Castro LR, bi-Gerges A, Rochais F, Jurevicius J, Leroy J, Vandecasteele G (2006) Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res 99:816–828
Galindo-Tovar A, Vargas ML, Kaumann AJ (2009) Phosphodiesterases PDE3 and PDE4 jointly control the inotropic effects but not chronotropic effects of (−)-CGP12177 despite PDE4-evoked sinoatrial bradycardia in rat atrium. Naunyn-Schmied Arch Pharmacol 379:379–384
Gergs U, Neumann J, Simm A, Silber R-E, Remmers FO, Läer S (2009) Phosphorylation of phospholamban and troponin I through 5-HT4 receptors in the isolated human atrium. Naunyn-Schmied Arch Pharmacol 379:349–359
Kalra PR, Clague JR, Bolger AP, Anker SD, Poole-Wilson PA, Struthers AD, Coats AJ (2003) Myocardial production of C-type natriuretic peptide in chronic heart failure. Circulation 107:571–573
Kaumann AJ, Levy FO (2006) 5-Hydroxytryptamine receptors in the human cardiovascular system. Pharmacol Ther 111:674–706
Kirstein M, Rivet-Bastide M, Hatem S, Benardeau A, Mercadier JJ, Fischmeister R (1995) Nitric oxide regulates the calcium current in isolated human atrial myocytes. J Clin Invest 95:794–802
Kjekshus JK, Torp-Pedersen C, Gullestad L, Køber L, Edvardsen T, Olsen IC, Sjaastad I, Qvigstad E, Skomedal T, Osnes JB, Levy FO (2009) Effect of piboserod, a 5-HT4 serotonin receptor antagonist, on left ventricular function in patients with symptomatic heart failure. Eur J Heart Fail 11:771–778
Kuhn M (2003) Structure, regulation, and function of mammalian membrane guanylyl cyclase receptors, with a focus on guanylyl cyclase-A. Circ Res 93:700–709
Layland J, Li JM, Shah AM (2002) Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol 540:457–467
Lohse MJ, Engelhardt S, Eschenhagen T (2003) What is the role of β-adrenergic signaling in heart failure? Circ Res 93:896–906
Massion PB, Feron O, Dessy C, Balligand JL (2003) Nitric oxide and cardiac function. Ten years after and continuing. Circ Res 93:388–398
Mukoyama M, Nakao K, Hosoda K, Suga S, Saito Y, Ogawa Y, Shirakami G, Jougasaki M, Obata K, Yasue H (1991) Brain natriuretic peptide as a novel cardiac hormone in humans. Evidence for an exquisite dual natriuretic peptide system, atrial natriuretic peptide and brain natriuretic peptide. J Clin Invest 87:1402–1412
Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, St Aubin C, Webster L, Rebeyka IM, Ross DB, Light PE, Dyck JR, Michelakis ED (2007) Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation 116:238–248
Pierkes M, Gambaryan S, Boknik P, Lohmann SM, Schmitz W, Potthast R, Holtwick R, Kuhn M (2002) Increased effects of C-type natriuretic peptide on cardiac ventricular contractility and relaxation in guanylyl cyclase A-deficient mice. Cardiovasc Res 53:852–861
Qvigstad E, Brattelid T, Sjaastad I, Andressen KW, Krobert KA, Birkeland JA, Sejersted OM, Kaumann AJ, Skomedal T, Osnes J-B, Levy FO (2005) Appearance of a ventricular 5-HT4 receptor-mediated inotropic response to serotonin in heart failure. Cardiovasc Res 65:869–878
Qvigstad E, Moltzau LR, Aronsen JM, Nguyen CH, Hougen K, Sjaastad I, Levy FO, Skomedal T, Osnes JB (2010) Natriuretic peptides increase β1-adrenoceptor signalling in failing hearts through phosphodiesterase 3 inhibition. Cardiovasc Res 85:763–772
Rochais F, Abi-Gerges A, Horner K, Lefebvre F, Cooper DM, Conti M, Fischmeister R, Vandecasteele G (2006) Specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ Res 98:1081–1088
Shah AM, MacCarthy PA (2000) Paracrine and autocrine effects of nitric oxide on myocardial function. Pharmacol Ther 86:49–86
Sjaastad I, Sejersted OM, Ilebekk A, Bjørnerheim R (2000) Echocardiographic criteria for detection of postinfarction congestive heart failure in rats. J Appl Physiol 89:1445–1454
Sjaastad I, Schiander I, Sjetnan A, Qvigstad E, Bøkenes J, Sandnes D, Osnes JB, Sejersted OM, Skomedal T (2003) Increased contribution of α1- vs. β-adrenoceptor-mediated inotropic response in rats with congestive heart failure. Acta Physiol Scand 177:449–458
Stangherlin A, Gesellchen F, Zoccarato A, Terrin A, Fields LA, Berrera M, Surdo NC, Craig MA, Smith G, Hamilton G, Zaccolo M (2011) cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ Res 108:929–939
Su J, Scholz PM, Weiss HR (2005) Differential effects of cGMP produced by soluble and particulate guanylyl cyclase on mouse ventricular myocytes. Exp Biol Med 230:242–250
Takimoto E, Belardi D, Tocchetti CG, Vahebi S, Cormaci G, Ketner EA, Moens AL, Champion HC, Kass DA (2007) Compartmentalization of cardiac β-adrenergic inotropy modulation by phosphodiesterase type 5. Circulation 115:2159–2167
Wang H, Kohr MJ, Traynham CJ, Ziolo MT (2009) Phosphodiesterase 5 restricts NOS3/soluble guanylate cyclase signaling to L-type Ca2+ current in cardiac myocytes. J Mol Cell Cardiol 47:304–314
Wen JF, Cui X, Jin JY, Kim SM, Kim SZ, Kim SH, Lee HS, Cho KW (2004) High and low gain switches for regulation of cAMP efflux concentration: distinct roles for particulate GC- and soluble GC-cGMP-PDE3 signaling in rabbit atria. Circ Res 94:936–943
Zaccolo M, Movsesian MA (2007) cAMP and cGMP signaling cross-talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ Res 100:1569–1578
Acknowledgements
This work was supported by The Norwegian Council on Cardiovascular Diseases, The Research Council of Norway, Anders Jahre's Foundation for the Promotion of Science, The Novo Nordisk Foundation, The Family Blix foundation and grants from the University of Oslo. The experiments were performed in accordance with all regulations concerning biomedical research in Norway.
Conflicts of interest
None.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Faraz Afzal and Eirik Qvigstad contributed equally to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Afzal, F., Qvigstad, E., Aronsen, J.M. et al. Agents increasing cyclic GMP amplify 5-HT4-elicited positive inotropic response in failing rat cardiac ventricle. Naunyn-Schmiedeberg's Arch Pharmacol 384, 543–553 (2011). https://doi.org/10.1007/s00210-011-0670-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-011-0670-6