
Abstract
The ATP-binding cassette transporter breast cancer resistance protein (BCRP/ABCG2) is supposed to be a major determinant of the multidrug resistance phenotype of tumors by extruding chemically diverse cytostatic drugs out of tumor cells. BCRP physically and possibly also functionally interacts with caveolin-1 (CAV1, encoded by Cav1), an integral membrane protein of lipid rafts important for signal transduction and membrane trafficking. Moreover, Cav1 is linked to an aggressive phenotype of cancer cells in various tumors. We therefore investigated whether Cav1 plays a functional role in the regulation of BCRP transport activity and in the resistance against chemotherapeutics that are BCRP substrates. As a cell model, we used the BCRP overexpressing cell line MDCKII-BCRP and the corresponding parental cell line MDCKII as a control. Cav1 expression was down-regulated using retrovirus-mediated RNA interference technology. BCRP activity was assessed by pheophorbide A efflux assay and the resistance towards cytostatic drugs was measured by proliferation assays. Efficient knockdown of Cav1 reduced Cav1 expression by 85–95% and BCRP activity by 35%. Concurrently, it reduced resistance towards the BCRP substrate mitoxantrone but not towards vincristine, a chemotherapeutic that is not extruded by BCRP. Western blot analysis of gradient ultracentrifugation fractions and immunofluorescence demonstrates that BCRP localization within the plasma membrane was largely unaltered in Cav1-deficient cells compared to controls. The diminished BCRP function after Cav1 knockdown is, thus, likely mediated by alterations in protein–protein interactions and suggests a positive regulation of BCRP function by CAV1.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer resistance protein (BCRP/ABCG2) is a member of the ATP-binding cassette transporter (ABC transporter) superfamily with 49 known members (Gillet et al. 2007). These transporters are integral membrane proteins that are capable to drive structurally diverse substrates across extra- and intracellular membranes under hydrolysis of ATP (Dean et al. 2001). Together with 12 other ABC transporters, BCRP has been associated with drug transport and consecutive drug resistance of cells expressing the transporter (Gillet et al. 2007). BCRP is expressed in the apical membrane of tissues with barrier function like the placenta, the small intestine, the liver, and the blood–brain barrier (Mao and Unadkat 2005). This localization suggests that the main physiological role of BCRP is to regulate the intestinal absorption and biliary excretion of potentially toxic compounds and to protect “sanctuary sites” of the human body, e.g., brain, stem cells, and fetus from xenobiotics (Mao and Unadkat 2005; Krishnamurthy and Schuetz 2006). Moreover, BCRP has been detected in drug resistant tumors and tumor cell lines (Mao and Unadkat 2005) and is supposed to be a major determinant of the multidrug resistance (MDR) phenotype (Szakács et al. 2006). Chemotherapeutics extruded by BCRP include topoisomerase inhibitors (topotecan, irinotecan, and its active metabolite SN-38), anthracyclines (doxorubicin and daunorubicin), the anthracene derivative mitoxantrone, and the tyrosine kinase inhibitor imatinib mesylate (Abbott 2006).
We have recently shown that BCRP is predominantly located in lipid rafts (Storch et al. 2007) representing small (10–200 nm) cholesterol- and sphingolipid-enriched highly structured membrane microdomains (Brown 2006). The particular lipid composition of these microdomains accounts for a tight packing resulting in reduced fluidity (liquid ordered phase) compared to the surrounding membrane (liquid disordered phase). A subgroup of lipid rafts are caveolae, which are omega-shaped cell surface invaginations characterized by the presence of special structure proteins, the caveolins (Hnasko and Lisanti 2003). Caveolins can interact with other proteins and modulate their activity by forming heterodimers or heterooligomers. The caveolin gene family comprises three members: caveolin (CAV, encoded by Cav) 1, 2, and 3. CAV1 and CAV2 are expressed in miscellaneous cell types and most abundantly in adipocytes, endothelial cells, and fibroblastic cell types, whereas CAV3 is a muscle-specific protein (Hnasko and Lisanti 2003). The importance of the membrane lipid composition and/or an interaction with CAV1 has been demonstrated for a number of relevant proteins involved in signal transduction (Schwencke et al. 2006), membrane trafficking (Kirkham and Parton 2005), and membrane transport (Orlowski et al. 2006; Hinrichs et al. 2004). Recent co-immunoprecipitation experiments of our group suggested a physical interaction between BCRP and CAV1 (Storch et al. 2007). Moreover, we and others have demonstrated that cholesterol, the most important lipid component of caveolae, modulates BCRP transport activity (Storch et al. 2007; Pál et al. 2007; Telbisz et al. 2007). The mechanism by which cholesterol impacts BCRP activity is yet unclear and it is not known whether the observed physical interaction between CAV1 and BCRP might also be of functional importance. However, this is a fundamental question given the fact that caveolae and caveolar constituents are up-regulated in multidrug-resistant cancer cells (Lavie et al. 1998). We therefore investigated the effect of Cav1 down-regulation on BCRP localization and activity as well as chemotherapeutic resistance against BCRP substrates.
Materials and methods
Materials
Culture media, fetal calf serum (FCS), medium supplements, antibiotics, glutamine, phosphate-buffered saline (PBS), and Lipofectamine™ 2000 were purchased from Invitrogen (Karlsruhe, Germany). Aprotinin, DMSO, polybrene, poly-d-lysine, gelatin, bovine serum albumin, 4′,6-diamidin-2-phenylindol (DAPI), and anti-β-actin (Clone AC-74) were from Sigma-Aldrich (Taufkirchen, Germany). Crystal violet, ethylene diamine tetraacetic acid (EDTA), sodium chloride (NaCl), 2-amino-2-(hydroxymethyl)-propan-1,3-diol (TRIS), Triton® X-100, sodium dodecyl sulfate (SDS), glycerol, Tween®20, β-mercaptoethanol, CaCl2, MgCl2, and paraformaldehyde (PFA) were from AppliChem (Darmstadt, Germany); 96-well microtiter plates and cell culturing bottles were from Nunc (Wiesbaden, Germany); pheophorbide A (PhA (3S-(3alpha,4beta,21beta))-14-ethyl-21-(methoxycarbonyl)-4,8,13,18-tetramethyl-20-oxo-9-vinylphorbine-3-propionic acid)) was from Frontier Scientific Europe (Carnforth, Lancashire, UK); pefabloc and collagen R were from Serva (Heidelberg, Germany); and leupeptin and pepstatin were from Biomol (Hamburg, Germany). Nitrocellulose membranes (Optitran BA-S 85) were obtained from Schleicher & Schuell BioScience (Dassel, Germany). Anti-BCRP antibody (BXP-21) was from Alexis Biochemicals (San Diego, USA), anti-BCRP antibody Alexa Fluor® 647 anti-human ABCG2 (clone 5D3) for immunofluorescence was from Biozol (Eching, Germany), anti-CAV1 antibody (C 37120) from Becton Dickinson (Heidelberg, Germany), and the Cy™5-conjugated AffiniPure donkey anti-mouse IgG (H+L) was from Jackson Immuno Research Laboratories (ordered via Dianova, Hamburg, Germany). Permafluor™ was obtained from Immunotech (Marseille, France). Fumitremorgin C (FTC) was kindly provided by the National Cancer Institute (Rockville, USA).
MDCKII and MDCKII-BCRP cells
MDCKII-BCRP cells were generated by transfection of the canine kidney epithelial cell line MDCKII with the human full-length wild-type ABCG2 cDNA inserted into the LZRS-IRES-GFP expression vector coding also for green fluorescence protein (GFP) (Pavek et al. 2005) and were kindly provided by Dr. A. H. Schinkel (Amsterdam, The Netherlands). The parental cell line MDCKII (available at ATCC, Manassas, USA) was used as a control. Both cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FCS, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Methods
Cav1 knockdown experiments
A retroviral RNAi vector generated by Schuck et al. (2004) was used (pRVH1-puro Cav1). As a control, a scrambled shRNA sequence was cloned into the pRVH1-puro vector (pRVH1-puro scr) (sense: 5′-GATCTCCTGGACCTTTAAAGGGCAAG TTCAAGAGACTTGCCCTTTAAAGGTCCATTTTTTGGAAC; antisense: 5′-TCGAG TTCCAAAAAATGGACCTTTAAAGGGCAAGTCTCTTGAACTTGCCCTTTAAAGGTCCAGGA-3′). Virus production: The human Phoenix gag-pol packaging cell line was kept in high-glucose DMEM (4.5 g/l) containing 10% FCS, 2 mM glutamine, and 100 U/ml penicillin and streptomycin. Approximately 80% confluent cells in six-well plates were transfected with 4 μg of pRVH1-puro Cav1, pRVH1-puro scr, or pRVH1-puro (as a negative control) and 0.4 μg of pVSV-G per well by using Lipofectamine™ 2000 according to the manufacturer’s protocol. Twenty-four hours post transfection, the medium was changed to low-glucose DMEM (1 g/l) with the same supplements as above (1 ml per well) and incubated at 32°C. Forty-eight hours post transfection, the medium was collected, centrifuged for 5 min at 400×g to remove cell debris, and used for infection. Batches of virus-containing supernatant were then collected every 24 h for up to 7 days. The supernatant was passed through a 0.45-μm syringe filter and frozen in liquid nitrogen. Target cell transduction: For infection, 1.5 × 105 cells were mixed with 450 μl of virus-containing supernatant in the presence of 4 mg/ml polybrene, seeded into a well of a 12-well plate, and incubated at 32°C. Twelve hours post infection, the medium was changed to normal culture medium, and cells incubated at 37°C. Thirty-six to 48 h post infection, cells were trypsinized and seeded into a well of a six-well plate in the presence of 10 μM puromycin. Selection was conducted for 48 h. The knockdown efficiency was assayed at the mRNA level by quantitative reverse transcriptase polymerase chain reaction (RT-PCR) and at the protein level by immunoblotting. BCRP activity was quantified by flow cytometry as described below.
RNA isolation and cDNA synthesis
Isolation of total RNA was performed with RNeasy Mini Kit from Qiagen (Hilden, Germany) according to the manufacturer’s instructions. Concentration and purity of the isolated mRNA were determined with a Biophotometer (Eppendorf, Hamburg, Germany). cDNA synthesis was conducted with the Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Science, Mannheim, Germany) according to the manufacturer’s instructions.
RT-PCR
mRNA expression of the ABCG2 and the Cav1 gene was quantified as described and validated previously (Schuck et al. 2004; Albermann et al. 2005) by RT-PCR with the LightCycler™ technology using canine glucuronidase-beta (GU) as housekeeping gene and the RelQuant Software Version 1.01. The software calculates the relative amount of the target gene and the reference gene based on the crossing point. Results were expressed as the target/reference ratio divided by the target/reference ratio of the calibrator. Moreover, the software uses an efficiency corrected calculation by using the determined PCR efficiencies of target and reference genes. The sequences of the primers used for human ABCG2 (Sauerbrey et al. 2002), canine Cav1 (Schuck et al. 2004), and the canine housekeeping gene GU (designed by LightCycler Probe Design software Version 1.0) are displayed in Table 1. For quantification of ABCG2, the FastStart DNA Master SYBR Green I Kit (Roche Applied Science, Mannheim, Germany) was used, and for quantification of Cav-1 and GU, the Absolute SYBR Capillary Mix (ABgene Advanced Biotechnologies, Epsom, Surrey, Great Britain) was used.
Protein assay
Total protein concentrations in cell lysates and in each fraction after sucrose density gradient centrifugation were determined with BCA®Protein Assay Kit from Pierce (Rockford, USA) according to the manufacturer’s instructions.
Cell counting
Cells were counted with an electronic cell counter (CASY®, Schärfe System, Reutlingen, Germany).
Western blot analysis
Knockdown efficiency at the protein level and distribution of proteins in the fractions of the density gradient after ultracentrifugation were analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting. Thirty-two microliters of cell lysate or fraction of the density gradient was mixed with 8 μl 5 × sample buffer (containing Tris–HCl, SDS, β-mercaptoethanol, bromphenol blue, and glycerol) and then subjected to a 12% SDS-PAGE and electrotransferred to nitrocellulose nitrate membranes. Blots were blocked by incubation for 1 h with 5% skim milk (w/v) in phosphate-buffered saline containing 0.1% Tween®20. Immunoblot analysis was carried out with murine monoclonal antibodies raised against human BCRP (BXP-21; diluted 1:250 in TRIS-buffered saline containing 0.1% Tween®20), canine CAV1 (C 37120; diluted 1:500), or β-actin (Clone AC-74; diluted 1:40,000). The blots were then washed extensively and incubated with horseradish peroxidase-linked secondary anti-mouse antibody (Amersham, Freiburg, Germany). Bands were visualized by enhanced chemiluminescence using the SuperSignal®West Pico Chemiluminescent Substrate Kit (Pierce, Rockford, USA).
BCRP/ABCG2 inhibition assay (flow cytometry efflux assay)
The BCRP inhibition assay was performed as described and validated previously (Storch et al. 2007). In brief, 106 cells were suspended in 500 μl incubation medium (RPMI with 2% FCS) containing 1 μM PhA and incubated at 37°C for 30 min on a rotary shaker (450 rpm). Cells were then washed once with 1 ml ice-cold incubation medium and resuspended in 500 μl incubation medium containing 10 μM of the specific BCRP inhibitor FTC (Rabindran et al. 1998). After incubation for 60 min at 37°C on a rotary shaker, cells were washed with 1 ml ice-cold PBS with 2% FCS, resuspended in ice-cold PBS with 2% FCS, and kept on ice until flow cytometry. Intracellular fluorescence was analyzed in a Becton Dickinson LSR II flow cytometer with a solid state coherent sapphire blue laser and a 530 bandpass filter for GFP and a 633-nm helium/neon laser and a 660 bandpass filter for PhA.
In each sample, 30,000 cells were counted. Cell debris was eliminated by gating the viable cells in the forward versus side scatter. BCRP-positive MDCKII-BCRP cells were additionally gated using their GFP signal. To quantify the activity of BCRP, the ratio between the MF with inhibitor (FTC) and without inhibitor during the efflux period was calculated and normalized to the effect observed in the corresponding parental cell line (Storch et al. 2007). Flow cytometry experiments were performed at least three times in untreated and transduced cells in independent experiments.
Proliferation assays
Cell proliferation was quantified by staining with crystal violet as described previously (Peters et al. 2006). In brief, 100 μl of each cell suspension at 5 × 105 cells/ml density was seeded onto collagen-coated 96-well microtiter plates and preincubated for 24 h. After addition of test compounds, cells were incubated for a further 48 h. Cells were washed with PBS and viable adherent cells were stained with crystal violet 0.5% (2.5 g in 100 ml methanol, diluted with 400 ml aqua bidest) for 15 min on a rotary shaker at 450 rpm and room temperature. The plates were washed with aqua bidest and dried for 4 h in a drying chamber at 37°C. To dissolve the dye, 200 μl/well methanol was added. Absorption was measured using a Multiskan RC photometer (ThermoLabsystems, Helsinki, Finland) with 555 nm excitation. Proliferation was expressed as proliferation index by calculating crystal violet absorption intensity as percentage relative to baseline (absorption intensity of medium-only wells [set to 0%]) and native proliferation (absorption intensity of untreated cells [set to 100%]). Each experiment was performed at least in triplicate with n = 6 wells for each concentration on different days. IC50 values were calculated to compare sensitivity of the cells to chemotherapeutics.
Isolation of detergent-resistant membranes (DRMs)
To investigate, whether Cav1 knockdown leads to displacement of BCRP within the plasma membrane, DRMs were isolated as described previously (Storch et al. 2007). Briefly, the cell pellet of four cell culture flasks (175 cm²; 80% confluence) was washed once with PBS and then lysed on ice in 2 ml TNE buffer (25 mM TRIS, 150 mM NaCl, 5 mM EDTA, pH 7.5), containing a mixture of protease inhibitors (1 mg/ml pefabloc, 5 μg/ml leupeptin, 1 μg/ml pepstatin, and 1 μg/ml aprotinin). Cells were homogenized 15 times through a 22-gauge needle. The total cell lysate was centrifuged at 3,000 rpm for 5 min at 1°C. Twenty microliters of Triton® X-100 (corresponding to 1% (v/v)) were added to the supernatant (2 ml). The extract was adjusted to 40% sucrose by adding 2 ml of an ice-cold solution of 80% sucrose in TNE buffer. This volume was placed at the bottom of an ultracentrifuge tube, and a discontinuous gradient was formed by adding 6 ml of an ice-cold 30% sucrose solution and 2 ml of ice-cold TNE buffer. After ultracentrifugation at 190,000×g for 18 h at 4°C in a SW-41 rotor, the gradient was fractionized into 12 fractions of 1 ml each (fraction 1 = bottom of the gradient, fraction 12 = top of the gradient). The fractions were kept on ice until analysis. For western blot analysis, equal amounts (16 μl) of each fraction were loaded onto the gel. Isolation of DRMs was performed in triplicate with MDCKII-BCRP pRVH1-puro and MDCKII-BCRP pRVH1-puro Cav1 cells.
Immunofluorescence procedure
To investigate whether the knockdown of Cav1 leads to an internalization of BCRP from the cell membrane to intracellular compartments, immunofluorescence experiments with untreated and treated MDCKII-BCRP cells were performed. Cells were seeded in 12-well plates on glass cover slips (13 mm diameter). Confluent monolayers were washed once with PBS containing 0.1 mM CaCl2 and 0.1 mM MgCl2 (PBS (+)) to preserve tight junctions and fixed with 3% PFA in PBS (+) for 30 min. PFA was quenched by incubating with 50 mM NH4Cl solution for 10 min. An additional neutralization step with 0.2% gelatin/PBS (+) for 10 min was added. Afterwards, the cover slips were washed three times for 5 min with PBS (+). After blocking for 30 min with 0.2% gelatin/PBS (+), the cell monolayer was incubated for 1 h with 30 μl of primary antibody (1:10 dilution of Alexa Fluor® anti-human ABCG2 clone 5D3 in 0.2% gelatin/PBS (+)) or 30 μl 0.2% gelatin/PBS (+) for negative control in a moisture box at room temperature. Cover slips were washed three times for 5 min with 0.2% gelatin/PBS (+) and then incubated for 1 h with 30 μl of secondary antibody (1:800 dilution of Cy™5-conjugated AffiniPure donkey anti-mouse IgG (H+L) in 0.2% gelatin/PBS (+)) in a moisture box. Cover slips were washed with 0.2% gelatin/PBS (+) for 5 min, then three times with PBS (+). For nucleus staining, the cells were incubated for 20 min with 30 μl of DAPI (1:500 dilution in aqua bidest) and washed three times with PBS (+). Afterwards, the cover slips were embedded with aqueous mounting medium on glass slides. BCRP and DAPI staining were analyzed with a DM IRE 2 TCS SP II confocal laser scanning microscope from Leica (Bensheim, Germany). For BCRP detection, a 633 He/Ne laser was used (25%, acousto optic tunable filter (AOTF) 750 V, offset −0.9 V) and for DAPI a 405 UV diode laser was used (15%, AOTF 750 V, offset −1.5 V). The objective used was a Leica HCX PL APO CS ×63 with an index of 1.4.
Statistical analysis
Data were analyzed using GraphPad Prism® Version 5.0 and InStat Version 3.05 (GraphPad Software, San Diego, CA, USA). ANOVA with Dunnett’s post hoc test was used for the comparison of Cav1 and ABCG2 mRNA expression in untreated cells and in cells after transduction, and ANOVA with Bonferroni’s multiple comparison post hoc test was applied for the comparison of IC50 values. Inhibition ratios of untreated and transduced cells were assessed with Mann–Whitney U test. Results are displayed as mean ± SD. A p value of ≤0.05 was considered significant.
Results
Verification and quantification of Cav1 knockdown efficiency
Knockdown of Cav1 by RNAi led to a significant reduction in Cav1 mRNA levels in both the parental cell line MDCKII and the BCRP/ABCG2 overexpressing cell line MDCKII-BCRP (Fig. 1). Cav1 mRNA levels in cells only transduced with the control vectors and untreated cells were similar. Knockdown efficiency was between 85% and 95% and stable over at least 30 days. ABCG2 mRNA levels in transduced and untreated cells were similar indicating that knockdown of Cav1 had no impact on ABCG2 mRNA levels (mRNA ABCG2/GU) in MDCKII-BCRP cells transduced with pRVH1-puro normalized to \( {\hbox{untreated control}} = 0.{6}\pm 0.{2} \); p > 0.05; mRNA ABCG2/GU in MDCKII-BCRP cells transduced with pRVH1-puro cav-1 normalized to \( {\hbox{untreated control}} = {1}.0\pm 0.{3} \); p > 0.05). Western blot analysis confirmed that CAV1 protein expression was significantly reduced in Cav1 knockdown cells and that transduction with the empty vector pRVH1-puro did not reduce CAV1 expression, but slightly increased it (Fig. 2). Transduction of MDCKII and MDCKII-BCRP cells with the scrambled control vector had no effect on CAV1 protein expression (data not shown).

Cav1 mRNA expression after transduction with anti-caveolin-1 RNAi vector (pRVH1-puro Cav1), empty vector (pRVH1-puro), or scrambled vector (pRVH1-puro scr) as negative controls. White bars: parental cell line MDCKII (17 days post transduction), black bars: BCRP overexpressing cell line MDCKII-BCRP (22 days post transduction). Results are displayed as mean ± SD of three independent experiments and are normalized to the untreated corresponding cell line. **p < 0.001. GU glucuronidase β

Representative CAV1 and BCRP western blot analysis in untreated cells and after transduction with anti-caveolin-1 RNAi vector (pRVH1-puro Cav1) and empty vector (pRVH1-puro) as a negative control. β-actin served as loading control
Impact of Cav1 knockdown on BCRP/ABCG2 protein expression
Knockdown of Cav1 did not change BCRP protein expression as demonstrated by western blot analysis (Fig. 2).
Impact of Cav1 knockdown on BCRP/ABCG2 transport activity
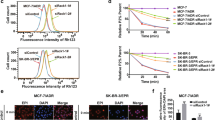
To investigate the influence of Cav1 knockdown on BCRP activity, BCRP transport activity measured by flow cytometry was compared between MDCKII-BCRP pRVH1-puro Cav1 cells and control cells. BCRP activity was significantly reduced by about 35% (p < 0.05) in Cav1 knockdown cells (\( {\hbox{inhibition ratio}} = {4}.{4}\pm 0.{8} \) in MDCKII-BCRP and 2.9 ± 0.9 in MDCKII-BCRP pRVH1-puro Cav1 cells), whereas in cells transduced with the empty vector pRVH1-puro or with the scrambled control pRVH1-puro scr BCRP/ABCG2, transport activity was unchanged (Fig. 3).

BCRP activity in untreated cells and after transduction with anti-caveolin-1 RNAi vector (pRVH1-puro Cav1) and the control vectors (pRVH1-puro and pRVH1-puro scr) as negative controls measured by PhA efflux assay. Overlay of flow cytometry histograms of cells incubated with 1 μM PhA in the absence (black) or presence of 10 μM of the selective BCRP inhibitor FTC (gray). A shift in fluorescence to higher values is provoked by PhA retention in the cells indicating inhibition of the efflux (by BCRP). BCRP activity was not influenced by any treatment in the parental cell line and by transduction with the control vectors in the MDCKII-BCRP cell line but significantly reduced (p < 0.05) in Cav1 knockdown MDCKII-BCRP cells. Depicted is one example (with 30,000 cells measured for each cell line) of a series of n = 3–6 independent experiments
Impact of Cav1 knockdown on chemotherapeutic sensitivity
Growth inhibition assays in untreated cells and in cells that had been transduced with pRVH1-puro Cav1, pRVH1-puro, or pRVH1-puro scr control vectors were performed to assess the sensitivity against a well-established BCRP chemotherapeutic substrate (mitoxantrone). If the knockdown of Cav1 has a negative impact on BCRP transport activity, Cav1 knockdown MDCKII-BCRP cells should be less resistant to antiproliferative effects provoked by a chemotherapeutic which is a BCRP substrate. Since mitoxantrone is not only a substrate of BCRP but also of P-glycoprotein (P-gp/ABCB1), proliferation assays with vincristine, which is not transported by BCRP but by P-gp, were performed as negative control.
Proliferation assays with mitoxantrone (Fig. 4) in untreated cells demonstrate that BCRP overexpression mediates mitoxantrone resistance (IC50 5.5 ± 1.1 μM in MDCKII cells and 53.7 ± 6.2 μM in MDCKII-BCRP cells, p < 0.0001). Knockdown of Cav1 in MDCKII-BCRP cells significantly reduced resistance of the cells against mitoxantrone (\( {\hbox{I}}{{\hbox{C}}_{{5}0}} = {7}.{2}\pm 0.{8} \); p < 0.001 vs. untreated MDCKII-BCRP cells). In the parental cell line, which served as a negative control, knockdown of Cav1 had no effect on mitoxantrone resistance. Transduction with the empty vector pRVH1-puro provoked a slight increase in mitoxantrone resistance in the MDCKII-BCRP cell line (\( {\hbox{I}}{{\hbox{C}}_{{5}0}} = {7}0.0\pm {11}.{3} \); p < 0.05), but had no significant effect on mitoxantrone resistance in the parental cell line. Transduction with the scrambled control vector pRVH1-puro scr had no effect on mitoxantrone resistance in the MDCKII-BCRP or parental cell line (data not shown).

Proliferation assay in the presence of mitoxantrone. Open symbols: parental cell line, black symbols: MDCKII-BCRP. Results are displayed as mean ± SD of n = 3 independent experiments with each drug concentration tested in octuplet
In contrast, the sensitivity of MDCKII-BCRP cells and the parental cell line MDCKII towards the P-gp substrate vincristine was similar, and the knockdown of Cav1 did not alter cellular resistance to vincristine in any cell line (Fig. 5). Transduction with the empty vector pRVH1-puro or pRVH1-puro scr (data not shown) also had no effect on resistance towards vincristine in any cell line. These results indicate a profound influence of CAV1 on BCRP-mediated drug resistance of tumor cells.

Proliferation assay in the presence of the P-gp substrate vincristine. Open symbols: parental cell line, black symbols: MDCKII-BCRP. Results are displayed as mean ± SD of n = 3 independent experiments with each drug concentration tested in octuplet
Characterization of density gradient ultracentrifugation fractions in Cav1 knockdown and control cells
DRMs were isolated and analyzed to investigate whether Cav1 knockdown leads to displacement of BCRP within the plasma membrane. Western blot analyses of density gradient ultracentrifugation fractions in MDCKII-BCRP pRVH1-puro Cav1 and the control cell line MDCKII-BCRP pRVH1-puro are displayed in Fig. 6 (one representative experiment out of three). The DRM fraction was found at the interface between 30% sucrose and TNE buffer as a cloudy band. BCRP was predominantly located in the DRM (raft) fractions (fractions 9, 10, and 11) and co-localized with CAV1. In the knockdown cell line, CAV1 expression was nearly abolished while the localization of BCRP within the membrane was similar to that in MDCKII-BCRP cells transduced with the control vector: Only a slight shift from raft to non-raft fraction was detected (Fig. 6b).

Representative CAV1 and BCRP western blot analysis of gradient ultracentrifugation fractions of MDCKII-BCRP pRVH1-puro (a) and MDCKII-BCRP pRVHA-puro Cav1 (b) cells. 1 = bottom of the ultracentrifugation tube, 12 = top of the ultracentrifugation tube. Of each fraction, equal amounts (16 μl) were loaded onto the gel
Immunofluorescence in Cav1 knockdown and control cells
Immunofluorescence of knockdown and control MDCKII-BCRP cells demonstrated that after Cav1 knockdown there was no evidence for changes in localization of BCRP (Fig. 7).

Immunofluorescence images of MDCKII-BCRP cells: a untreated cells and b the corresponding negative control without primary antibody; c cells transduced with the pRVH1-puro vector and d the corresponding negative control; e cells transduced with the pRVH1-puro scr vector and f the corresponding negative control; g Cav1 knockdown cells and h the corresponding negative control
Discussion
BCRP and CAV1 play an important role in drug resistance of cancer cells. MDR cancer cells often express very high levels of CAV1 and the surface density of caveolae is high (Lavie et al. 2001). Moreover, CAV1 is linked to an aggressive phenotype of cancer cells in different tumor entities (Shatz and Liscovitch 2008). The mechanisms, however, on how CAV1 affects cancer cell phenotype and ultimately survival, are still poorly understood.
BCRP is one important ABC transporter mediating MDR and like CAV1 also BCRP is a prognostic marker in certain tumors (Diestra et al. 2002; Candeil et al. 2004; Yoh et al. 2004; Benderra et al. 2005). In addition, it is frequently expressed in stem cell populations including cancer stem cells where it is believed to protect cancer tissues from chemotherapeutic drugs (Dean et al. 2005).
We have recently demonstrated that BCRP co-localizes with CAV1 in DRMs, and co-immunoprecipitation experiments suggest a physical and possibly functional interaction between BCRP and CAV1 (Storch et al. 2007). We therefore aimed to clarify whether CAV1 is of functional importance for the transport activity of BCRP and for drug resistance mediated by BCRP.
We opted for retrovirus-mediated knockdown of Cav1 to obtain high and long-lasting knockdown efficiency. Our results clearly reproduce the results of Schuck et al. (2004), confirming the efficiency of the pRVH1-puro Cav1 vector in the Cav1 knockdown. The resulting reduced Cav1 expression had two functional consequences in MDCKII-BCRP cells: (1) BCRP function was significantly decreased as demonstrated by PhA efflux assay and (2) resistance towards the BCRP substrate mitoxantrone was reduced as tested by proliferation assays. This effect can be attributed to Cav1 knockdown because transduction with the empty control vector even slightly increased the resistance in MDCKII-BCRP cells possibly due to the slightly increased CAV1 protein expression (Fig. 2), and transduction with the scrambled control vector did not change resistance towards mitoxantrone. Moreover, transduction of the parental cell line with the control vectors or with the pRVH1-puro Cav1 vector had no influence on the sensitivity towards mitoxantrone. Because mitoxantrone is both a BCRP and a P-gp substrate, we confirmed the selectivity of the effect by testing the sensitivity of untreated and transduced cells towards the P-gp substrate vincristine. In MDCKII-BCRP cells, transduction with the control vectors or with pRVH1-puro Cav1 did not change the sensitivity towards vincristine, suggesting that the mitoxantrone effects are specific for BCRP.
Western blot analysis of total cell lysates confirmed that BCRP protein expression is not changed after Cav1 knockdown, and western blot analysis of the gradient ultracentrifugation fractions indicates that Cav1 knockdown neither displaces BCRP within the membrane nor extracts the transporter from the plasma membrane because the highest concentration of BCRP was still found in the DRM fraction. Therefore, the diminished BCRP function after Cav1 knockdown is likely caused by a disruption of the protein–protein interaction between CAV1 and BCRP. Interestingly, in contrast to BCRP, for P-gp all published data so far indicate that CAV1 negatively regulates this ABC transporter leading to increased P-gp function in Cav1 knockdown cells and to diminished P-gp function and higher sensitivity toward chemotherapeutics in cells with CAV1 overexpression (Cai and Chen 2004; Barakat et al. 2008). This, however, implies that Cav1 overexpression, which is observed in many MDR cells, would partly compensate resistance towards cytostatic drugs rather than increasing MDR.
The limitations of this study are as follows: (1) In our cell model, the interaction between canine CAV1 and human BCRP was investigated. However, bioinformatic comparison revealed that CAV1 is a highly conserved protein in mammalians, with a protein sequence identity of 96.6% between Homo sapiens and Canis familiaris (www.ncbi.nlm.nih.gov/homologene). Moreover, the putative binding region for BCRP, the scaffolding domain, features 100% sequence identity between the two species (Couet et al. 1997). Therefore, it can be assumed that these results can be extrapolated to humans. (2) It has to be considered that not only direct interaction between CAV1 and BCRP, but also indirect effects might lead to effects of Cav1 knockdown on BCRP transport activity. CAV1 has been demonstrated to bind to cholesterol (Murata et al. 1995) and it facilitates the transport of newly synthesized cholesterol from the endoplasmatic reticulum to the plasma membrane (Smart et al. 1996). Our previous study associated the cholesterol content of the plasma membrane with the activity of BCRP (Storch et al. 2007). It might therefore be possible that Cav1 knockdown leads to decreased availability of cholesterol in the caveolae, thus provoking reduced BCRP activity. (3) We have used a cell model with profound overexpression of BCRP. It still has to be demonstrated that the effects observed also translate to cells with lower expression levels of BCRP.
In conclusion, our study argues for a positive regulation of BCRP function by CAV1. Cav1 knockdown leads to diminished activity of BCRP and to a reduction of MDR towards the BCRP substrate mitoxantrone. These findings may be relevant for the understanding of the role of CAV1 in tumor progression and modulation of the MDR phenotype. Indeed, our findings suggest that CAV1 expression might be a more relevant marker for MDR because it may impair BCRP activity even when large amounts of this transporter are expressed. Hence, high expression of BCRP might be irrelevant in the absence of CAV1.
Abbreviations
- ABC:
-
ATP-binding cassette
- AOTF:
-
Acousto optic tunable filter
- BCRP:
-
Breast cancer resistance protein
- Cav:
-
Caveolin
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- DMSO:
-
Dimethyl sulfoxide
- DRM:
-
Detergent-resistant membrane
- EDTA:
-
Ethylene diamine tetraacetic acid
- FTC:
-
Fumitremorgin C
- GFP:
-
Green fluorescence protein
- MDCK:
-
Madin–Darby canine kidney
- MDR:
-
Multidrug resistance
- MF:
-
Median fluorescence
- MRP:
-
Multidrug resistance-related protein
- PBS:
-
Phosphate-buffered saline
- PBS (+):
-
PBS containing 0.1 mM CaCl2 and 0.1 mM MgCl2
- PFA:
-
Paraformaldehyde
- P-gp:
-
P-glycoprotein
- PhA:
-
Pheophorbide A (3S-(3alpha,4beta,21beta))-14-ethyl-21-(methoxycarbonyl)-4,8,13,18-tetramethyl-20-oxo-9-vinylphorbine-3-propionic acid)
- RNAi:
-
RNA interference
- SDS:
-
Sodium dodecyl sulfate
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TNE buffer:
-
TRIS–NaCl–EDTA buffer
- TRIS:
-
2-Amino-2-(hydroxymethyl)-propane-1,3-diol
References
Abbott BL (2006) ABCG2 (BCRP) a cytoprotectant in normal and malignant stem cells. Clin Adv Hematol Oncol 4:63–72
Albermann N, Schmitz-Winnenthal FH, Z’graggen K, Volk C, Hoffmann MM, Haefeli WE, Weiss J (2005) Expression of the drug transporters MDR1/ABCB1, MRP1/ABCC1, MRP2/ABCC2, BCRP/ABCG2, and PXR in peripheral blood mononuclear cells and their relationship with the expression in intestine and liver. Biochem Pharmacol 70:949–958
Barakat S, Turcotte S, Demeule M, Lachambre M-P, Régina A, Baggetto LG, Béliveau R (2008) Regulation of brain endothelial cell migration and angiogenesis by P-glycoprotein/caveolin-1 interaction. Biochem Biophys Res Commun 372:440–446
Benderra Z, Faussat AM, Sayada L, Perrot JY, Tang R, Chaoui D, Morjani H, Marzac C, Marie JP, Legrand O (2005) MRP3, BCRP, and P-glycoprotein activities are prognostic factors in adult acute myeloid leukemia. Clin Cancer Res 11:7764–7772
Brown D (2006) Lipid rafts, detergent-resistant membranes, and raft targeting signals. Physiology 21:430–439
Cai C, Chen J (2004) Overexpression of caveolin-1 induces alteration of multidrug resistance in Hs578T breast adenocarcinoma cells. Int J Cancer 111:522–529
Candeil L, Gourdier I, Peyron D, Vezzio N, Copois V, Bibeau F, Orsetti B, Scheffer GL, Ychou M, Khan QA, Pommier Y, Pau B, Martineau P, Del Rio M (2004) ABCG2 overexpression in colon cancer cells resistant to SN38 and in irinotecan-treated metastases. Int J Cancer 109:848–854
Couet J, Shengwen L, Okamoto T, Ikezu T, Lisanti MP (1997) Identification of peptide and protein ligands for the caveolin-scaffolding domain. J Biol Chem 272:6525–6533
Dean M, Rzhetsky A, Allikmets R (2001) The human ATP-binding cassette (ABC) transporter superfamily. Genome Res 11:1156–1166
Dean M, Fojo T, Bates S (2005) Tumour stem cells and drug resistance. Nat Rev Cancer 5:275–284
Diestra JE, Scheffer GL, Català I, Maliepaard M, Schellens JH, Scheper RJ, Germà-Lluch JR, Izquierdo MA (2002) Frequent expression of the multi-drug resistance-associated protein BCRP/MXR/ABCP/ABCG2 in human tumours detected by the BXP-21 monoclonal antibody in paraffin-embedded material. J Pathol 198:213–219
Hinrichs JW, Klappe K, Hummel I, Kok JW (2004) ATP-binding cassette transporters are enriched in non-caveolar detergent-insoluble glycosphingolipid-enriched membrane domains (DIGs) in human multidrug-resistant cancer cells. J Biol Chem 279:5734–5738
Hnasko R, Lisanti MP (2003) The biology of caveolae: lessons from caveolin knockout mice and implications for human disease. Mol Interv 3:445–464
Gillet JP, Efferth T, Remacle J (2007) Chemotherapy-induced resistance by ATP-binding cassette transporter genes. Biochim Biophys Acta 1775:237–262
Kirkham M, Parton RG (2005) Clathrin-independent endocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophys Acta 1746:350–363
Krishnamurthy P, Schuetz JD (2006) Role of ABCG2/BCRP in biology and medicine. Annu Rev Pharmacol Toxicol 46:381–410
Lavie Y, Fiucci G, Liscovitch M (1998) Up-regulation of caveolae and caveolar constituents in multidrug-resistant cancer cells. J Biol Chem 273:32380–32383
Lavie Y, Fiucci G, Liscovitch M (2001) Upregulation of caveolin in multidrug resistance cancer cells: functional implications. Adv Drug Deliv Rev 49:317–323
Mao Q, Unadkat JD (2005) Role of the breast cancer resistance protein (ABCG2) in drug transport. AAPS J 7:E118–E133
Murata M, Peranen J, Schreiner R, Weiland F, Kurzchalia T, Simons K (1995) VIP21/caveolin is a cholesterol-binding protein. Proc Natl Acad Sci USA 92:10339–10343
Orlowski S, Martin S, Escargueil A (2006) P-glycoprotein and ‘lipid rafts’: some ambiguous mutual relationships (floating on them, building them or meeting them by chance?). Cell Mol Life Sci 63:1038–1059
Pál Á, Méhn D, Molnár É, Gedey S, Mészáros P, Nagy T, Glavinas H, Janáky T, von Richter O, Báthori G, Szente L, Krajcsi P (2007) Cholesterol potentiates ABCG2 activity in a heterologous expression system—improved in vitro model to study function of human ABCG2. J Pharmacol Exp Ther 321:1085–1094
Pavek P, Merino G, Wagenaar E, Bolscher E, Novotna M, Jonker JW, Schinkel AH (2005) Human breast cancer resistance protein: interactions with steroid drugs, hormones, the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo(4, 5-b)pyridine, and transport of cimetidine. J Pharmacol Exp Ther 312:144–152
Peters T, Lindenmaier H, Haefeli WE, Weiss J (2006) Interaction of the mitotic kinesin Eg5 inhibitor monastrol with P-glycoprotein. Naunyn-Schmiedeberg’s Arch Pharmacol 372:291–299
Rabindran SK, He H, Singh M, Brown E, Collins KI, Annable T, Greenberger LM (1998) Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res 58:5850–5858
Sauerbrey A, Sell W, Steinbach D, Voigt A, Zintl F (2002) Expression of the BCRP gene (ABCG2/MXR/ABCP) in childhood acute lymphoblastic leukaemia. Br J Haematol 118:147–150
Schuck S, Manninen A, Honsho M, Füllekrug J, Simons K (2004) Generation of single and double knockdowns in polarized epithelial cells by retrovirus-mediated RNA interference. Proc Natl Acad Sci USA 101:4912–4917
Schwencke C, Braun-Dullaeus RC, Wunderlich C, Strasser RH (2006) Caveolae and caveolin in transmembrane signaling: implications for human disease. Cardiovasc Res 70:42–49
Shatz M, Liscovitch M (2008) Caveolin-1: a tumor-promoting role in human cancer. Int J Radiat Biol 84:177–189
Smart EJ, Ying YS, Donzell WC, Anderson RGW (1996) A role for caveolin in transport of cholesterol from the endoplasmatic reticulum to plasma membrane. J Biol Chem 271:29427–29435
Storch CH, Ehehalt R, Haefeli WE, Weiss J (2007) Localization of the human breast cancer resistance protein (BCRP/ABCG2) in lipid rafts/caveolae and modulation of its activity by cholesterol in vitro. J Pharmacol Exp Ther 323:257–264
Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM (2006) Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5:219–234
Telbisz A, Müller M, Özvegy-Laczka C, Homolya L, Szente L, Váradi A, Sarkadi B (2007) Membrane cholesterol selectively modulates the activity of the human ABCG2 multidrug transporter. Biochim Biophys Acta 1768:2698–2713
Yoh K, Ishii G, Yokose T, Minegishi Y, Tsuta K, Goto K, Nishiwaki Y, Kodama T, Suga M, Ochiai A (2004) Breast cancer resistance protein impacts clinical outcome in platinum-based chemotherapy for advanced non-small cell lung cancer. Clin Cancer Res 10:1691–1697
Acknowledgments
We thank Dr. A. H. Schinkel (Amsterdam, The Netherlands) for generously providing the cell line MDCKII-BCRP and the National Cancer Institute (Rockville, USA) for providing FTC. Moreover, we would like to thank Richard Sparla for his help with transfection and transduction.
The project was funded by the Wilhelm-Sander Stiftung (#2008.005.1). Caroline Henrike Storch was supported by a fellowship of the Ernst Schering Foundation for doctoral students.
Author information
Authors and Affiliations
Corresponding author
Additional information
Melanie Herzog and Caroline Henrike Storch contributed equally to this work.
Rights and permissions
About this article
Cite this article
Herzog, M., Storch, C.H., Gut, P. et al. Knockdown of caveolin-1 decreases activity of breast cancer resistance protein (BCRP/ABCG2) and increases chemotherapeutic sensitivity. Naunyn-Schmied Arch Pharmacol 383, 1–11 (2011). https://doi.org/10.1007/s00210-010-0568-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-010-0568-8