
Abstract
The purpose of this study was to apply the target-mediated drug disposition (TMDD) pharmacokinetic (PK) model to describe binding, internalization, and turnover of erythropoietin receptor (EPOR). This model allows one to determine from free drug (C) PK data not only parameters describing linear disposition of EPO such as the elimination rate constant (k el) and volume of distribution (V c), but also the total receptor concentration (R tot0), drug–receptor complex (RC) internalization rate constant (k int), as well as synthesis and degradation rate constants (k syn and k deg) for the receptor turnover. The previously published data on PK of recombinant EPO (rHuEPO) in humans and the results of EPOR binding studies were used for analysis. The estimated PK parameters were used to simulate time courses of free and bound EPOR after IV administration of clinically relevant rHuEPO doses. The estimates of k el = 0.106 h−1 and V c = 0.032 l/kg are consistent with reported in the literature values of rHuEPO linear disposition parameters. The determined value of R tot0 was 66.35 pM and the half-life for EPOR degradation was 8.8 h. Computer simulations showed a very rapid binding phase in the EPOR time profile followed by a decline to a nadir, and a subsequent return to the baseline. The nadir values decreased with increasing doses and resulted in the maximum values of the bound fractions of the total EPOR in the ranges 33–99%. At the baseline conditions, only 3.1% of EPOR were occupied. The saturation of EPOR was correlated with the time C remained above the K D level. In conclusion, the time courses of serum rHuEPO concentrations contain information about internalization and turnover of EPOR. Kinetics of EPOR can be utilized to determine the relationship between the pharmacologic effect and exposure to rHuEPO.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Erythropoietin (EPO) is a 30.4-kD glycoprotein hormone that is the primary regulator of red blood cell production. EPO is predominantly produced by peritubal cells in the kidneys in response to tissue hypoxia. For clinical use, EPO is obtained by recombinant DNA technology. Recombinant human erythropoietin (rHuEPO) is derived from a cloned human EPO gene and it has the same 165 amino acid sequence that differs only in the glycosylation pattern. rHuEPO has been indicated for the treatment of renal failure anemia as well as anemias induced by chemotherapy of cancer and acquired immunodeficiency syndrome (AIDS) patients. rHuEPO stimulates production of red blood cells by binding to erythropoietin receptors expressed on progenitor cells in bone marrow and initiating intracellular signaling pathways leading to inhibition of cell apoptosis and enhancement of proliferation and differentiation (Fisher 2003).
Erythropoietin receptor (EPOR) is a 185-kD member of the class 1 cytokine receptor superfamily. It is expressed mainly on erythroid progenitor cells, but also on epicardium, neurons, liver, gut, and endothelium. EPOR homodimerizes upon binding to EPO and activates JAK2 tyrosine kinase. Subsequently, different intracellular pathways are activated including Ras/MAP kinase, phosphatidylinositol 3-kinase and STAT signaling pathways (Lacombe and Mayeux 1998). However, the exact mechanisms by which the proliferation and/or differentiation of erythroid cells are regulated after EPO stimulation are not known. EPOR expressed on human erythroblasts exhibit a single high-affinity (K D 100 pM) binding site for EPO and 100–300 receptors per cell (Broudy et al. 1991). On the other hand, high-affinity (K D ∼ 1 nM) and low-affinity (K D ∼ 1 μM) binding sites for EPO have been demonstrated in the soluble extracellular domain of EPOR (Philo et al. 1996). Target disruption of both EPO and EPOR showed that EPO is not involved in the commitment of the erythroid lineage and it seems to act mainly as a survival factor. EPO binding to its receptor induces ubiquitination, internalization, and degradation of EPOR by proteosomes and lysosomes. Few or any EPOR are recycled back to the membrane (Walrafen et al. 2005).
Pharmacologic target-mediated drug disposition (TMDD) is a term used to describe the phenomenon in which a drug is bound with high affinity to its pharmacologic target site (e.g., receptor) such that this interaction is reflected in the pharmacokinetic (PK) characteristics of the drug (Levy 1994). Drug-receptor binding and dissociation, endocytotic internalization followed by degradation in lysosomes are the major components of such TMDD PK models (Sugiyama and Hanano 1989). A general pharmacokinetic model for drugs exhibiting TMDD has been developed (Mager and Jusko 2001). This model describes drug plasma concentrations in terms of first-order elimination and distribution processes combined with non-linear saturable receptor binding clearance augmented with drug-receptor complex internalization and receptor turnover. Receptor-mediated disposition has been postulated as a major clearance pathway for rHuEPO (Chapel et al. 2001).
The objective of this report is to determine PK of EPOR upon intravenous administration of rHuEPO to healthy volunteers. An approach taken is to apply a TMDD PK model to describe serum rHuEPO concentrations, obtain the model parameters, and use computer simulations to predict the time courses of EPOR expression (PK) as well as concentration of the EPO–EPOR complex, an entity initiating pharmacological effect (PD).
Materials and methods
PK model of receptor-mediated disposition of rHuEPO
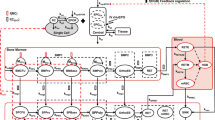
A previously introduced general TMDD PK model has been adopted to describe the disposition of rHuEPO (Mager and Jusko 2001). Its schematic diagram is shown in Fig. 1. Briefly, the model consists of two compartments: serum (C) and nonspecific tissue binding site (A T) that account for linear drug disposition. rHuEPO in the central compartment can be eliminated at the first-order rate k el or bind nonspecifically to tissues at first-order rates k pt and k tp. Additionally, free rHuEPO can bind to free EPOR (R) at the second-order rate constant k on forming a drug–receptor complex RC. The bound drug can dissociate from its receptor at the first-order rate k off or be internalized at the first-order rate k int via endocytosis. The receptor turnover is described by the zero-order synthesis rate k syn and first-order degradation rate k deg. The EPO endogenous production is described by the zero-order rate constant k EPO. Free drug C, free receptor R, and drug–receptor complex RC are expressed in molar concentrations, whereas A T denotes amount (moles) of nonspecifically tissue bound drug. The model equations are as follows (Mager and Jusko 2001):
where V c denotes the volume of distribution for rHuEPO. The initial conditions for the above system are defined by the steady-state (baseline) values:
assuming that the free drug is administered as an IV bolus dose. At the steady-state there are no changes in the model variables, and consequently the following relationships hold:

Schematic diagram of the TMDD PK model. The model variables, parameters, and processes they describe are explained in Materials and methods
PK data
The TMDD PK model Eqs. 1-5 was used to fit the mean rHuEPO serum concentrations after IV bolus injection of epoetin beta to healthy volunteers. The data were obtained from Flaharty et al. (1990). Single doses of 10, 50, 150, 500, and 1,000 IU/kg were intravenously injected to healthy men. Samples of whole blood were collected just before and at 0.25, 0.5, 1, 2, 4, 8, 12, 16, 24, and 48 h. Serum erythropoietin concentrations were determined by a radioimmunoassay. Because the original mean data were corrected by the endogenous EPO levels, the baseline value of C 0 = 15 IU/l was added to the concentrations (Cheung et al. 1998). The conversion from IU/l to pM was made assuming that 7.7 μg of rHuEPO is equivalent to 1,000 IU and the molecular weight of rHuEPO of 30.4 kD (Jelkmann 1992).
Estimation of model parameters
Because the TMDD PK model contained a number of parameters that could not be resolved by fitting the model to the data, results from in vitro receptor–ligand binding studies have been used to assign some of the receptor-binding parameters values. The binding constant k off = 10.1 h−1 has been obtained from studies on binding kinetics of hexahistidine-tagged EPOR (EPOR-6H) anchored to a nitrilotriacetic acid ditetradecylamine membrane sprayed on a BIAcore sensor interacting with rHuEPO (Altin et al. 2001). The K D value of 100 pM has been reported for the high affinity EPO binding site on EPOR (Broudy et al. 1991). The relationship k on = k off / K D provided the value of k on = 0.101 pM−1 h−1. The internalization rate constant k int = 2 h−1 was obtained from the initial slope of the radioactive signal vs time curve describing degradation of 125I-rHuEPO from the membrane of Friend Virus-infected erythroid cells (Sawyer et al. 1987). The parameters actually estimated by fitting the data were k el, V c, k pt, k tp, R 0, and k deg. The remaining model parameters (secondary parameters) were calculated from the baseline relationships Eqs. 6–9. The estimates were obtained by minimizing the maximum likelihood objective function with the variance model
where Y was the model predicted value and a and b were the variance parameters. The fittings were preformed by ADAPT II (D’Argenio and Schumitzky 1997). The same program was used for simulations of time courses of various model variables.
Results
The study was performed in two stages. First, the TMDD PK model was fitted to the clinical data obtained from Flaharty et al. (1990). The fittings provided estimates of the model parameters that were not available in the literature and the values for secondary parameters. They are presented in Table 1. Second, a series of computer simulations was performed to generate time courses of EPOR, rHuEPO-EPOR complex, fraction of bound EPOR, and percent of rHuEPO clearance due to receptor binding for various IV doses of rHuEPO. The parameter values obtained in the first stage were used for simulations. The time courses of the mentioned above variables are informative about PK and PD of rHuEPO and EPOR in healthy humans.
Serum concentrations of rHuEPO
Fittings of the TMDD PK model to the mean serum concentrations of rHuEPO in healthy volunteers who received 10, 50, 150, 500, and 1,000 IU/kg IV bolus doses are shown in Fig. 2. The estimated parameters were k el, V c, k pt, k tp, R 0, and k deg. Both goodness of fit criteria (r 2 = 0.98) and the precision of parameter estimates (CV% range 5–28%) indicated that the model described the data reasonably well. The V c estimated value of 0.032 l/kg approximates the plasma volume in a 70-kg human. This indicates that majority of rHuEPO remains in the circulation, which is consistent with the previous finding (Spivak and Cotes 1991). For the data used in this study, the presence of the baseline rHuEPO concentrations prevents an immediate observation of the terminal slopes from the plots. Simulations of (C–C 0) variables in the semilogarithmic scale up to 100 h (data not shown) resulted in the slopes of 0.074, 0.076, 0.076, 0.079, and 0.086 h−1 for corresponding doses. These translate to the terminal half-lives in the range of 8.1–9.4 h. The relatively similar values of k el and terminal half-lives may indirectly indicate that receptor-mediated elimination of rHuEPO is very fast as it does not influence considerably the terminal portion of the concentration vs time curve.

Mean serum rHuEPO concentrations (symbols) in healthy subjects administered an IV dose. The solid lines represent the TMDD PK model predicted values. The data were obtained from Flaharty et al. (1990)
Kinetics of EPOR
The purpose of modeling of the rHuEPO serum concentration data was to identify parameters that describe receptor binding, turnover, and internalization. Some of these were obtained from previously published studies (k off, k on, and k int). The remaining parameters (R 0, k deg, and k syn) were estimated from the fitting. The availability of the rHuEPO concentrations data alone prohibits determination of the volume of distribution for EPOR. Consequently, information carried by values of R 0, k syn, and R is relative to serum rHuEPO concentrations. The TMDD model allows to predict the time courses of free (unbound) EPOR concentrations shown in Fig. 3. The dose-dependent rapid drop of the initial R is dictated by the second-order binding process with the slope \({\left( {{\text{in the logarithmic scale}}} \right)} - k_{{{\text{on}}}} {\left( {C_{{\text{0}}} + {{\text{Dose}}} \mathord{\left/ {\vphantom {{{\text{Dose}}} {V_{{\text{c}}} }}} \right. \kern-\nulldelimiterspace} {V_{{\text{c}}} }} \right)}\). The extent of it is determined by the fraction of bound receptors at equilibrium discussed in the next section. The subsequent slower phase is controlled by the internalization and degradation processes. After reaching the nadir, the free receptor concentrations gradually return to the baseline as a consequence of the balance between the constant production rate k syn and decreasing with time receptor loss rates. The duration of the nadir in the free receptor concentrations is controlled by the time the receptors are saturated by rHuEPO, which depends on the levels of C relative to the K D value. The free receptor degradation half-life is 8.8 h.

Simulated by the TMDD PK model concentrations of the free EPOR for indicated doses of rHuEPO. The broken line denotes the baseline EPOR value R 0. The model parameters used for simulations are presented in Table 1
Kinetics of EPO–EPOR complex
The model variable that account for the EPO–EPOR bound species is RC. The time courses of RC corresponding to various rHuEPO doses are shown in Fig. 4. The time profiles exhibit many phases: an initial very rapid rise due to receptor binding, sharp peak followed by a rapid decline due to internalization, prolonged plateau (for higher doses) duration of which correlates with the time C is above K D level, and relatively slow return to the baseline due to exhaustion of free rHuEPO. The peak times for the RC variable were 8.7, 4.2, 2.1, 0.9, and 0.6 min, respectively, to increasing doses. The plateau region at the level of 4.6 pM was observed for doses 150, 500, and 1,000 IU/kg, and started approximately at 3.5 h. Based on the simulations of the time course of rHuEPO serum concentrations performed up to 100 h for all doses (data not shown), the times the K D level was reached by the C variable were 7.5, 22.0, 43.1, and 55.6 h and can be considered as the end of the plateau phase (see Fig. 4). The plateau level RC plateau can be calculated from the model by assuming that dR / dt ∼ 0, dRC / dt ∼ 0, and R ∼ 0. Then, by adding Eqs. 4 and 5, one can obtain
This relationship explains the plateau phase as a consequence of the balance between the receptor synthesis and receptor internalization at low receptor levels.

Simulated by the TMDD PK model concentrations of the rHuEPO-EPOR complex for indicated doses of rHuEPO. The left panel illustrates the time courses from 0 to 2 h, whereas the right panel covers the time range from 2 to 100 h. The solid symbols represent RC values at times when C = K D. The broken lines denote the baseline bound EPOR value RC 0. The model parameters used for simulations are presented in Table 1
Pharmacologically interesting is the kinetics of the fraction of occupied receptors, because only these species initiate the intracellular signaling pathways leading to the pharmacological effect. The bound fraction of receptors can be defined in terms of the model variables as
where R tot = R + RC is the total concentration of bound and free EPOR. The plot of f B vs time is shown in Fig. 5. The baseline value of R tot can be determined from Eq. 6

Simulated fractions of the bound EPOR for indicated doses of rHuEPO. The f B values were calculated according to Eq. 12. The left panel illustrates the time courses from 0 to 1 h, whereas the right panel covers the time range from 1 to 100 h. The broken lines indicate time course of the drug effect E described by the E max model Eq. 14. The baseline value for f B is denoted by the bold broken line. The dotted lines mark the 50% level
The parameter R tot0 can be interpreted as a receptor capacity. At baseline conditions, the fraction of occupied receptors was 3.1%. Upon drug administration, it rapidly increased in a dose-dependent manner to reach a peak value followed by a gradual decrease to the baseline that coincided with the decrease of serum rHuEPO concentrations. The peak times 12, 8.4, 4.2, 1.8, and 0.9 min were delayed with respect to the RC peak times. The corresponding peak values were 33.5, 76.4, 91.6, 97.5, and 98.7%. The times f B reached the 50% level were 6.4, 19.8, 40.6, 53.2 h. The differences between these times and times at which C reached the K D level are noticeable because C = K D yields f B = 50% only if the equilibrium between receptor binding and dissociation is achieved at all times. The profiles of the fraction of bound receptors simulated under the assumption of equilibrium between the binding and dissociation (E) calculated according to the equation
show little differences when compared with f B vs time profiles (see Fig. 5).
Sensitivity of the model to k on and k off
As it was not possible to estimate k on and k off directly from the data used for the analysis, the values of k off and K D were obtained from the in vitro studies reported in literature and k on was calculated as a secondary parameter (see Materials and methods). To acquire insight into the influence of these parameters on PK and PD of rHuEPO, the TMDD model was used to simulate rHuEPO and EPO–EPOR complex concentration profiles with various values of k off for the dose of 150 IU/kg (Fig. 6). As it can be seen from this figure, the time course of EPO–EPOR is significantly influenced by k off when this parameter values range from 0.01 to 10 h−1 and this effect is minimal for the values higher than 10 h−1. Similar tendency may be observed for rHuEPO profiles (Fig. 6, inset), however the influence of this parameter on PK is much less pronounced. Thus, it seems that k off value is an important factor determining the time course of EPO-EPOR and in consequence the intensity of pharmacologic response. Besides, it has been shown that receptor binding may also influence PK of rHuEPO, especially when k off is lower than 1 h−1. Therefore, when binding constants can not be estimated from the data available, significant care should be taken to ensure that the most appropriate values of k on or k off and K D have been selected for the analysis using TMDD model.

Sensitivity analysis for k off at a dose of 150 IU/kg. The k off values ranged from 0.01 to 100 h−1 corresponding to the k on of 0.0001 to 1 pM−1 h−1. The left panel illustrates the time courses from 0 to 2 h, whereas the right panel covers the time range from 2 to 60 h. The broken lines denote the baseline bound EPOR value RC 0. The remaining model parameters used for simulations are presented in Table 1. Inset shows the influence of k off values on concentration vs time profile of rHuEPO for the dose of 150 IU/kg
Receptor-mediated rHuEPO serum clearance
The nonlinear binding process makes the clearance of rHuEPO from serum time and dose-dependent. The TMDD allows one to evaluate the clearance process. The drug is cleared from the plasma by the first-order process k el, distributed to tissues by the first-order process k pt, and binding to EPOR according to the second-order process k on. Therefore, the total clearance of rHuEPO from serum can be written as (Rowland and Tozer 1995)
where the latter term in Eq. 15 can be identified as a receptor-mediated clearance Clbind. The fraction Clbind / Cltot describes the contribution of receptor binding to the total clearance of rHuEPO. The plots of this fraction against time are shown in Fig. 7. At the baseline conditions, the receptor binding accounts for 97.4% of the total clearance. Qualitatively, the time courses of Clbind / Cltot resemble that of R with a rapid initial decline, followed by a slower decrease to a nadir with a subsequent return to the baseline. The nadir values were 93.6, 62.5, 32.0, 11.8, and 6.2% for the doses 10, 50, 150, 500, and 1,000 IU/kg, respectively, and the nadir times occurred within 2.5–3.2 h range. The initial reductions in the contribution of the receptor binding to the overall clearance were approximately 97, 89, 75, 48, and 32% and they are probably attributed to the saturation of the free EPOR. Consequently, for the lowest dose of 10 IU/kg majority (>90%) of the drug was cleared from the serum by binding to the receptors whereas for the highest dose 1,000 IU/kg up to 27 h the receptor-mediated clearance constituted between 6 and 32% of the total clearance.

Simulated fractions of the total clearance due to receptor-mediated clearance for indicated doses of rHuEPO. The fraction was calculated as the ratio of Clbind = k on R V c and Cltot that was defined by Eq. 15. The broken line indicates the fraction of the total clearance at the baseline conditions
Discussion
The applied TMDD PK model describing rHuEPO serum concentrations does not specify a particular organ or tissue expressing EPOR. However, it does assume that the R compartment is well perfused by the circulating blood, so there is very rapid distribution and redistribution of rHuEPO to this compartment. The studies on the distribution rate of rHuEPO to the bone marrow report a delay between the peaks of serum and bone marrow concentrations (Kinoshita et al. 1992). Also, expression of EPOR in various tissues is not homogeneous and representing it by a single compartment is a simplification. The model assumes straightforward mechanisms for receptor turnover with zero-order production and first-order elimination rates. According to the model, exposure to rHuEPO does not change these processes, and consequently there is no upregulation or downregulation except for internalization and degradation upon binding to rHuEPO. The model ignores a possible increase in EPOR level due to an increase in the number of progenitor cells expressing EPOR which is the physiological effect of rHuEPO. There is no recycling of rHuEPO nor EPOR, which is consistent with the literature (Walrafen et al. 2005). The above-mentioned simplifications pose limits on interpretation of the presented results. On the other hand, scarcity of the available data prohibited including additional processes into the model. Also, a more complicated model would result in difficulties with interpretation of the simulated time courses of the key model variables.
Among various attempts to model nonlinear pharmacokinetics of EPO, the Michaelis–Menten equation has been probably the most frequently used (Ramakrishnan et al. 2003; Veng-Pedersen et al. 2003). Although it has a capability to account for a saturation of the receptor-mediated clearance, this equation is not reflective of the drug internalization and receptor turnover by a virtue of limited number of parameters (V max and K m) it provides. The TMDD model offers an alternative description of the drug kinetics that is more mechanistic. A natural question arises about the differences and similarities of the presented model for TMDD and the Michealis–Menten equation. Because of the larger number of model parameters, the TMDD model is more flexible in describing receptor-mediated drug concentration data. Under some assumptions these two models are identical, but in this report we did not attempt to specify these conditions. There is not a straightforward answer to this question and further studies are necessary to provide a quantitative answer.
Some studies have demonstrated that levels of endogenous EPO may undergo a circadian rhythm both in healthy (Pasqualetti and Casale 1996) and disease (Pasqualetti et al. 2000). However, as can be seen in Fig. 2, the contribution of endogenous EPO to the total concentration observed after rHuEPO administration is negligible and it is not likely to influence significantly the results of PK analysis. Despite this, if sufficient data are available, this phenomenon may be incorporated into the model by substituting a zero-order rate constant (k EPO) in Eq. 1 into a more sophisticated equation (e.g., the cosine function or Fourier series).
The comparison of the binding phases in the time courses of rHuEPO, EPOR, and rHuEPO–EPOR concludes that the kinetics of EPOR precedes changes in time profiles of the bound EPOR. The effect of binding can be observed in the plots of these two species earlier than in the kinetic profiles of rHuEPO. This indicates that R and RC drive the time behavior of C. Two binding parameters K D and R tot0 play particularly important roles. The former controls the degree to which the EPOR are saturated and the latter determines how many rHuEPO molecules will bind to EPOR. Therefore, R tot0 can serve as a measure of binding capacity.
The classical Clark’s receptor occupancy theory assumes that the pharmacological effect is proportional to the number of receptor occupied by drug (Kenakin 1987). From this point of view, the fraction of occupied receptors f B can serve as a PD marker of the pharmacological effect of rHuEPO. The simulation shows that f B exhibits a time profile that parallels one for the E max model with E max = 100% and EC50 = K D that is commonly used to describe pharmacological effects (Mager et al. 2003). Using the K D value as a reference, the calculated by the TMDD model times when the rHuEPO serum is above this level, or similarly the times when f B is greater than 50%, can be used as qualitative measures of the duration of receptor activation yielding a noticeable response. This would hold true if other than internalization and degradation mechanisms of EPOR inactivation did not take place. However, EPOR can be inactivated via different mechanisms that include recruitment of the SHP-1 (domain containing protein-tyrosine phosphates-1) phosphatase to EPOR (Klingmuller et al. 1995) and possible others.
In recent studies on healthy subjects, the application of a catenary model based on the life-span concept allowed to estimate the plasma concentration of rHuEPO producing 50% of maximal stimulation (SC50) when reticulocyte count was used as a PD marker (Ramakrishnan et al. 2004; Krzyzanski et al. 2005). The values of this parameter were very similar in both reports and were 14.69 pM (Krzyzanski et al. 2005) or ranged from 6.84 to 15.45 pM (Ramakrishnan et al. 2004). Thus, the SC50 was lower than K D = 100 pM value used in the present study for the analysis or than those reported by others (Philo et al. 1996; Sawyer et al. 1987). This means that rHuEPO may be an efficacy-driven drug (Kenakin 1987) and as such it should be insensitive to changes in receptor upregulation and downregulation. However, it must be stressed that the data used for the analysis in the present work were collected after intravenous administration of epoetin beta, whereas in both studies cited above, healthy subjects received subcutaneous doses of epoetin alfa. The results of the study performed by Halstenson et al. (1991) indicated that there are differences in PK and PD between these two forms of recombinant EPO. In addition, when all patient data were fitted individually, the median PD parameters were higher by more than threefold from the values estimated using the mean data (Krzyzanski et al. 2005). Thus, further studies are warranted to clarify whether rHuEPO is efficacy or affinity driven agonist.
Receptor-mediated clearance is considered as a major mechanism of rHuEPO elimination (Chapel et al. 2001) while other investigators consider it as moderate for clinically relevant concentrations (Ramakrishnan et al. 2004). The simulated profiles of the fraction of the total clearance attributed to the receptor binding demonstrate that it is highly dependent on availability of free EPOR that is limited for higher rHuEPO serum concentrations and in plenty at the baseline levels. This observation may have significant clinical implications. It has been shown that endogenous EPO levels are significantly increased in cancer patients receiving chemotherapy (Piroso et al. 1989; Glaspy et al. 2005) and AIDS patients treated with zidovudine (Spivak et al. 1989). Both anticancer drugs and zidovudine cause downregulation o EPOR expression in bone marrow (Chapel et al. 2001; Gogu et al. 1992). Thus, it is expected that in these two patient populations, the fraction of the total clearance attributed to the receptor binding should be significantly reduced in parallel with the decreasing number of free receptors. This, in turn, may influence the concentration of EPO–EPOR complex and corresponding pharmacologic response.
In summary, a PK model implementing receptor-mediated disposition of rHuEPO was applied to integrate available data to reconstruct time evolution of free and bound EPOR species in humans receiving IV doses of rHuEPO. Although limited by simplifying assumptions and somewhat arbitrary selection of the model parameters, the model permitted simulations of time courses of EPOR expressions that are very difficult, if not impossible, to obtain experimentally at present. The kinetics of EPOR in response to doses of rHuEPO (PK) and kinetics of bound EPOR (PD) are driving forces for pharmacokinetics and pharmacodynamics of rHuEPO itself.
References
Altin JG, White FAJ, Easton CJ (2001) Synthesis of the chelator lipid nitrilotriacetic acid ditetradecylamine (NTA-DTDA) and its use with the IAsys biosensor to study receptor-ligand interactions on model membranes. Biochem Biophys Acta 1513:131–148
Broudy VC, Lin N, Brice M, Nakamoto B, Papayannopoulou T (1991) Erythropoietin receptor characteristics on primary human erythroid cells. Blood 77:2583–2590
Chapel S, Veng-Pedersen P, Hohl RJ, Schmidt RL, McGuire EM, Widness JA (2001) Changes in erythropoietin pharmacokinetics following busulfan-induced bone marrow ablation in sheep: evidence for bone marrow as a major erythropoietin elimination pathway. J Pharmacol Exp Ther 298:820–824
Cheung K, Goon BL, Guilfoyle MC, Wacholtz MC (1998) Pharmacokinetics and pharmacodynamics of recombinant human erythropoietin after single and multiple subcutaneous doses to healthy subjects. Clin Pharmacol Ther 64:412–423
D’Argenio DZ, Schumitzky A (1997) ADAPT II User’s Guide: Pharmacokinetic/Pharmacodynamic System Analysis Software. Biomedical Simulations Resource, Los Angeles
Fisher JW (2003) Erythropoietin: physiology and pharmacology update. Exp Biol Med 228:1–14
Flaharty KK, Caro J, Erslev A, Whalen JJ, Morris EM, Bjornsson TD, Vlasses PH (1990) Pharmacokinetics and erythropoietic response to human recombinant erythropoietin in healthy men. Clin Pharmacol Ther 47:557–564
Glaspy J, Henry D, Patel R, Tchekmedyian S, Applebaum S, Berdeaux D, Lloyd R, Berg R, Austin M, Rossi G, the Darbepoetin Alfa 20010162 Study Group (2005) Effects of chemotherapy on endogenous erythropoietin levels and the pharmacokinetics and erythropoietic response of darbepoetin alfa: a randomized clinical trial of synchronous versus asynchronous dosing of darbepoetin alfa. Eur J Caner 41:1140–1149
Gogu SR, Malter JS, Agrawal KC (1992) Zidovudine induced blockade of the expression and function of the erythropoietin receptor. Biochem Pharmacol 44:1009–1012
Halstenson CE, Macres M, Katz SA, Schnieders JR, Watanabe M, Sobota JT, Abraham PA (1991) Comparative pharmacokinetics and pharmacodynamics of epoetin alfa and epoetin beta. Clin Pharmacol Ther 50:702–712
Jelkmann W (1992) Erythropoietin: structure, control of production, and function. Physiol Rev 72:449–489
Kenakin TP (1987) Pharmacologic analysis of drug-receptor interaction. Raven Press, New York
Kinoshita H, Ohishi N, Kato M, Tokura S, Okazaki A (1992) Pharmacokinetics and distribution of recombinant human erythropoietin in rats. Arzneim-Forsch/Drug Res 42:174–178
Klingmuller U, Lorenz U, Cantley LC, Neel BG, Lodish HF (1995) Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell 80:729–38
Krzyzanski W, Jusko WJ, Wacholtz MC, Minton N, Cheung WK (2005) Pharmacokinetic and pharmacodynamic modeling of recombinant human erythropoietin after multiple subcutaneous doses in healthy subjects. Eur J Pharm Sci 26:295–306
Lacombe C, Mayeux P (1998) Biology of erythropoietin. Haematologica 83:724–732
Levy G (1994) Pharmacologic target-mediated drug disposition. Clin Pharmacol Ther 56:248–52
Mager DE, Jusko WJ (2001) General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn 28:507–532
Mager DE, Wyska E, Jusko WJ (2003) Diversity of mechanism-based pharmacodynamic models. Drug Metab Dispos 31:510–18
Pasqualetti P, Casale R (1996) Circadian rhythm of serum erythropoietin in healthy subjects. Riv Eur Sci Med Farmacol 18:91–93
Pasqualetti P, Collacciani A, Casale R (2000) Circadian rhythm of serum erythropoietin in myelodysplastic syndroms. Riv Eur Sci Med Farmacol 4:111–115
Philo JS, Aoki KH, Arakawa T, Owers Narhi L, Wen J (1996) Dimerization of the extracellular domain of the erythropoietin (EPO) receptor by EPO: one high-affinity and one low-affinity interaction. Biochemistry 35:1681–1691
Piroso E, Erslev AJ, Caro J (1989) Inappropriate increase in erythropoietin titers during chemotherapy. Am J Hematol 32:248–254
Ramakrishnan R, Cheung WK, Farrell F, Joffee L, Jusko WJ (2003) Pharmacokinetic and pharmacodynamic modeling of recombinant human erythropoietin after intravenous and subcutaneous dose administration in cynomolgus monkeys. J Pharmacol Exp Ther 306:324–331
Ramakrishnan R, Cheung WK, Wacholtz MC, Winton N, Jusko WJ (2004) Pharmacokinetic and pharmacodynamic modeling of recombinant human erythropoietin after single and multiple doses in healthy volunteers. J Clin Pharmacol 44:991–1002
Rowland M, Tozer TN (1995) Clinical pharmacokinetics. Concepts and applications. Williams & Wilkins, Philadelphia
Sawyer ST, Krantz SB, Goldwasser E (1987) Binding and receptor-mediated endocytosis of erythropoietin in Friend virus-infected erythroid cells. J Biol Chem 262:5554–5562
Sugiyama Y, Hanano M (1989) Receptor-mediated transport of peptide hormones and its importance in the overall hormone disposition in the body. Pharm Res 6:192–202
Spivak JL, Cotes PM (1991) The pharmacokinetics and metabolism of erythropoietin. In: Erslev AJ, Adamson JW, Eschbach JW, Winearls CG (eds) Erythropoietin, molecular, cellular, and clinical biology. The John Hopkins University Press, Baltimore, MD
Spivak JL, Barnes DC, Fuchs E, Quinn TC (1989) Serum immunoreactive erythropoietin in HIV-infected patients. J Am Med Assoc 210:3104–3107
Veng-Pedersen P, Chapel S, Al-Huniti NH, Schmidt RL, Sedars EM, Hohl RJ, Widness JA (2003) A differential pharmacokinetic analysis of the erythropoietin receptor population in newborn and adult sheep. J Pharmacol Exp Ther 306:532–537
Walrafen P, Verdier F, Kadri Z, Chretien S, Lacombe C, Mayeux P (2005) Both proteosome and lysosome degrade the activated erythropoietin receptor. Blood 105:600–608
Acknowledgments
This work was supported by grant GM57980 from the National Institutes of Health and funds from the University at Buffalo and Pfizer Strategic Alliance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Krzyzanski, W., Wyska, E. Pharmacokinetics and pharmacodynamics of erythropoietin receptor in healthy volunteers. Naunyn-Schmied Arch Pharmacol 377, 637–645 (2008). https://doi.org/10.1007/s00210-007-0225-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-007-0225-z