
Abstract
The complexity of cardiovascular responses produced by 5-hydroxytryptamine (5-HT, serotonin), including bradycardia or tachycardia, hypotension or hypertension, and vasodilatation or vasoconstriction, has been explained by the capability of this monoamine to interact with different receptors in the central nervous system (CNS), on the autonomic ganglia and postganglionic nerve endings, on vascular smooth muscle and endothelium, and on the cardiac tissue. Depending, among other factors, on the species, the vascular bed under study, and the experimental conditions, these responses are mainly mediated by 5-HT1, 5-HT2, 5-HT3, 5-HT4, 5-ht5A/5B, and 5-HT7 receptors as well as by a tyramine-like action or unidentified mechanisms. It is noteworthy that 5-HT6 receptors do not seem to be involved in the cardiovascular responses to 5-HT. Regarding heart rate, intravenous (i.v.) administration of 5-HT usually lowers this variable by eliciting a von Bezold-Jarisch-like reflex via 5-HT3 receptors located on sensory vagal nerve endings in the heart. Other bradycardic mechanisms include cardiac sympatho-inhibition by prejunctional 5-HT1B/1D receptors and, in the case of the rat, an additional 5-ht5A/5B receptor component. Moreover, i.v. 5-HT can increase heart rate in different species (after vagotomy) by a variety of mechanisms/receptors including activation of: (1) myocardial 5-HT2A (rat), 5-HT3 (dog), 5-HT4 (pig, human), and 5-HT7 (cat) receptors; (2) adrenomedullary 5-HT2 (dog) and prejunctional sympatho-excitatory 5-HT3 (rabbit) receptors associated with a release of catecholamines; (3) a tyramine-like action mechanism (guinea pig); and (4) unidentified mechanisms (certain lamellibranch and gastropod species). Furthermore, central administration of 5-HT can cause, in general, bradycardia and/or tachycardia mediated by activation of, respectively, 5-HT1A and 5-HT2 receptors. On the other hand, the blood pressure response to i.v. administration of 5-HT is usually triphasic and consists of an initial short-lasting vasodepressor response due to a reflex bradycardia (mediated by 5-HT3 receptors located on vagal afferents, via the von Bezold-Jarisch-like reflex), a middle vasopressor phase, and a late, longer-lasting, vasodepressor response. The vasopressor response is a consequence of vasoconstriction mainly mediated by 5-HT2A receptors; however, vasoconstriction in the canine saphenous vein and external carotid bed as well as in the porcine cephalic arteries and arteriovenous anastomoses is due to activation of 5-HT1B receptors. The late vasodepressor response may involve three different mechanisms: (1) direct vasorelaxation by activation of 5-HT7 receptors located on vascular smooth muscle; (2) inhibition of the vasopressor sympathetic outflow by sympatho-inhibitory 5-HT1A/1B/1D receptors; and (3) release of endothelium-derived relaxing factor (nitric oxide) by 5-HT2B and/or 5-HT1B/1D receptors. Furthermore, central administration of 5-HT can cause both hypotension (mainly mediated by 5-HT1A receptors) and hypertension (mainly mediated by 5-HT2 receptors). The increasing availability of new compounds with high affinity and selectivity for the different 5-HT receptor subtypes makes it possible to develop drugs with potential therapeutic usefulness in the treatment of some cardiovascular illnesses including hypertension, migraine, some peripheral vascular diseases, and heart failure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Serotonin, chemically known as 5-hydroxytryptamine (5-HT), is a biogenic monoamine with a molecular weight of 176Da. In mammals, 5-HT is mainly found in the platelets, enterochromaffin cells, and in the central nervous system (CNS), where it plays an important role as a neurotransmitter (see Barnes and Sharp 1999). Thanks to the development of selective 5-HT receptor agonists and antagonists, many unsuspected functions of 5-HT in the CNS and in the periphery have been elucidated. Accordingly, these findings originated a new era in the development of drugs with therapeutic usefulness in the treatment of diseases such as anxiety, schizophrenia, hypertension, migraine, peripheral vascular diseases, etc.
5-HT in the nineteenth century
Long before the identification of serotonin as 5-hydroxytryptamine, pharmacologists and physiologists already knew that a substance active on blood vessels and heart appears in serum when blood is allowed to clot. The first set of publications describing objectively that blood serum is by no means an innocuous substance dates back to the nineteenth century, when researchers such as Creite (1869), Rummo and Bordoni (1889), and Weiss (1896) reported that a diminution in the amount of urine, albuminuria, an increase in both respiratory frequency and heart rate, and an increase in blood pressure followed by a sudden decrease (and eventually death) were induced when serum was injected by the subcutaneous or i.v. route into an animal. Of particular interest are the cardiovascular effects observed by Weiss (1896), namely, that “if serum was continuously but slowly injected into a rabbit, cat, or dog until it caused death, the heart rate was increased and at first the beat was stronger but gradually failed, although it persisted until the respiration had ceased; in addition, an increase in the peristalsis of the small intestines and death due to paralysis of the respiratory and vasomotor centers was noticed.
5-HT in the early twentieth century
Subsequently, Brodie (1900) described in an extensive study that the i.v. injection of blood serum from any source into the cat produced a vagally mediated reflex resulting in a reversible bradycardia, hypotension, and arrest of the respiration, whereas injection of either blood plasma or boiled serum did not show this effect. Simultaneously, the vasoconstrictor properties of blood serum on perfused organs and excised arterial strips were being described and investigated as to its nature and origin. With respect to the latter, Janeway et al. (1918) reported some findings of particular relevance, namely: (1) that either the ox uncoagulated blood or the citrated plasma has no constrictor action on the excised strip of the ox carotid, in contrast to the marked constrictor action exerted by serum to which citrate had been subsequently added; (2) that the extract of platelets from either dog, ox, or pig shows marked vasoconstrictor properties, but not that of erythrocytes or leucocytes; and (3) that either the “platelet substance(s)” or the blood serum causes a constriction of the ox coronary artery, whereas adrenaline causes relaxation. Therefore, Janeway et al. (1918) concluded that the vasoconstrictor substance of clotted blood is derived from the disintegration of platelets and that it is not adrenaline. Based on this conclusion, Hirose (1918) confirmed and extended these findings by establishing for the first time a direct correlation between the platelet count of human blood and its vasoconstrictor action after clotting in the ox carotid artery. From all of the above findings, it is quite clear that the basic cardiovascular effects of blood serum could be characterized before the chemical nature of the substance involved could have been elucidated. There was no further substantial advance in connection with the latter until the early 1930s, when Vialli and Erspamer (1933) isolated a substance from the stomach enterochromaffin cells of rabbits. Then, Ersparmer, by a diazo reaction, noted that this substance contained a phenolic structure linked to an amino group and called it enteramine (Erspamer 1940a, b, c). In the late 1940s, Page, who was searching for humoral pressor agents that might explain arterial hypertension, in collaboration with Green and Rapport, could isolate this vasoconstrictor substance as a crystalline complex and named it serotonin, as it could indeed increase the vascular tone (Rapport et al. 1948a, b); shortly thereafter, Rapport (1949) deduced that the active moiety of this complex (for which he retained the name serotonin) was 5-HT. This compound, when prepared synthetically by Hamlin and Fischer (1951) and others, proved to have all the properties of natural serotonin, including those in the gastrointestinal system (Erspamer and Asero 1952). It is therefore not surprising that the introduction of synthetic 5-HT in 1951 (Hamlin and Fischer 1951) touched off an explosion of research (e.g., Erspamer and Asero 1952; Page 1958; Page and McCubbin 1953).
5-HT in the late twentieth century
By the late twentieth century, it was already known that 5-HT elicits complex responses in the cardiovascular system comprising bradycardia or tachycardia, hypotension or hypertension, and vasodilatation or vasoconstriction (see Saxena and Villalón 1990). The eventual response depends, among other factors, on the species, the vascular bed under study, the dose employed, the experimental conditions, and most importantly, the nature of the receptor(s) involved. Considering the operational, transductional, and molecular criteria for classification of 5-HT receptors, it is nowadays clear that the effects of 5-HT seem to be mediated by at least seven main families of receptors (see Hoyer et al. 1994; Saxena et al. 1998; Villalón et al. 1997b).
Classification and nomenclature of 5-HT receptors
The first vital step towards characterizing 5-HT receptors was undoubtedly that undertaken by Gaddum and Picarelli (1957), reporting the existence of musculotropic ‘D’ and neurotropic ‘M’ receptors for 5-HT in the guinea pig ileum. However, subsequent evidence showed that some musculotropic responses to 5-HT could not be placed within this scheme (Saxena 1972). Then, Peroutka and Snyder (1979) challenged this classification showing the existence of 5-HT1 (low affinity for [3H]spiperone) and 5-HT2 (high affinity for [3H]spiperone) binding sites in cerebral membranes. Significantly, the advent of ketanserin (Van Nueten et al. 1981) confirmed that the 5-HT2 site corresponded to most ‘D’ receptors.
Hence, the International Union of Pharmacology (IUPHAR) 5-HT receptor nomenclature committee built upon the above information (Bradley et al. 1986) and based upon the conjunction of structural (gene and receptor structural sequences for their nucleotide and amino acid components, respectively), transductional (receptor-effect coupling events), and operational (drug-related characteristics) criteria (see Hoyer and Martin 1997; Saxena et al. 1998), 5-HT receptors can now be categorized into seven main types (see Table 1), namely:
5-HT 1 , which corresponds to some ‘D’ receptors and 5-HT1 binding sites and can be subdivided into functional subtypes including, 5-HT1A, 5-HT1B\({\left( {{\text{previously }}5 - {\text{HT}}1{\text{D $ \beta $ }}} \right)}\) and 5-HT1D\( {\left( {{\text{previously }}5 - {\text{HT}}1{\text{D $ \alpha $ }}} \right)} \); negatively coupled to adenylyl cyclase.
5-HT2, which corresponds to most ‘D’ receptors and 5-HT2 binding sites and can be subdivided into 5-HT2A, 5-HT2B and 5-HT2C subtypes; positively coupled to phospholipase C.
5-HT3, which is equivalent to ‘M’ receptors. It is unique, not just among 5-HT receptors but also among mono- and diamine neurotransmitter receptors, in forming a ligand-gated Na+/K+ ion channel analogous to nicotinic and glycine receptors; thus, it does not directly activate second messenger systems.
5-HT4, positively coupled to adenylyl cyclase.
5-ht5, which includes 5-ht5A (preferentially coupled to Gi/o; it could also be coupled to inwardly rectifying potassium channels) and 5-ht5B (transductional system unknown; see Thomas 2006).
5-HT6, positively coupled to adenylyl cyclase.
5-HT7, positively coupled to adenylyl cyclase.
Thus, information about whether the receptor is G-protein-coupled (i.e., 5-HT1, 5-HT2, 5-HT4, 5-ht5A, 5-HT6, and 5-HT7 receptor types) or integral to an ion channel (i.e., 5-HT3 receptor type) immediately indicates what superfamily the receptor belongs to and some of its functional characteristics (see Hoyer et al. 1994, 2002). It must be highlighted that the above classification also includes certain receptors waiting definitive characterization, such as: (1) recombinant 5-ht1E, 5-ht1F, 5-ht5A, and 5-ht5B receptors, which are written in lower case letters to indicate that their functional role is not clear in whole tissues due to the lack of selective agonists and antagonists (see Table 1); and (2) “orphan” receptors, which have a functional role in whole tissues (e.g., depolarization in rat motoneurons, inhibition of noradrenaline release in pig coronary artery, and the slow depolarization of myenteric neurons), but do not correlate with any of the above 5-HT receptor types (see Hoyer et al. 1994, 2002; Martin 1994; Saxena et al. 1998; Villalón et al. 1997b).
Moreover, it is noteworthy that the advent of sumatriptan (Humphrey et al. 1988) led to the subdivision of 5-carboxamidotryptamine (5-CT)-sensitive “5-HT1-like” receptors into 5-HT1X (sumatriptan-sensitive) and 5-HT1Y (sumatriptan-insensitive) receptors (see Saxena and Villalón 1990). Utilizing their differential coupling to adenylyl cyclase and employing cloning techniques, it was subsequently shown that 5-HT1X (vasoconstrictor) and 5-HT1Y (vasodilator) receptors are different and seem to correspond to, respectively, 5-HT1B/1D (Villalón et al. 1996, 2001) and 5-HT7 (Villalón et al. 1997a, b) receptors.
As shown in Table 1, 5-HT1 receptors are heterogeneous in nature as five 5-HT1 subtypes (5-HT1A, 5-HT1B, 5-HT1D, 5-ht1E, and 5-ht1F) have been recognized. Moreover, heterogeneity cannot be ruled out for the 5-HT2 receptors, as they include 5-HT2A, 5-HT2B, and 5-HT2C receptors. Although several splice variants for 5-HT3 (5-HT3a and 5-HT3b), 5-HT4 (5-HT4a, 5-HT4b, 5-HT4c, and 5-HT4d) and 5-HT7 (5-HT7a, 5-HT7b, 5-HT7c, and 5-HT7d) receptors have been reported, it is unknown whether these variants show differences on their operational/transductional characteristics and, most importantly, whether they play a differential functional role.
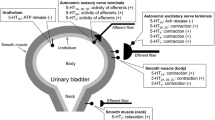
The physiological role of 5-HT, other than as a neurotransmitter in the central and, perhaps, in the enteric nervous system, is at best debatable (see Saxena and Villalón 1990). However, 5-HT and related agonists exert a variety of responses via the stimulation of different 5-HT receptors (see Table 1) located in the CNS, on autonomic nerves, autonomic ganglia, vascular endothelium, and on various smooth muscle containing tissues.
Activation of sensory afferents by 5-HT in the cardiovascular system
I.v. administration of 5-HT can elicit a wide variety of cardiovascular reflexes by acting mainly on the carotid body arterial chemoreceptors, cardiopulmonary receptors, and pulmonary J (deflation) receptors. These reflex responses, which basically consist of bradycardia and hypotension, are (1) mimicked by the 5-HT3 receptor agonists phenylbiguanide or 2-methyl-5-HT; and (2) selectively antagonized by the 5-HT3 receptor antagonists MDL 72222 or tropisetron (see Table 1). Therefore, the receptor involved is of the 5-HT3 type, which is located on vagal (and other) afferents (for references see Saxena and Villalón 1990). Moreover, an i.v. bolus injection of 5 HT in the dog induces a hypertensive chemoreceptor reflex, which increases efferent vagal and sympathetic nerve activities, leading to an initial transient decrease in heart rate, followed by tachycardia (see Saxena and Villalón 1990); the role of 5 HT3 receptors in this reflex response is established because of antagonism by tropisetron (Berthold et al. 1989).
Actions of 5-HT at sympathetic ganglia
I.v. administration of 5 HT and related agonists can result in excitatory and/or inhibitory actions at sympathetic ganglia (see Fozard 1984), which may in turn translate into sympatho-excitation and/or sympatho-inhibition and, consequently, into vasopressor, vasodepressor, tachycardic, and/or bradycardic responses (see below; Saxena and Villalón 1990). In this respect, some studies suggest that 5-HT1 receptors may be mediating inhibition (5-HT- and 5-CT-induced hyperpolarization) of the sympathetic ganglionic transmission (Ireland and Jordan 1987; Jones et al. 1995). These inhibitory ganglionic 5-HT1 receptors closely resemble the 5-HT1A subtype in rats (Ireland and Jordan 1987) or the 5-HT1B/1D, but not the 5-HT1A subtypes in cats (Jones et al. 1995). Although this apparent discrepancy may be due to species differences, it must be highlighted that the possible role of inhibitory ganglionic 5 HT1B/1D receptors in rats will be unequivocally proven by the use of selective agonists and antagonists at 5-HT1B and 5-HT1D receptor subtypes (see Table 1).
Heart rate responses to 5-HT
I.v. or central administration of 5-HT can elicit bradycardia and/or tachycardia. As described below, the nature of the 5-HT receptors and the underlying mechanisms involved in the heart rate responses to 5-HT have been generally well worked out (see Saxena and Villalón 1990; Villalón et al. 1997b).
Bradycardic action
The overwhelming effect of an i.v. bolus injection 5-HT in different species with intact CNS and vagi is an intense, but transient, bradycardia, which is abolished by ganglion blockade, vagotomy, atropine, or spinal section (see Saxena and Villalón 1990) and is therefore due to a von Bezold-Jarisch-like reflex originating from depolarization of afferent cardiac neurons (McQueen 1990). However, bradycardia can be also obtained via a central or a presynaptic action on sympathetic (inhibition) and/or cholinergic (stimulation) neurons.
von Bezold-Jarisch-like reflex
As reviewed by Saxena and Villalón (1990), in anesthetized rats, rabbits, cats, ferrets, dogs, and guinea pigs i.v. bolus injections of 5-HT, phenylbiguanide, and 2-methyl-5-HT elicit a short-lasting bradycardia that can be (1) blocked by bilateral vagotomy or atropine, and (2) effectively antagonized by metoclopramide, MDL 72222, tropisetron and a variety of other 5-HT3 receptor antagonists (see Table 1). Overall, these findings clearly show that the bradycardia due to 5-HT mainly results from an effect on cardiac sensory receptors (producing the von Bezold-Jarisch-like reflex) belonging to the 5-HT3 type.
Prejunctional inhibition of the cardiac sympathetic outflow
In vagotomized pithed rats, selective preganglionic stimulation of the sympathetic cardioaccelerator nerves results in an increase in heart rate, which can be dose-dependently inhibited by i.v. 5-HT (Villalón et al. 1999a). Subsequent pharmacological analysis (Sánchez-López et al. 2003) showed that this 5-HT-induced cardiac sympatho-inhibition was (1) unaltered after saline or the antagonists GR 127935 (5-HT1B/1D), the combination of WAY 100635 (5-HT1A) plus GR 127935, ritanserin (5-HT2), tropisetron (5-HT3/4), LY215840 (5-HT7), or a cocktail of antagonists/inhibitors consisting of yohimbine (α2), prazosin (α1), ritanserin, GR 127935, WAY 100635, and indomethacin (cyclo-oxygenase); (2) abolished by methiothepin (5-HT1/2/6/7 and recombinant 5-ht5A/5B); (3) mimicked by the agonists 5-CT (5-HT1/7 and recombinant 5-ht5A/5B), CP 93,129 (r5-HT1B), sumatriptan (5-HT1B/1D), PNU-142633 (5-HT1D), and ergotamine (5-HT1B/1D and recombinant 5-ht5A/5B), but not by indorenate (5-HT1A) and LY344864 (5-ht1F). Interestingly, 5-CT-induced cardiac sympatho-inhibition was abolished by methiothepin, the cocktail of antagonists/inhibitors, GR 127935, or the combination of SB224289 (5-HT1B) plus BRL15572 (5-HT1D), but remained unchanged when SB224289 or BRL15572 were given separately.
The above and other findings led to the conclusion that 5-HT-induced cardiac sympatho-inhibition, being unrelated to 5-HT2, 5-HT3, 5-HT4, 5-HT6, 5-HT7 receptors, α1/2-adrenoceptors or prostaglandins synthesis, seems to be primarily mediated by: (1) 5-HT1B/1D receptors (Sánchez-López et al. 2004), and (2) a novel mechanism antagonized by methiothepin that, most likely, involves putative 5-ht5A/5B receptors (Sánchez-López et al. 2003).
Stimulation of cholinergic neurons
In the isolated perfused heart obtained from reserpine-treated rabbits, 5-HT elicits a MDL 72222-susceptible bradycardia, which has been ascribed to the presence of 5-HT3 receptors on parasympathetic ganglia and postganglionic cholinergic nerve endings (see Saxena and Villalón 1990). Consistent with this view, in pithed rats pretreated with atenolol, ketanserin, and methiothepin, 5-HT and m-chlorophenyl-biguanide enhanced the bradycardia induced by electrical stimulation of vagi nerves. As this effect was blocked by MDL72222 (Morán et al. 1994), it was suggested that 5-HT3 receptors enhanced the parasympathetic outflow.
Tachycardic action
As previously reviewed by Saxena and Villalón (1990), the tachycardic response produced by i.v. 5-HT in vagotomized animals is notoriously species-dependent and may be mediated by a wide variety of receptors/mechanisms.
Tyramine-like action
In the spinal guinea pig, i.v. 5-HT elicits a tachycardic response resistant to methiothepin, ketanserin, or MDL 72222, but is antagonized by β-adrenoceptor antagonists (propranolol and atenolol) or by the 5-HT-uptake inhibitor indalpine. Reserpine pre-treatment did not affect the first challenge with 5-HT, but the responses to the subsequent doses showed rapid tachyphylaxis (Dhasmana et al. 1988). In the guinea pig isolated atrium, results similar to those in spinal animals were reported except that reserpine was not able to influence the responses to 5-HT (Eglen and Whiting 1989). From these findings, it appears that the tachycardic action of 5-HT in the spinal guinea pig is predominantly via a release of catecholamines by a mechanism similar, but not identical, to that of tyramine. Interestingly, in isolated guinea pig atrium, other mechanisms seem to be involved (see below).
5-HT2A receptor stimulation
Previous studies in pithed rats have shown that i.v. 5-HT increases heart rate and blood pressure and that these responses are effectively antagonized by cyproheptadine but, in contrast, another 5-HT2 receptor antagonist, pirenperone, suppressed the pressor response but not the tachycardia (Krstic and Katusic 1982). Subsequently, Saxena and Lawang (1985) showed that 5-CT, in contrast to 5-HT, failed to increase heart rate in ganglion-blocked rats, and the tachycardia elicited by 5-HT was blocked by ketanserin or cyproheptadine, suggesting the involvement of 5-HT2 receptors.
Accordingly, in pithed normotensive and/or spontaneously hypertensive rats, i.v. 5-methoxytryptamine and 5-HT, but not 5-CT, 8-OH-DPAT, ipsapirone, RU 24969, 5-methoxy-N,N-dimethyltryptamine, TFMPP or DOI, cause tachycardia, which is amenable to blockade by ketanserin, LY53857, methysergide, or methiothepin but not by MDL 72222, propranolol, or desipramine (Göthert et al. 1986; Docherty 1988; Dabiré et al. 1989a, b; Chaouche-Teyara et al. 1993; 1994). It is noteworthy that i.v. DOI, which exhibited a partial agonist action at 5-HT2 receptors mediating vasopressor responses, neither elicited tachycardia nor antagonized the tachycardic action of 5-HT (Chaouche-Teyara et al. 1994). The ineffectiveness of DOI (Chaouche-Teyara et al. 1994) and pirenperone (Krstic and Katusic 1982) suggested that the 5-HT2 receptors mediating tachycardia in the rat were “atypical” in nature. However, at high doses (100μg/kg and above) i.v. 5-HT can also increase heart rate probably by a tyramine-like action, which can be reduced by desipramine or propranolol (Docherty 1988). To shed further light on the receptors/mechanisms involved, Centurión et al. (2002) reported in reserpinized pithed rats that i.v. 5-HT produced a dose-dependent tachycardia, which was selectively and dose-dependently blocked by antagonists at 5-HT2A receptors, but unaffected by antagonists at 5-HT2B or 5-HT2C receptors. These results (1) demonstrate that the above “atypical” 5-HT2 receptors actually correlate with the 5-HT2A receptor subtype and (2) are consistent with the increase in phosphoinositide hydrolysis induced by 5-HT in rat atria (El Rawadi et al. 1994).
In addition, several investigators have reported that i.v. 5-HT elicits tachycardia in anesthetized dogs (see Saxena and Villalón 1990); this effect is (1) accompanied by an increase in noradrenaline concentration in the coronary sinus and vena caval blood, (2) absent after autonomic blockade, and (3) associated with a release of noradrenaline and adrenaline into the blood in ganglion-blocked dogs and suppressed not only by 5-HT2 receptor antagonists but also by a catecholamine depleting agent and bilateral adrenalectomy. It therefore appears that 5-HT elicits tachycardia by a tyramine-like action and by a 5-HT2 receptor-mediated release of catecholamines from the adrenomedullary chromaffin cells.
5-HT3 receptor stimulation
The tachycardic response to 5-HT in the rabbit isolated atria is mediated by noradrenaline release, and detailed pharmacological analysis in the perfused heart has revealed that reserpine, propranolol, cocaine, MDL 72222, or tropisetron, but not desipramine, methiothepin, or methysergide, inhibit this response (for references see Saxena and Villalón 1990). Therefore, the cardiostimulatory effect of 5-HT in the rabbit is due to a 5-HT3 receptor-mediated release of noradrenaline from postganglionic cardiac sympathetic neurons.
Moreover, in conscious dogs, i.v. 5-HT and 2-methyl-5-HT elicited a tachycardia, which was dose-dependently blocked by tropisetron, zacopride, or GR 38032F but not by propranolol or LY53857. Thus, activation of 5-HT3 receptors mediates this response (Wilson et al. 1990).
Similarly, in isolated guinea pig atrium, 5-HT produced positive inotropic and chronotropic responses insensitive to propranolol or imipramine, implying that an indirect mechanism is not involved. Interestingly, tropisetron, granisetron, ondansetron, cisapride, or zacopride blocked the response to 5-HT, suggesting that 5-HT3 receptors are involved (Nishio et al. 1996).
5-HT4 receptor stimulation
Villalón et al. (1990, 1991) have reported that the tachycardia induced by i.v. 5-HT in the anesthetized pig is mainly mediated by 5-HT4 receptors, as this response: (1) was not antagonized by the antagonists methiothepin (5-HT1/2), ketanserin (5-HT2), and/or MDL72222 (5-HT3); (2) was not mimicked by the selective agonists 5-CT (5-HT1/7) or phenylbiguanide (5-HT3); and (3) was dose-dependently blocked by high doses of tropisetron. Likewise, the 5-HT4 receptor also seems to be involved in the positive inotropic action of 5-HT on human atria, as this response is not modified by ketanserin, methysergide, lysergide, methiothepin, yohimbine, (±)propranolol, (−)pindolol, or MDL 72222 but was blocked by a high concentration (2μM) of tropisetron (Kaumann et al. 1989, 1990) or by the selective 5-HT4 receptor antagonist, GR 113808 (Kaumann 1993).
Interestingly, in rats with chronic heart failure, i.v. 5-HT produced a positive inotropic response amenable to blockade by the 5-HT4 receptor antagonist, GR 113808 (Qvigstad et al. 2005a). Consistent with these findings, an increase in messenger RNA (mRNA) for the 5-HT4 receptor in ventricles and an increase in cAMP in cardiomyocytes were found in hearts from rats with chronic heart failure (Qvigstad et al. 2005a).
5-HT7 receptor stimulation
In a detailed analysis in spinal cats (see Saxena and Villalón 1990), it was established that the i.v. 5-HT-induced tachycardia is not—or only slightly—susceptible to blockade by guanethidine, propranolol, burimamide, 5-HT2 receptor antagonists (cyproheptadine, ketanserin, ritanserin, pizotifen and mianserin), or bilateral adrenalectomy. Interestingly, Villalón et al. (1997c) further showed that this response was (1) mimicked by several compounds with a rank order of agonist potency of 5-CT>5-methoxytryptamine>clozapine; (2) resistant to the agonist action of sumatriptan; and (3) effectively blocked by methiothepin (non-selective 5-HT receptor antagonist), methysergide (5-HT1/2/7), mesulergine (5-HT2/7), clozapine (5-HT2/7), and lisuride (5-HT7) but not by GR 127935 (5-HT1B/1D). Thus, this response is mainly mediated by 5-HT7 receptors (Villalón et al. 1997c).
Activation of non-adrenergic non-cholinergic (CGRPergic) sensory neurons
As previously discussed, 5-HT3 receptors mediate the tachycardia to 5-HT in isolated guinea pig atrium (Nishio et al. 1996). Interestingly, the above 5-HT-induced tachycardia was (1) completely inhibited by ruthenium red; (2) attenuated in the capsaicin pre-treated atrium as well as in the presence of capsaicin; (3) potentiated by thiorphan, an inhibitor of peptide degeneration; (4) blocked by the CGRP1 receptor antagonist, CGRP[8–37] (which also blocked the tachycardia to i.v. CGRP); and (5) resistant to tetrodotoxin (excluding the involvement of serotonergic interneurons). These findings suggest that, in isolated guinea pig atrium, activation of 5-HT3 receptors induces the release of CGRP from sensory neurons, which in turn, activates cardiac CGRP1 receptors (Tramontana et al. 1993; Nishio et al. 2002).
Unidentified mechanisms
The hearts of certain lamellibranch and gastropod species (including Mercenaria mercenaria, Tapes watlingi, Patella vulgata, Helix aspersa, Aplysia, etc) are extremely sensitive to 5-HT (threshold concentration 0.1nM; for references see Saxena and Villalón 1990). Interestingly, in the Mercenaria heart, 5-HT has been shown to be an excitatory neurotransmitter (Kuwasawa and Hill 1997) whose effects are antagonized by methysergide and 2-bromolysergide, but lysergide mimics 5-HT with an incredible potency (threshold concentration 1fM; Saxena and Villalón 1990). Moreover, in Aplysia heart, 5-HT increases cyclic AMP (cAMP) accumulation (Marinesco et al. 2004a, b). However, to the best of our knowledge, the receptors/mechanisms involved in the positive inotropic and chronotropic effects of 5-HT in these species have not been fully identified (atypical receptors/mechanisms).
Bradycardia and tachycardia by central mechanisms
In general, central 5-HT containing pathways regulating the cardiovascular system involve two main receptors: 5-HT1A receptors (which mostly mediate sympatho-inhibition) and 5-HT2 receptors (which mostly mediate sympatho-excitation; see McCall and Clement 1994; Ramage 2001). Furthermore, central administration of 5-HT can elicit complex—and, sometimes, apparently contradictory—responses which depend, among other factors, on the species (see below), the experimental conditions (anaesthetized or conscious animals), the exact site of central application, the receptor involved, the drug used, and the dose employed. For instance, in anesthetized rats, intracerebroventricular (i.c.v.) application of 5-HT elicits a biphasic response in heart rate, namely, a brief bradycardia mediated by 5-HT2 receptors followed by tachycardia due to activation of 5-HT1A receptors (Anderson et al. 1992). This tachycardia, associated with sympatho-excitation by 5-HT1A receptors, was also observed at low doses of several 5-HT1A receptor agonists including N-N-dipropyl-5-CT while, at high doses, hypotension occurred (Anderson et al. 1992). Interestingly, in anesthetized cats, fourth ventricle application of 5-HT and several 5-HT1A receptor agonists decreased sympathetic nerve activity and blood pressure without affecting heart rate (Shepheard et al. 1994); however, in the presence of cinanserin (a 5-HT2 receptor antagonist), these compounds produced a significant bradycardia. In contrast, i.c.v. application of 5-HT increased blood pressure, heart rate, and cardiac and splanchnic sympathetic nerve activity (Anderson et al. 1995). These effects are mainly mediated by 5-HT2 receptors, as they were (1) mimicked by the 5-HT2 receptor agonist, DOI; and (2) blocked by the 5-HT2 receptor antagonist, cinanserin (Anderson et al. 1995).
In addition, central stimulation of 5-HT1A receptors delays the hypotension, bradycardia, and sympatho-inhibition induced by hemorrhagic shock in anesthetized rats (Scrogin et al. 2000; Scrogin 2003), implying that central activation of these receptors may be useful during hypovolemic shock. Interestingly, a possible role of 5-HT7 receptors in cardiovascular regulation has been also suggested, as SB-269970, a 5-HT7 receptor antagonist, blocked the bradycardia evoked by cardiopulmonary reflex, baroreflexes, and chemoreflex (Damaso et al. 2007; Kellett et al. 2005; see Table 1).
Blood pressure responses to 5-HT
I.v. 5-HT has a triphasic effect on arterial blood pressure, comprising an initial intense, but brief, vasodepressor response followed by a moderate vasopressor response, and finally, a longer-lasting vasodepressor response (see Page and McCubbin 1953; Saxena and Villalón 1990; Villalón et al. 1997b). The vasopressor phase varies quantitatively depending upon the species and the experimental conditions; for instance, rabbits, cats, and pigs exhibit a poor vasopressor phase, while it is prominent in the dog, particularly after ganglion blockade (see Saxena and Villalón 1990). As described below, these differences may be due to the type of receptors involved and/or their distribution in the different species.
Initial transient vasodepressor response
The initial vasodepressor response to i.v. 5-HT is the result of an abrupt bradycardia (and the consequent decrease in cardiac output) after stimulation of 5-HT3 receptors on cardiac vagal afferents. This response is observed in intact rats, rabbits, cats, and dogs (see Saxena and Villalón 1990). Interestingly, a short-lasting i.v. 5-HT-induced vasodilatation in the human forearm, blocked by tropisetron (ICS 205–930), may also be mediated by 5-HT3 receptors, which may induce an axon-like reflex (Blauw et al. 1988).
Vasopressor response
The vasopressor response to 5-HT, being blocked by 5-HT2 receptor antagonists such as ketanserin, cyproheptadine, pizotifen, and methysergide, is due to activation of vascular 5-HT2 receptors in several species including the rat, cat, and dog (see Martin 1994; Saxena and Villalón 1990; Villalón et al. 1997b); in the latter, a release of catecholamines by adrenomedullary 5-HT2 is also involved (Kimura and Satoh 1983). Accordingly, the contractile effects on both arteries and veins from rats, rabbits, cats, dogs, calf, monkey, and humans are generally mediated by 5-HT2 receptors (see Hoyer et al. 1994; Martin 1994). However, in cranial blood vessels of different species, including humans, pigs, and dogs, as well as in other specific blood vessels (e.g., the saphenous vein and external/internal carotid arterial bed of the dog), 5-HT1 receptors (stimulated by sumatriptan and blocked by methiothepin) mediate the vasoconstrictor responses. These receptors are negatively coupled to adenylyl cyclase and are selectively blocked by the 5-HT1B/1D receptor antagonist, GR 127935 (see Martin 1994; Skingle et al. 1996; Villalón et al. 1997b). With the advent of the selective antagonists SB224289 (5-HT1B) and BRL15572 (5-HT1D; Hagan et al. 1997), it was subsequently shown that these vasoconstrictor 5-HT1B/1D receptors, being selectively blocked by the former but resistant to the latter, pharmacologically correlate with the 5-HT1B rather than the 5-HT1D, subtype (De Vries et al. 1998a; Saxena et al. 1998; Centurión et al. 2001b).
In some cases, both 5-HT1B and 5-HT2 receptors seem to mediate vasoconstriction in the same blood vessel of several species (see Martin 1994; Villalón et al. 1997b), for example, in the canine internal carotid arterial bed (Centurión et al. 2001a, b) and, perhaps to a lesser extent, in the pig carotid arteriovenous anastomoses (see Saxena and Villalón 1990; Saxena et al. 1998). In rarer instances, 5-HT may act directly on α-adrenoceptors in isolated rabbit ear and external carotid arteries (Apperley et al. 1976; Van Nueten 1983).
Late long-lasting vasodepressor response
The most prominent hemodynamic response to i.v. 5-HT in anesthetized animals is vasodepression, first described by Page and McCubbin (1953) as the tertiary component of the triphasic response to 5-HT. Several attempts were made in the past to identify the pharmacological profile of the receptors involved in this vasodepressor response, initially described as mediated by “5-HT1-like” receptors (see above; Saxena and Villalón 1990), which were resistant to agonists at 5-HT1A (8-OH-DPAT, indorenate), 5-HT1B/1D (sumatriptan), and other receptors. With the subsequent advent of selective agonists and antagonists for the different 5-HT receptor (sub)types, this response was clearly shown to be mediated by 5-HT7 receptors (Centurión et al. 2004; Saxena et al. 1998). Basically, the pharmacological profile of the 5-HT7 receptors mediating vasodepressor/direct vasodilator responses to 5-HT in several species includes the following characteristics: (1) mimicked by 5-CT and 5-methoxytryptamine, with a rank order of agonist potency of 5-CT>>5-HT≥5-methoxytryptamine, but not by sumatriptan (5-HT1B/1D), cisapride (5-HT4) or agonists of other 5-HT receptors; (2) resistant to blockade by 5-HT1A (pindolol, WAY 100635), 5-HT1B/1D (GR 127935), 5-HT2 (ketanserin, cyproheptadine or ritanserin), 5-HT3 (MDL72222 or granisetron), and 5-HT3/4 (tropisetron) receptor antagonists; and (3) blocked by methysergide, metergoline, methiothepin, lisuride, clozapine, and/or mesulergine (see Martin 1994; Saxena and Villalón 1990; Saxena et al. 1998; Villalón et al. 1997b). It is noteworthy that (1) the rank order of agonist potency of 5-CT>>5-HT≥5-methoxytryptamine (with sumatriptan and other 5-HT1 receptor agonists inactive) is a pharmacological fingerprint for the 5-HT7 receptor type, an order which is reversed for the other 5-HT receptor types (see Table 1; Hoyer et al. 1994); (2) methiothepin, methysergide, metergoline, lisuride, and clozapine have a high affinity for 5-HT6 and 5-HT7 binding sites/receptors (see Hoyer et al. 1994); and (3) mesulergine displays an almost 300-fold selectivity for 5-HT7 receptors (pK D = 8.15) over 5-HT6 receptors (pK D = 5.76) and does not interact with the 5-HT1 receptor family (see Hoyer et al. 1994).
Moreover, there is a good correlation between the vasodepressor activity of tryptamines and their affinity for the 5-HT7 binding site in anesthetized vagotomized rats (De Vries et al. 1997) and cats (Villalón et al. 2000). Further evidence clearly indicates that the late vasodepressor phase is primarily mediated by 5-HT7 receptors (Centurión et al. 2004). Nevertheless, it must be admitted that several mechanisms may probably contribute to different degrees in different experimental conditions and species. These mechanisms may include the following.
Direct vascular relaxation
The direct vasorelaxation to 5-HT also resembles the pharmacological profile of the 5-HT7 receptor (see above) in several blood vessels/vascular beds, for example: (1) in vitro in the cat saphenous vein, rabbit jugular vein, rabbit femoral vein, sheep pulmonary vein, canine coronary artery, canine femoral vein, and neonatal pig vena cava (see Hoyer et al. 1994; Martin 1994; Saxena et al. 1998; Villalón et al. 1997b); and (2) in vivo in the dog external/internal carotid vascular bed circulations (Villalón et al. 1997a; Centurión et al. 2000). Moreover, in the blood vessels where both 5-HT7 and 5-HT2 receptors are present and elicit opposing effects (relaxation by 5-HT7 receptors and contraction by 5-HT2 receptors), the ultimate response to 5-HT depends upon the pre-existing vascular tone, the dose employed, and the proportions in which the two receptors types are distributed. In vivo studies in our laboratory have shown that intracarotid infusions of 5-HT in anesthetized dogs produced (1) external carotid vasodilatation (Villalón et al. 1993) by both postsynaptic 5-HT7 (Villalón et al. 1997a) and prejunctional sympatho-inhibitory 5-HT1B (Villalón et al. 2001) receptors and (2) external carotid vasoconstriction by postjunctional 5-HT1B receptors (De Vries et al. 1998a). These—and other (see Saxena and Villalón 1990)—results indicate that the density of 5-HT7 and 5-HT2 receptors varies in different segments of a vascular bed. The vasodilator 5-HT7 receptor is located primarily on resistance blood vessels (De Vries et al. 1999), the vasoconstrictor 5-HT1B receptor on non-nutrient blood vessels (arteriovenous anastomoses; De Vries et al. 1998b), while the vasoconstrictor 5-HT2 receptor is mainly present on large conducting blood vessels (see Saxena and Villalón 1990). Therefore, 5-HT redistributes arterial blood flow in such a way that despite a decrease in the total blood flow, the arteriolar component, particularly in the skin, increases (see Saxena and Villalón 1990). The segmental distribution of 5-HT receptors is also, at least partly, responsible for the fact that in vitro studies, performed mainly with large conducting vessels, generally show a 5-HT2 receptor-mediated contractile response. In vivo, where presynaptic (see above) and reflex effects of 5-HT may modify the vascular responses, the amine causes vasodilatation in some vascular beds, and vasoconstriction in others. Interestingly, in the sheep pulmonary vein, 5-HT4 receptors mediate vasorelaxation (Cocks and Arnold 1992).
Prejunctional inhibition of vascular sympathetic neurons
The inhibitory action of 5-HT and related agonists on transmitter release from postganglionic sympathetic neurons has now been confirmed in many organs of different species including: (1) in vitro preparations such as the canine (Humphrey et al. 1988) and human (Molderings et al. 1990) saphenous veins, the canine (Cohen 1985) and porcine (Molderings et al. 1989) coronary arteries, as well as the rat kidney (Bond et al. 1989) and vena cava (Molderings et al. 1987); and (2) in vivo preparations such as the canine external carotid circulation (Villalón et al. 1993, 2001) as well as the rat cardiac (Villalón et al. 1999a; Sánchez-López et al. 2003, 2004) and vasopressor (Villalón et al. 1995a) sympathetic outflow.
With the exception of 5-HT-induced inhibition of the pig coronary sympathetic neurons, in which a “novel” receptor may exist (Molderings et al. 1989), the above 5-HT-induced vascular sympatho-inhibition is mainly mediated by 5-HT1 receptors. In this respect, for example, the i.v. 5-HT-induced sympatho-inhibition of the rat vasopressor sympathetic outflow is mediated by sympatho-inhibitory 5-HT1 receptors (Villalón et al. 1995b) on the basis that this response was (1) resistant to blockade by ritanserin, MDL7222, and tropisetron; (2) blocked by methysergide; and (3) potently mimicked by 5-CT. A subsequent pharmacological analysis using more selective agonists and antagonists revealed that these sympatho-inhibitory 5-HT1 receptors correlated with the 5-HT1A, 5-HT1B, and 5-HT1D subtypes, as the sympatho-inhibition to i.v. 5-HT was (1) mimicked by 8-OH-DPAT, indorenate, and sumatriptan and (2) blocked by WAY 100635 and GR 127935 (Villalón et al. 1998).
Obviously, the interference with the sympathetic neurotransmission by 5-HT will modify its direct cardiovascular effects. For example, in anesthetized dogs, 5-HT produces vasodilatation in the external carotid vascular bed when the sympathetic vascular tone is high and vasoconstriction when it is low (Villalón et al. 1993). This carotid vasodilatation is mediated, at least in part, by sympatho-inhibitory 5-HT1B receptors and, to a certain extent, by 5-HT7 receptors located on vascular smooth muscle (Villalón et al. 2001), while the carotid vasoconstriction is mainly mediated by 5-HT1B (rather than 5-HT1D) receptors (De Vries et al. 1998a).
Endothelium dependent vasorelaxation
In the absence of endothelium, the relaxant effect of 5-HT is attenuated, while the contractions are exaggerated in isolated blood vessels of the pig, dog, chick, horse, and rabbit (see Martin 1994; Saxena and Villalón 1990; Sumner 1991; Obi et al. 1994). These findings indicate that 5-HT can release endothelial nitric oxide, and this effect is mainly mediated by 5-HT1 receptors (Cohen et al. 1983; Martin et al. 1987; Houston and Vanhoutte 1988; Cocks and Arnold 1992). Interestingly, in porcine isolated blood vessels, the 5-HT-induced endothelium-dependent vasorelaxation is mediated by (1) 5-HT1B/1D receptors (stimulated by sumatriptan) in coronary arteries (Schoeffter and Hoyer 1990) or (2) 5-HT2B receptors in pulmonary arteries (Glusa and Richter 1993; Glusa and Pertz 2000). In vivo, the 5-HT-induced endothelium-dependent vasodilatation in dogs has thus far been studied indirectly by showing that the contractile responses to 5-HT but not to angiotensin II or phenylephrine, in the left anterior descending coronary artery, are enhanced after endothelial damage (Lamping et al. 1985).
Considering the above findings, it is noteworthy that the 5-HT2B receptor is also involved in the 5-HT-induced endothelium-dependent vasorelaxation in the rat jugular vein (Ellis et al. 1995); therefore, this mechanism may also be involved in the late vasodepressor response to i.v. 5-HT. Nevertheless, Centurión et al. (2004) reported that this is unlikely, as the selective 5-HT2B receptor agonist, BW723C86 produced dose-dependent vasopressor (rather than vasodepressor) responses and failed to affect the late vasodepressor responses to 5-HT. Consistent with this view, the nitric oxide synthetase inhibitor, NG-nitro-l-arginine methyl ester (L-NAME), failed to attenuate i.v. 5-HT- or 5-CT-induced vasodepressor responses in pithed rats (Van Gelderen and Saxena 1992). Although we have no clear-cut explanation for the above vasopressor responses to i.v. BW723C86, one cannot ignore that 5-HT2B receptors mediate vasoconstriction in rat mesenteric arteries (Watts and Fink 1999). Nevertheless, a central mechanism is unlikely, as i.c.v. administration of BW723C86 failed to increase blood pressure and heart rate in rats (Knowles and Ramage 2000). Therefore, the complete blockade produced by the 5-HT7 receptor antagonist SB269970 on both 5-HT- and 5-CT-induced vasodepressor responses in rats reconfirms the involvement of 5-HT7 receptors without a discernible participation of other mechanisms (Centurión et al. 2004). Admittedly, this conclusion (1) is based on the assumption that species differences between the affinities of SB269970 and BW723C86 do not play a major role and (2) cannot be categorically extrapolated to other species.
Vasodepressor and vasopressor responses by central mechanisms
Injection of 5-HT into the CNS has been reported to cause depressor, pressor, or biphasic responses (see Saxena and Villalón 1990; McCall and Clement 1994). The magnitude and the nature of the responses (pressor, depressor, or biphasic) to centrally administered 5-HT largely depend upon the exact site of application, the species, the dose employed, and/or whether conscious or anesthetized and normotensive or hypertensive animals are used (see Saxena and Villalón 1990; McCall and Clement 1994). This discrepancy may be due to the fact that 5-HT neurons in different brain areas have divergent effects; that is, dorsal and median raphe, anterior hypothalamus, and ventrolateral medullary raphe areas seem to be associated with pressor effects, whereas midline medullary raphe nuclei produce either pressor or depressor effects (see McCall and Clement 1994). These central pressor and depressor effects of 5-HT seem to be mediated via different 5-HT receptors in the CNS (Ramage 2001). Now, it is clear that the control of the cardiovascular system by central 5-HT neurons involves two main receptors, namely, 5-HT1A and 5-HT2 receptors. The former produces sympatho-inhibition, hypotension, and bradycardia while the latter produces sympatho-excitation and hypertension (see Ramage 2001).
As previously discussed, microinjections in the rostral ventrolateral medulla or i.v. bolus injections of 8-OH-DPAT produced bradycardia and hypotension susceptible to blockade by 5-HT1A receptor antagonists including WAY 100635 (McCall and Clement 1994). Thus, activation of 5-HT1A receptors produces central sympatho-inhibition. Indeed, renal nerve activity is decreased by i.v. injection of the 5-HT1A receptor agonist, 8-OH-DPAT (see McCall and Clement 1994). Hypotension, along with bradycardia and a decrease in the renal, cardiac, and splanchnic nerve activity, has also been shown to occur with the administration of 8-OH-DPAT, DP-5-CT, 5-CT, and 5-HT into the fourth ventricle in the cat (Shepheard et al. 1994). In addition, stimulation of 5-HT1A receptors in vagal preganglionic neurons located in the nucleus ambiguous and dorsal motor vagal nucleus increased cardiac vagal tone (see Ramage 2001).
5-HT also seems to act at the spinal level. In this respect, sympathetic preganglionic neurons located in the intermediolateral cell column of the spinal cord receive a dense serotonergic input (Thor et al. 1993). 5-HT2 receptors may be involved in the inhibitory effect on the sympathetic nerve activity because such an effect is mimicked by intrathecal administration of α-methyl-5-HT and antagonized by ketanserin but unaffected by prazosin, MDL 72222, or tropisetron (Yusof and Coote 1988).
Potential use of 5-HT receptor ligands in cardiovascular diseases
The cardiovascular pharmacology of 5-HT suggests that compounds acting on 5-HT receptors can be employed for therapeutic use in the treatment of several (cardio)vascular diseases/disorders including, among others, migraine, systemic, pulmonary, and portal hypertension, cardiac disorders, some peripheral vascular diseases, cerebral ischemia, etc (Robertson 1990, 1991; Saxena 1995; Saxena and Ferrari 1996; Saxena and Villalón 1990; Villalón et al. 1997b). In particular, the recognition of the 5-HT7 receptor as a functional receptor will undoubtedly disclose more therapeutic possibilities.
Migraine
Pathophysiology
Although the precise mechanisms behind migraine still remain elusive, several theories have been proposed to explain its pathophysiology (see Saxena 1972; Peroutka 2005; Villalón et al. 2002). As reviewed by Peroutka (2005), the “neurogenic inflammation theory of migraine” predicts that inhibitors of dural neurogenic inflammation in animal models should be effective in the acute treatment of migraine. Neurogenic inflammation (NI), however, consists of two major physiological components, namely: (1) plasma protein extravasation (PPE), mainly mediated by tachykinins and endothelin-3 and (2) neurogenic vasodilatation (NV), mediated predominantly by calcitonin gene-related peptide (CGRP) effects on vascular smooth muscle. Notwithstanding, as described below: (1) selective inhibitors of PPE (without affecting NV) in animal models, including lanepitant, GR 205171, L-758-298, FK888, dapitant, CP,122-288, 4991W93, and bosentan, were ineffective in the acute treatment of migraine (see Peroutka 2005); and (2) a single drug (BIBN4096 BS; a CGRP receptor antagonist) that specifically blocks CGRP-induced NV was effective in the acute treatment of migraine (Olesen et al. 2004). These findings suggest that the PPE component of NI may be far more relevant to lower mammal physiology than to humans. Thus, the dural “NI theory of migraine,” specifically as it predicts the induction of dural PPE in humans during a migraine attack, is no longer tenable (see below). In contrast, the CGRP-induced NV component of NI remains a molecular pathway that may play a key role in migraine pathophysiology (see Peroutka 2005).
Interaction with 5-HT receptors: an approach to antimigraine treatment
Stimulation of 5-HT1B, 5-HT1D and/or 5-ht1F receptors
Migraine treatment has evolved from the realms of the supernatural into the scientific arena (see Villalón et al. 2002). Many studies have conclusively shown that sumatriptan, a 5-HT1B/1D receptor agonist (Humphrey and Feniuk 1991) with moderate affinity for 5-ht1F receptors (Hoyer et al. 1994) produces constriction of cranial large arteries and carotid arteriovenous anastomoses (Centurión et al. 2001b; De Vries et al. 1998a, b) and is effective in aborting migraine attacks (see Saxena and Ferrari 1996; Villalón et al. 2002). The success of this drug has prompted a large number of pharmaceutical companies to develop novel 5-HT1B/1D receptor agonists (see Saxena 1995; Villalón et al. 1997b, 2002). In particular, efforts have been directed towards developing more lipid-soluble and selective compounds to improve oral bioavailability and to avoid coronary artery vasoconstriction. Although the new 5-HT1B/1D receptor agonists, e.g., rizatriptan, zolmitriptan, naratriptan, and eletriptan, appear to have a better oral bioavailability (see Saxena and Ferrari 1996; Villalón et al. 2002), they do not seem to differ with respect to their coronary side-effect potential (Maassen Van Den Brink et al. 1997).
To our knowledge, no study has yet reported the development of a selective 5-HT1B receptor agonist. Notwithstanding, several studies (using selective antagonists at 5-HT1B and 5-HT1D receptors as well as agonists at 5-ht1F receptors) support the notion that mainly 5-HT1B receptors are involved in sumatriptan-induced vasoconstriction of extracerebral (intra- and extracranial) blood vessels (Centurión et al. 2001b; De Vries et al. 1998a, b), which appear to be dilated during migraine (see NV above; Peroutka 2005). Unfortunately, the 5-HT1B receptor, being not exclusively confined to cranial blood vessels, is most likely also responsible for the moderate hypertension and coronary constriction noticed with triptans (Maassen Van Den Brink et al. 1997). In an attempt to avoid vasoconstriction, two additional avenues have been explored for potential antimigraine action within the bounds of serotonergic mechanisms, namely, selective agonists at 5-HT1D and 5-ht1F receptors.
PNU-142633 is a selective 5-HT1D receptor agonist (see Table 1), which: (1) was more potent than sumatriptan in preventing dural PPE (McCall et al. 2002); and (2) failed to produce vasoconstriction in the extracranial carotid circulations of anesthetized cats (McCall et al. 2002) and dogs (Muñoz-Islas et al. 2006). However, PNU-142633 was ineffective in the acute treatment of migraine (Gómez-Mancilla et al. 2001).
Moreover, LY334370 is a selective 5-ht1F receptor agonist (see Table 1) which: (1) potently inhibited dural PPE (Johnson et al. 1997); (2) failed to produce vasoconstriction in the rabbit saphenous vein (Cohen and Schenck 2000); and (3) was clinically effective to abort migraine attacks (Goldstein et al. 2001). Although, as described above, dural PPE is not important in the pathophysiology of migraine (Peroutka 2005), it has to be emphasized that LY334370 displayed antimigraine activity at doses that may have interacted with extracranial vasoconstrictor 5-HT1B receptors (Goldstein et al. 2001). Consistent with the latter notion: (1) some compounds, for example, neurokinin NK1 and endothelin ETA/B receptor antagonists and CP-122,288, all of which potently inhibit PPE (Gupta et al. 1995; Shepheard et al. 1995; Brandli et al. 1996), failed to show clinical efficacy in migraine (Goldstein et al. 1996; May et al. 1996); (2) sumatriptan (pK i, 7.63 and 7.94) has a higher affinity than ergotamine (pK i, 6.76) for the 5-ht1F receptor (Adham et al. 1993) and yet sumatriptan is a less potent antimigraine agent on the basis of the parenteral doses used in migraine (sumatriptan, 6 mg, s.c.; ergotamine, 0.25–0.5mg, i.m.); (3) the inhibitory action of sumatriptan on PPE, but not of CP-122,288, is blocked by GR 127935 (Yu et al. 1997); (4) GR 127935 has a higher affinity for 5-HT1B/1D receptors (pK i, 9.9/8.9; Skingle et al. 1996) than for the 5-ht1F receptor (pK i, 7.1; Hoyer et al. 1994); (5) the 5-HT1B/1D receptor agonists rizatriptan and alniditan, which are effective in migraine (Goldstein et al. 1996), have little affinity for the 5-ht1F receptor (Leysen et al. 1996); and (6) the vasoconstriction of the human middle meningeal artery (Razzaque et al. 1999) and canine extracranial external carotid circulation (De Vries et al. 1998a) by sumatriptan is mainly mediated by the 5-HT1B (rather than the 5-HT1D or 5-ht1F) receptor, reinforcing the view that the antimigraine activity of sumatriptan and second generation triptans is dependent upon their interaction with the 5-HT1B receptor. Thus, in the absence of the importance of dural PPE in migraine (Peroutka 2005), further experiments will be needed to explain the antimigraine efficacy of LY334370.
Blockade of 5-HT2A, 5-HT2B, and/or 5-HT2C receptors
Some antimigraine drugs are potent antagonists at 5-HT2A receptors (e.g., methysergide, pizotifen, ergotamine, and dihydroergotamine), but many other such agents (e.g., ketanserin, cyproheptadine, mianserin, methiothepin) are not of much use in migraine therapy (Saxena 1995). It has also been proposed that 5-HT2B/2C receptors may be involved in the initiation of migraine attacks (Kalkman 1994; Fozard and Kalkman 1994; Fozard 1995). Thus, selective antagonism of these receptors, in particular, the 5-HT2B receptor, which may mediate nitric oxide release from vascular endothelium (Hoyer et al. 1994; Fozard 1995), would be effective in migraine prophylaxis (Kalkman 1994; Fozard and Kalkman 1994; Fozard 1995). However, as already discussed elsewhere (Saxena 1995), several 5-HT2B/2C receptor antagonists, including mianserin and cyproheptadine, are not very effective antimigraine agents. It seems, therefore, that additional properties, including the vasoconstriction in the extracranial external carotid circulation in the case of methysergide (Villalón et al. 1999b), ergotamine (Valdivia et al. 2004), and dihydroergotamine (Villalón et al. 2004) partly via 5-HT1B receptors and the antidepressant action in the case of pizotifen, may be necessary for therapeutic antimigraine action (Villalón et al. 1997b).
Blockade of 5-HT3 receptors
As no clear-cut antimigraine effect has been found with any 5-HT3 receptor antagonist (Ferrari 1991; Ferrari et al. 1991), this receptor does not appear to play a major role in migraine.
Blockade of novel receptors
Lastly, it may be pointed out that the constriction of porcine carotid arteriovenous anastomoses elicited by ergotamine, dihydroergotamine, or 5-HT (the latter in the presence of ketanserin) is not very susceptible to blockade by GR 127935. This suggests the involvement of a receptor other than the 5-HT2A and 5-HT1B/1D receptors (De Vries et al. 1998b). Hence, it will be interesting to characterize this receptor further and explore whether it can be a target for developing novel antimigraine drugs.
Systemic, portal, and pulmonary hypertension
Systemic hypertension
Both urapidil (5-HT1A receptor agonist) and ketanserin (5-HT2A receptor antagonist) have been approved for the treatment of systemic hypertension (Villalón et al. 1997b). Indeed, it is claimed that these drugs decrease blood pressure by stimulating 5-HT1A receptors located centrally in the rostral ventrolateral medulla (urapidil) or by blocking 5-HT2A receptors mediating peripheral vasoconstriction (ketanserin). However, as discussed in detail elsewhere (see Saxena 1995; Saxena and Villalón 1990), it seems that such effects are not involved to a significant degree in the clinical effects of these drugs; both urapidil and ketanserin have a potent α1-adrenoceptor antagonist activity, which can adequately explain their antihypertensive effect.
In the complex setting of cardiac surgery and cardiopulmonary bypass, 5-HT has been shown to be one of the mediators involved in the origin of systemic hypertension during and after cardiac surgery; indeed, subthreshold or threshold doses of 5-HT amplify the vasoconstrictor responses to, for example, adrenaline and noradrenaline, the levels of which are significantly elevated under these conditions (see Reneman and van der Starre 1990). That 5-HT plays a role through its amplifying effect is supported by the finding that ketanserin effectively lowers arterial blood pressure in patients with systemic postoperative hypertension by combined blockade of 5-HT2A receptors and α1-adrenoceptors (see Reneman and van der Starre 1990).
Moreover, in view of the hypotension mediated by vascular 5-HT7 receptors (Centurión et al. 2004; De Vries et al. 1997), it may be worthwhile exploring selective 5-HT7 receptor agonists as antihypertensive agents. In this regard, however, one has to be aware of the competition with excellent drugs already available.
Portal hypertension
5-HT may also be involved in some cases of portal hypertension, where higher levels of plasma 5-HT are found in the portal venous circulation (Robertson 1991). This view is reinforced by the decrease in portal pressure produced by: (1) ketanserin, a 5-HT2A and α1-adrenoceptor antagonist, which also decreased portal-systemic collateral blood flow in cirrhotic patients, whereas systemic blood pressure slightly decreased (see Lebrec 1990); or (2) ritanserin, a 5-HT2A/2B/2C receptor antagonist (at doses without α1-adrenoceptor blockade), which produced no systemic hemodynamic changes in both cirrhotic patients (see Lebrec 1990) and portal-hypertensive rats (Nevens et al. 1991). These findings support a role for the actions of 5-HT via 5HT2 receptors in portal hypertension and add 5-HT2 receptor antagonists as a group of drugs for its therapeutic treatment.
Pulmonary hypertension
Pulmonary arterial hypertension (PAH) is a rare and often fatal disease characterized by an increase in pulmonary artery pressure associated with abnormal vascular proliferation and irreversible pulmonary remodeling (see Montani et al. 2004). Several lines of evidence have suggested a role for 5-HT in the etiology of PAH, as this monoamine may have a dual effect on the pulmonary circulation, contributing to both vasoconstriction and vascular remodeling (see MacLean 1999). Indeed, platelet and plasma 5-HT levels are increased in both primary PAH and PAH that is secondary to (1) cardiovascular (e.g., left ventricular failure) and/or pulmonary (e.g., chronic lung obstruction) diseases; (2) hypoxic conditions; or (3) use of anorectic agents (see Fishman 1998; MacLean 1999). The latter drugs (e.g., dexfenfluramine and fenfluramine) produce 5-HT release from neurons and platelets and inhibit 5-HT reuptake (Fishman 1999; Hervé et al. 1995).
In pharmacological experiments, Morecroft et al. (1999) have shown that 5-HT-induced contraction in the human pulmonary artery was potently mimicked by sumatriptan (5-HT1B/1D receptor agonist), a response that was (1) blocked by SB224289 (a 5-HT1B receptor antagonist; pK B = 8.4) and (2) resistant to blockade by BRL15572 (a 5-HT1D receptor antagonist). These findings indicate that the 5-HT1B receptor mediates contraction in the human pulmonary artery and may explain why ketanserin has been of limited use in the treatment of PAH (see MacLean 1999).
Interestingly, the 5-HT2B receptor has also been involved in the pathogenesis of PAH. In this respect, Launay et al. (2002) have reported, using the chronic-hypoxic-mouse model of PAH, that (1) hypoxia-dependent increase in pulmonary blood pressure and lung remodeling are associated with an increase in vascular proliferation; (2) the increase is vascular proliferation is potentiated by dexfenfluramine, whose active metabolite (nor-dexfenfluramine) is a selective 5-HT2B receptor agonist (Fitzgerald et al. 2000); and (3) a genetic deficiency in 5-HT2B receptors or selective blockade of 5-HT2B receptors (using 1mg/kg per day of RS-127445; see Table 1) manifested no change in vascular proliferation. Thus, Launay et al. (2002) have shown that (1) a substantial increase in 5-HT2B receptor expression is induced in the pulmonary arteries of mice with PAH; (2) selective blockade of 5-HT2B receptors prevented PAH; and (3) the 5-HT2B receptor is a limiting factor in chronic hypoxia-induced pulmonary vascular proliferation.
In summary, all of the above data, taken together, clearly suggest that selective antagonists at 5-HT1B and/or 5-HT2B receptors may have potential therapeutic usefulness in the treatment of PAH.
Cardiac disorders
As previously discussed, i.v. 5-HT-induced tachycardia in the healthy rat is mediated by 5-HT2A receptors (Centurión et al. 2002). However, after congestive heart failure, the cardiostimulation produced by 5-HT is mediated by 5-HT4 and 5-HT2A receptors (Qvigstad et al. 2005a, b). These findings raise the possibility that changes in the density/expression of 5-HT receptors may also occur during congestive human heart failure (see Kaumann and Levy 2006).
5-HT4 receptors have been shown to mediate increases in the rate and contractility in the human atrium (Kaumann 1993; Kaumann and Levy 2006; Kaumann et al. 1989, 1990). As 5-HT can induce arrhythmias in the human isolated atrium, it is proposed that 5-HT4 receptor antagonists could be useful in the treatment of cardiac arrhythmias (see Kaumann and Levy 2006). However, the role of 5-HT, if any, in the pathogenesis of cardiac arrhythmias has not been definitely established. Thus, it seems unlikely that such drugs will be effective in this disorder.
On the other hand, the increase in atrial contractility and arrhythmic effect by 5-HT4 receptor agonists may suggest that these drugs could have application in the therapy of heart failure (see Kaumann and Levy 2006), but one has to be aware of the competition with excellent drugs already available. Interestingly, functional 5-HT4 receptors are absent in human healthy ventricles (Jahnel et al. 1992; Schoemaker et al. 1993) but are present in human failing ventricles (Brattelid et al. 2004). In this respect, Qvigstad et al. (2005a, b) have observed a positive inotropic response to 5-HT in the left ventricle of rats with congestive heart failure. This response (1) was not observed in control (healthy) hearts; (2) became apparent after large myocardial infarctions and also in the absence of congestive heart failure; and (3) was mediated through the 5-HT4 receptor, which also mediates this response in the human failing ventricle (Brattelid et al. 2004). This 5-HT4 receptor-mediated response was characterized by an increase in contractile force as well as by a more rapid relaxation, features similar to those of the β-adrenoceptor-mediated contractile response.
The 5-HT4 receptor is known to activate adenylyl cyclase through the stimulating G protein Gs, and signals by increasing cAMP levels (see Table 1). This was also found in the failing myocardium of humans and rats (Brattelid et al. 2004; Qvigstad et al. 2005a) and parallels the signaling mechanism for the β-adrenoceptor system. Taken together, these findings suggest that the abnormal expression of the 5-HT4 receptor after large myocardial infarctions could be maladaptive, for example, through induction of myocardial remodeling. On this basis, Birkeland et al. (2007) hypothesized that the upregulation of the 5-HT4 receptor in post-infarction congestive heart failure is maladaptive and that chronic blockade of the 5-HT4 receptor would reduce myocardial remodeling and improve cardiac function. Indeed, these authors found that treatment with a 5-HT4 receptor antagonist to some extent improved in vivo cardiac function in rats. In addition, ex vivo β-adrenoceptor responsiveness increased, and 5-HT4 receptor-mediated signaling decreased, consistent with some beneficial effects of this treatment (Birkeland et al. 2007).
Peripheral vascular diseases
5-HT has been implicated in the pathophysiology of some peripheral vascular diseases, but the evidence does not seem to be compelling (Coffman 1991). Although some clinical trials show a moderate efficacy of ketanserin (Coffman 1991), this drug is not universally registered for this indication. It is however interesting to point out that one of the prominent cardiovascular effects of i.v 5-HT is its ability to produce vasodilatation via stimulation of 5-HT7 receptors (Centurión et al. 2004; De Vries et al. 1997; Villalón et al. 1997a). Therefore, selective agonists at vascular 5-HT7 receptors may be expected to enhance capillary blood flow and be useful in the treatment of peripheral vascular diseases, including trophic skin ulcers. This approach might also have potential applications, as yet unexplored, in the medical treatment of skin grafts and baldness.
Cerebral ischemia
Buspirone and ipsapirone, anxiolytic agents with an action at central 5-HT1A receptors (Saxena 1995), have been proven to decrease the infarct size in animal models of focal (Bielenberg and Burkhardt 1990) and global (Bode-Greuel et al. 1990) cerebral ischemia. Notwithstanding, the involvement of 5-HT1A receptors is questionable because these drugs are non-selective agents and, most significantly, 8-OH-DPAT, which is also a potent 5-HT1A receptor agonist (see Table 1), was ineffective in the above experimental models (Silver et al. 1996).
Concluding remarks
Research in the field of 5-HT has been boosted by the advent of potent and selective agonists and antagonists. These drug tools and the increasing understanding of the transduction mechanisms and the structure of the receptor protein have enabled the characterization and nomenclature of 5-HT receptors in a more meaningful way (see Table 1). Indeed, the molecular cloning and expression of a growing number of 5-HT receptors in host cells now offer the possibility to screen new molecules easily. This will help recognize selective ligands, which could in turn provide access to better drugs for a more efficient treatment of human ailments.
References
Adham N, Kao HT, Schechter LE, Bard J, Olsen M, Urquhart D, Durkin M, Hartig PR, Weinshank RL, Branchek TA (1993) Cloning of another human serotonin receptor (5-HT1F): a fifth 5-HT1 receptor subtype coupled to the inhibition of adenylate cyclase. Proc Nat Acad Sci U S A 90:408–412
Anderson IK, Martin GR, Ramage AG (1992) Central administration of 5-HT activates 5-HT1A receptors to cause sympathoexcitation and 5-HT2/5-HT1C receptors to release vasopression in anaesthetized rats. Br J Pharmacol 107:1020–1028
Anderson IK, Martin GR, Ramage AG (1995) Evidence that activation of 5-HT2 receptors in the brain of anaesthetized cats causes sympathoexcitation. Br J Pharmacol 116:1751–1756
Apperley E, Humphrey PP, Levy GP (1976) Receptors for 5-hydroxytryptamine and noradrenaline in rabbit isolated ear artery and aorta. Br J Pharmacol 58:211–221
Barnes NM, Sharp T (1999) A review of central 5-HT receptors and their function. Neuropharmacology 38:1083–1152
Berthold H, Scholtysik G, Engel G (1989) Inhibition of the 5-HT-induced cardiogenic hypertensive chemoreflex by the selective 5-HT3 receptor antagonist ICS 205–930. Naunyn-Schmiedeberg’s Arch Pharmacol 339:259–262
Bielenberg GW, Burkhardt M (1990) 5-hydroxytryptamine1A agonists. A new therapeutic principle for stroke treatment. Stroke 21:IV161–IV163
Birkeland JA, Sjaastad I, Brattelid T, Qvigstad E, Moberg ER, Krobert KA, Bjornerheim R, Skomedal T, Sejersted OM, Osnes JB, Levy FO (2007) Effects of treatment with a 5-HT4 receptor antagonist in heart failure. Br J Pharmacol 150:143–152
Blauw GJ, van Brummelen P, van Zwieten PA (1988) Serotonin induced vasodilatation in the human forearm is antagonized by the selective 5-HT3 receptor antagonist ICS 205–930. Life Sci 43:1441–1449
Bode-Greuel KM, Klisch J, Horvath E, Glaser T, Traber J (1990) Effects of 5-hydroxytryptamine1A-receptor agonists on hippocampal damage after transient forebrain ischemia in the Mongolian gerbil. Stroke 21:IV164–IV166
Bond RA, Craig DA, Charlton KG, Ornstein AG, Clarke DE (1989) Partial agonistic activity of GR43175 at the inhibitory prejunctional 5-HT1-like receptor in rat kidney. J Auton Pharmacol 9:201–210
Bradley PB, Engel G, Feniuk W, Fozard JR, Humphrey PP, Middlemiss DN, Mylecharane EJ, Richardson BP, Saxena PR (1986) Proposals for the classification and nomenclature of functional receptors for 5-hydroxytryptamine. Neuropharmacology 25:563–576
Brandli P, Loffler BM, Breu V, Osterwalder R, Maire JP, Clozel M (1996) Role of endothelin in mediating neurogenic plasma extravasation in rat dura mater. Pain 64:315–322
Brattelid T, Qvigstad E, Lynham JA, Molenaar P, Aass H, Geiran O, Skomedal T, Osnes JB, Levy FO, Kaumann AJ (2004) Functional serotonin 5-HT4 receptors in porcine and human ventricular myocardium with increased 5-HT4 mRNA in heart failure. Naunyn-Schmiedeberg’s Arch Pharmacol 370:157–166
Brodie TG (1900) The immediate action of an intravenous injection of blood-serum. J Physiol (London) 26:48–71
Centurión D, Glusa E, Sánchez-López A, Valdivia LF, Saxena PR, Villalón CM (2004) 5-HT7, but not 5-HT2B, receptors mediate hypotension in vagosympathectomized rats. Eur J Pharmacol 502:239–242
Centurión D, Ortiz MI, Sánchez-López A, De Vries P, Saxena PR, Villalón CM (2001a) Evidence for 5-HT1B/1D and 5 HT2A receptors mediating constriction of the canine internal carotid circulation. Br J Pharmacol 132:983–990
Centurión D, Ortiz MI, Saxena PR, Villalón CM (2002) The atypical 5-HT2 receptor mediating tachycardia in pithed rats: pharmacological correlation with the 5-HT2A receptor subtype. Br J Pharmacol 135:1531–1539
Centurión D, Sánchez-López A, De Vries P, Saxena PR, Villalón CM (2001b) The GR127935-sensitive 5-HT1 receptors mediating canine internal carotid vasoconstriction: resemblance to the 5-HT1B, but not to the 5-HT1D or 5-ht1F receptor subtype. Br J Pharmacol 132:991–998
Centurión D, Sánchez-López A, Ortiz MI, De Vries P, Saxena PR, Villalón CM (2000) Mediation of 5-HT-induced internal carotid vasodilatation in GR127935-and ritanserin-pretreated dogs by 5-HT7 receptors. Naunyn-Schmiedeberg’s Arch Pharmacol 362:169–176
Chaouche-Teyara K, Fournier B, Safar M, Dabiré H (1993) Vascular and cardiac effects of alpha-methyl-5-HT and DOI are mediated by different 5-HT receptors in the pithed rat. Eur J Pharmacol 250:67–75
Chaouche-Teyara K, Fournier B, Safar M, Dabiré H (1994) Systemic and regional haemodynamic effects of 1-(2,5-dimethoxy-4-iodo-phenyl)-2-aminopropane (DOI) and alpha-methyl-5-HT, in the anaesthetised rat. Clin Exp Hypertens 16:779–798
Cocks TM, Arnold PJ (1992) 5-Hydroxytryptamine (5-HT) mediates potent relaxation in the sheep isolated pulmonary vein via activation of 5-HT4 receptors. Br J Pharmacol 107:591–596
Coffman JD (1991) Raynaud’s phenomenon. An update. Hypertension 17:593–602
Cohen ML, Schenck K (2000) Contractile responses to sumatriptan and ergotamine in the rabbit saphenous vein: effect of selective 5-HT(1F) receptor agonists and PGF(2alpha). Br J Pharmacol 131:562–568
Cohen RA (1985) Serotonergic prejunctional inhibition of canine coronary adrenergic nerves. J Pharmacol Exp Ther 235:76–80
Cohen RA, Shepherd JT, Vanhoutte PM (1983) Inhibitory role of the endothelium in the response of isolated coronary arteries to platelets. Science 221:273–274
Creite A (1869) Versuche über die Wirkung des Serumeiweisses nach Injection in das Blut. Zeitschrift für Rationelle Medicin 36:90–108
Dabiré H, Chaouche-Teyara K, Cherqui C, Fournier B, Laubie M, Schmitt H (1989a) Characterization of DOI, a putative 5-HT2 receptor agonist in the rat. Eur J Pharmacol 168:369–374
Dabiré H, Chaouche-Teyara K, Cherqui C, Fournier B, Schmitt H (1989b) DOI is a mixed agonist-antagonist at postjunctional 5-HT2 receptors in the pithed rat. Eur J Pharmacol 170:109–111
Damaso EL, Bonagamba LG, Kellett DO, Jordan D, Ramage AG, Machado BH (2007) Involvement of central 5-HT7 receptors in modulation of cardiovascular reflexes in awake rats. Brain Res 1144:82–90
De Vries P, De Visser PA, Heiligers JPC, Villalón CM, Saxena PR (1999) Changes in systemic and regional haemodynamics during 5-HT7 receptor-mediated depressor responses in rats. Naunyn-Schmiedeberg’s Arch Pharmacol 359:331–338
De Vries P, Sánchez-López A, Centurión D, Heiligers JP, Saxena PR, Villalón CM (1998a) The canine external carotid vasoconstrictor 5-HT1 receptor: blockade by 5-HT1B (SB224289), but not by 5-HT1D (BRL15572) receptor antagonists. Eur J Pharmacol 362:69–72
De Vries P, Villalón CM, Heiligers JP, Saxena PR (1997) Nature of 5-HT1-like receptors mediating depressor responses in vagosympathectomized rats; close resemblance to the cloned 5-ht7 receptor. Naunyn-Schmiedeberg’s Arch Pharmacol 356:90–99
De Vries P, Villalón CM, Heiligers JP, Saxena PR (1998b) Characterization of 5-HT receptors mediating constriction of porcine carotid arteriovenous anastomoses; involvement of 5-HT1B/1D and novel receptors. Br J Pharmacol 123:1561–1570
Dhasmana KM, De Boer HJ, Banerjee AK, Saxena PR (1988) Analysis of the tachycardiac response to 5-hydroxytryptamine in the spinal guinea-pig. Eur J Pharmacol 145:67–73
Docherty JR (1988) Investigations of cardiovascular 5-hydroxytryptamine receptor subtypes in the rat. Naunyn-Schmiedeberg’s Arch Pharmacol 337:1–8
Eglen RM, Whiting RL (1989) Comparison of the positive chronotropic response to 5-hydroxytryptamine with beta-adrenoceptor agonists on the guinea pig atria in vitro. J Cardiovasc Pharmacol 13:45–51
Ellis E, Byrne C, Murphy OE, Tilford NS, Baxter GS (1995) Mediation by 5-hydroxytryptamine2B receptors of endothelium-dependent relaxation in rat jugular vein. Br J Pharmacol 114:400–404
El Rawadi C, Davy M, Midol-Monnet M, Cohen Y (1994) Biochemical characterization of the mechanisms involved in the 5-hydroxytryptamine-induced increase in rat atrial rate. Biochem Pharmacol 48:683–688
Erspamer V (1940a) Pharmakologische studien über Enteramine. I. Miteilung: Über die Wirkung von Acetonextrakten der Kaninchenmagenschleimhaut auf den Blutdruck und auf isolierte überlebende Organe. Arch Exper Path U Pharmakol 196:343
Erspamer V (1940b) Pharmakologische studien über Enteramine. II. Mitteilung: Über eigenschaften des Enteramins, sovie über die Abgrenzung des Enteramins von der anderen Kreislaufwirksamen Gewebsprodukten. Arch Exper Path U Pharmakol 196:366
Erspamer V (1940c) Pharmakologische studien über Enteramine. III. Mitteilung: Über das vorhandensein eines enteraminähnlichen Stoffes in Milzextrakten. Arch Exper Path U Pharmakol 196:391
Erspamer V, Asero B (1952) Identification of enteramine, the specific hormone of the enterochromaffin cell system, as 5-hydroxytryptamine. Nature 169:800–801
Ferrari MD (1991) 5-HT3 receptor antagonists and migraine therapy. J Neurol 238(Suppl 1):S53–S56
Ferrari MD, Wilkinson M, Hirt D, Lataste X, Notter M (1991) Efficacy of ICS 205–930, a novel 5-hydroxytryptamine3 (5-HT3) receptor antagonist, in the prevention of migraine attacks. A complex answer to a simple question. ICS 205-930 Migraine Study Group. Pain 45:283–291
Fishman AP (1998) Etiology and pathogenesis of primary pulmonary hypertension: a perspective. Chest 114(Suppl 3):242S–247S
Fishman AP (1999) Aminorex to fen/phen: an epidemic foretold. Circulation 99:156–161
Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay MH, Valentine PA, Sun JH, Link JR, Abbaszade I, Hollis JM, Largent BL, Hartig PR, Hollis GF, Meunier PC, Robichaud AJ, Robertson DW (2000) Possible role of valvular serotonin 5-HT2B receptors in the cardiopathy associated with fenfluramine. Mol Pharmacol 57:75–81
Fozard JR (1984) Neuronal 5-HT receptors in the periphery. Neuropharmacology 23:1473–1486
Fozard JR (1995) The 5-hydroxytryptamine-nitric oxide connection: the key link in the initiation of migraine. Arch Int Pharmacodyn Ther 329:111–119
Fozard JR, Kalkman HO (1994) 5-Hydroxytryptamine (5-HT) and the initiation of migraine: new perspectives. Naunyn-Schmiedeberg’s Arch Pharmacol 350:225–229
Gaddum JH, Picarelli ZP (1957) Two kinds of tryptamine receptors. Br J Pharmacol 12:323–328
Glusa E, Pertz HH (2000) Further evidence that 5-HT-induced relaxation of pig pulmonary artery is mediated by endothelial 5-HT2B receptors. Br J Pharmacol 130:692–698
Glusa E, Richter M (1993) Endothelium-dependent relaxation of porcine pulmonary arteries via 5- HT1C-like receptors. Naunyn-Schmiedeberg’s Arch Pharmacol 347:471–477
Goldstein J, Dählof CG, Diener HC, Olesen J, Schellens R, Senard JM, Simard D, Steiner TJ (1996) Alniditan in the acute treatment of migraine attacks: a subcutaneous dose-finding study. Subcutaneous Alniditan Study Group. Cephalalgia 16:497–502
Goldstein DJ, Roon KI, Offen WW, Ramadan NM, Phebus LA, Johnson KW, Schaus JM, Ferrari MD (2001) Selective serotonin1F (5-HT1F) receptor agonist LY334370 for acute migraine: a randomised controlled trial. Lancet 358:1230–1234
Gómez-Mancilla B, Cutler NR, Leibowitz MT, Spierings EL, Klapper JA, Diamond S, Goldstein J, Smith T, Couch JR, Fleishaker J, Azie N, Blunt DE (2001) Safety and efficacy of PNU-142633, a selective 5-HT1D agonist, in patients with acute migraine. Cephalalgia 21:727–732
Göthert M, Schlicker E, Kollecker P (1986) Receptor-mediated effects of serotonin and 5-methoxytryptamine on noradrenaline release in the rat vena cava and in the heart of the pithed rat. Naunyn-Schmiedeberg’s Arch Pharmacol 332:124–130
Gupta P, Brown D, Butler P, Ellis P, Grayson KL, Land GC, Macor JE, Robson SF, Wythes MJ, Shepperson NB (1995) The in vivo pharmacological profile of a 5-HT1 receptor agonist, CP-122,288, a selective inhibitor of neurogenic inflammation. Br J Pharmacol 116:2385–2390
Hagan JJ, Slade PD, Gaster L, Jeffrey P, Hatcher JP, Middlemiss DN (1997) Stimulation of 5-HT1B receptors causes hypothermia in the guinea pig. Eur J Pharmacol 331:169–174
Hervé P, Launay JM, Scrobohaci ML, Brenot F, Simonneau G, Petitpretz P, Poubeau P, Cerrina J, Duroux P, Drouet L (1995) Increased plasma serotonin in primary pulmonary hypertension. Am J Med 99:249–254
Hamlin KE, Fischer FE (1951) The synthesis of 5-hydroxytryptamine. J Am Chem Soc 73:5007–5008
Hirose K (1918) Relation between the platelet count of human blood and its vasoconstrictor action after clotting. Arch Int Med 21:604–612
Houston DS, Vanhoutte PM (1988) Comparison of serotonergic receptor subtypes on the smooth muscle and endothelium of the canine coronary artery. J Pharmacol Exp Ther 244:1–10
Hoyer D, Clarke DE, Fozard JR, Hartig PR, Martin GR, Mylecharane EJ, Saxena PR, Humphrey PP (1994) International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (Serotonin). Pharmacol Rev 46:157–203
Hoyer D, Hannon JP, Martin GR (2002) Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol Biochem Behav 71:533–554
Hoyer D, Martin G (1997) 5-HT receptor classification and nomenclature: towards a harmonization with the human genome. Neuropharmacology 36:419–428
Humphrey PP, Feniuk W (1991) Mode of action of the anti-migraine drug sumatriptan. Trends Pharmacol Sci 12:444–446
Humphrey PP, Feniuk W, Perren MJ, Connor HE, Oxford AW, Coates LH, Butina D (1988) GR43175, a selective agonist for the 5-HT1-like receptor in dog isolated saphenous vein. Br J Pharmacol 94:1123–1132
Ireland SJ, Jordan CC (1987) Pharmacological characterization of 5-hydroxytryptamine-induced hyperpolarization of the rat cervical ganglion. Br J Pharmacol 92:417–427
Jahnel U, Rupp J, Ertl R, Nawrath H (1992) Positive inotropic response to 5-HT in human atrial but not in ventricular heart muscle. Naunyn- Schmiedeberg’s Arch Pharmacol 346:482–485
Janeway TC, Richardson HB, Park EA (1918) Experiments on the vasoconstrictor action of blood serum. Arch Int Med 21:565–603
Johnson KW, Schaus JM, Durkin MM, Audia JE, Kaldor SW, Flaugh ME, Adham N, Zgombick JM, Cohen ML, Branchek TA, Phebus LA (1997) 5-HT1F receptor agonists inhibit neurogenic dural inflammation in guinea pigs. Neuroreport 8:2237–2240
Jones JF, Martin GR, Ramage AG (1995) Evidence that 5-HT1D receptors mediate inhibition of sympathetic ganglionic transmission in anaesthetized cats. Br J Pharmacol 116:1715–1717
Kalkman HO (1994) Is migraine prophylactic activity caused by 5-HT2B or 5-HT2C receptor blockade. Life Sci 54:641–644
Kaumann AJ (1993) Blockade of human atrial 5-HT4 receptors by GR 113808. Br J Pharmacol 110:1172–1174
Kaumann AJ, Levy FO (2006) 5-Hydroxytryptamine receptors in the human cardiovascular system. Pharmacol Ther 111:674–706
Kaumann AJ, Murray KJ, Brown AM, Sanders L, Brown MJ (1989) A receptor for 5-HT in human atrium. Br J Pharmacol 98:664
Kaumann AJ, Sanders L, Brown AM, Murray KJ, Brown MJ (1990) A 5-hydroxytryptamine receptor in human atrium. Br J Pharmacol 100:879–885
Kellett DO, Ramage AG, Jordan D (2005) Central 5-HT7 receptors are critical for reflex activation of cardiac vagal drive in anaesthetized rats. J Physiol 563:319–331
Kimura T, Satoh S (1983) Presynaptic inhibition by serotonin of cardiac sympathetic transmission in dogs. Clin Exp Pharmacol Physiol 10:535–542
Knowles ID, Ramage AG (2000) Evidence that activation of central 5-HT2B receptors causes renal sympathoexcitation in anaesthetised rats. Br J Pharmacol 129:177–183
Krstic MK, Katusic ZS (1982) Divergent effects of cyproheptadine and R 50 656, a 5-HT2 antagonist, on the cardiovascular response to 5-hydroxytryptamine in rats. Eur J Pharmacol 85:225–227
Kuwasawa K, Hill R (1997) Evidence for cholinergic inhibitory and serotonergic excitatory neuromuscular transmission in the heart of the bivalve Mercenaria mercenaria. J Exp Biol 200:2123–2135
Lamping KG, Marcus ML, Dole WP (1985) Removal of the endothelium potentiates canine large coronary artery constrictor responses to 5-hydroxytryptamine in vivo. Circ Res 57:46–54
Launay JM, Herve P, Peoc’h K, Tournois C, Callebert J, Nebigil CG, Etienne N, Drouet L, Humbert M, Simonneau G, Maroteaux L (2002) Function of the serotonin 5-HT2B receptor in pulmonary hypertension. Nat Med 8:1129–1135
Lebrec D (1990) Portal hypertension: serotonin and pathogenesis. Cardiovasc Drugs Ther 4(Suppl 1):33–35
Leysen JE, Gommeren W, Heylen L, Luyten WH, van de Weyer I, Vanhoenacker P, Haegeman G, Schotte A, van Gompel P, Wouters R, Lesage AS (1996) Alniditan, a new 5-hydroxytryptamine1D agonist and migraine-abortive agent: ligand-binding properties of human 5-hydroxytryptamine1D alpha, human 5-hydroxytryptamine1D beta, and calf 5-hydroxytryptamine1D receptors investigated with [3H]5-hydroxytryptamine and [3H]alniditan. Mol Pharmacol 50:1567–1580
Maassen Van Den Brink A, Reekers M, Bax WA, Ferrari MD, Saxena PR (1997) Current and future anti-migraine drugs in the human isolated coronary artery. Cephalalgia 17:244–391
MacLean MR (1999) Pulmonary hypertension, anorexigens and 5-HT: pharmacological synergism in action. Trends Pharmacol Sci 20:490–495
Marinesco S, Kolkman KE, Carew TJ (2004a) Serotonergic modulation in aplysia. I. Distributed serotonergic network persistently activated by sensitizing stimuli. J Neurophysiol 92:2468–2486
Marinesco S, Wickremasinghe N, Kolkman KE, Carew TJ (2004b) Serotonergic modulation in aplysia. II. Cellular and behavioral consequences of increased serotonergic tone. J Neurophysiol 92:2487–2496
Martin GR (1994) Vascular receptors for 5-hydroxytryptamine: distribution, function and classification. Pharmacol Ther 62:283–324
Martin GR, Leff P, Cambridge D, Barrett VJ (1987) Comparative analysis of two types of 5-hydroxytryptamine receptor mediating vasorelaxation: differential classification using tryptamines. Naunyn-Schmiedeberg’s Arch Pharmacol 336:365–373
May A, Gijsman HJ, Wallnofer A, Jones R, Diener HC, Ferrari MD (1996) Endothelin antagonist bosentan blocks neurogenic inflammation, but is not effective in aborting migraine attacks. Pain 67:375–378
McCall RB, Clement ME (1994) Role of serotonin1A and serotonin2 receptors in the central regulation of the cardiovascular system. Pharmacol Rev 46:231–243
McCall RB, Huff R, Chio CL, Tenbrink R, Bergh CL, Ennis MD, Ghazal NB, Hoffman RL, Mimeisheri K, Higdon NR, Hall E (2002) Preclinical studies characterizing the anti migraine and cardiovascular effects of the selective 5 HT1D receptor agonist PNU-142633. Cephalalgia 22:799–806
McQueen DS (1990) Cardiovascular reflexes and 5-hydroxytryptamine. In: Saxena PR, Wallis DI, Wouters W, Bevan P (eds) Cardiovascular pharmacology of 5-hydroxytryptamine, prospective therapeutic applications. Kluwer, Dordrecht, pp 233–245
Molderings GJ, Fink K, Schlicker E, Göthert M (1987) Inhibition of noradrenaline release via presynaptic 5-HT1B receptors of the rat vena cava. Naunyn-Schmiedeberg’s Arch Pharmacol 336:245–250
Molderings GJ, Göthert M, Fink K, Roth E, Schlicker E (1989) Inhibition of noradrenaline release in the pig coronary artery via a novel serotonin receptor. Eur J Pharmacol 164:213–222
Molderings GJ, Werner K, Likungu J, Göthert M (1990) Inhibition of noradrenaline release from the sympathetic nerves of the human saphenous vein via presynaptic 5-HT receptors similar to the 5-HT 1D subtype. Naunyn-Schmiedeberg’s Arch Pharmacol 342:371–377
Montani D, Jais X, Ioos V, Sitbon O, Simonneau G, Humbert M (2004) Treatments for pulmonary arterial hypertension. Rev Med Interne 25:720–731
Morán A, Velasco C, Martín ML, San Román L (1994) Pharmacological characterization of 5-HT receptors in parasympathetic innervation of rat heart. Eur J Pharmacol 252:161–166
Morecroft I, Heeley RP, Prentice HM, Kirk A, MacLean MR (1999) 5-hydroxytryptamine receptors mediating contraction in human small muscular pulmonary arteries: importance of the 5-HT1B receptor. Br J Pharmacol 128:730–734
Muñoz-Islas E, Gupta S, Jiménez-Mena LR, Lozano-Cuenca J, Sánchez-López A, Centurión D, Mehrotra S, Maassen Van Den Brink A, Villalón CM (2006) Donitriptan, but not sumatriptan, inhibits capsaicin-induced canine external carotid vasodilatation via 5-HT1B rather than 5-HT1D receptors. Br J Pharmacol 149:82–91
Nevens F, Pizcueta MP, Fernandez M, Bosch J, Rodes J (1991) Effects of ritanserin, a selective and specific S2-serotonergic antagonist, on portal pressure and splanchnic hemodynamics in portal hypertensive rats. Hepatology 14:1174–1178
Nishio H, Fujii A, Nakata Y (1996) Re-examination for pharmacological properties of serotonin-induced tachycardia in isolated guinea-pig atrium. Behav Brain Res 73:301–304
Nishio H, Morimoto Y, Hisaoka K, Nakata Y, Watanabe T (2002) 5-HT-induced, 5-HT3 receptor-mediated, and ruthenium red- and capsaicin-sensitive positive chronotropic effects in the isolated guinea pig atrium. Jpn J Pharmacol 89:242–248
Obi T, Kabeyama A, Nishio A (1994) Equine coronary artery responds to 5-hydroxytryptamine with relaxation in vitro. J Vet Pharmacol Ther 17:218–225
Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, Pollentier S, Lesko LM (2004) Calcitonin gene-related peptide receptor antagonist BIBN4096 BS for the acute treatment of migraine. N Engl J Med 350:1104–1110
Page IH (1958) Serotonin (5-hydroxytryptamine); the last four years. Physiol Rev 38:227–335
Page IH, McCubbin JW (1953) Modification of vascular response to serotonin by drugs. Am J Physiol 174:436–444
Peroutka SJ (2005) Neurogenic inflammation and migraine: implications for the therapeutics. Mol Interv 5:304–311
Peroutka SJ, Snyder SH (1979) Multiple serotonin receptors: differential binding of . [. 3H]-5-hydroxytryptamine, [3H]-lysergic acid diethylamide and [3H]-spiroperidol. Mol Pharmacol 16:687–699
Qvigstad E, Brattelid T, Sjaastad I, Andressen KW, Krobert KA, Birkeland JA, Sejersted OM, Kaumann AJ, Skomedal T, Osnes JB, Levy FO (2005a) Appearance of a ventricular 5-HT4 receptor-mediated inotropic response to serotonin in heart failure. Cardiovasc Res 65:869–878
Qvigstad E, Sjaastad I, Brattelid T, Nunn C, Swift F, Birkeland JA, Krobert KA, Andersen GO, Sejersted OM, Osnes JB, Levy FO, Skomedal T (2005b) Dual serotonergic regulation of ventricular contractile force through 5-HT2A and 5-HT4 receptors induced in the acute failing heart. Circ Res 97:268–276
Ramage AG (2001) Central cardiovascular regulation and 5-hydroxytryptamine receptors. Brain Res Bull 56:425–439
Rapport MM (1949) Serum vasoconstrictor (serotonin). V. The presence of creatinine in the complex: a proposed study of the vasoconstrictor principle. J Biol Chem 180:961–969
Rapport MM, Green AA, Page IH (1948a) Partial purification of the vasoconstrictor in beef serum. J Biol Chem 174:735–741
Rapport MM, Green AA, Page IH (1948b) Serum vasoconstrictor (serotonin). IV. Isolation and characterization. J Biol Chem 176:1243–1251
Razzaque Z, Heald MA, Pickard JD, Maskell L, Beer MS, Hill RG, Longmore J (1999) Vasoconstriction in human isolated middle meningeal arteries: determining the contribution of 5-HT1B- and 5-HT1F-receptor activation. Br J Clin Pharmacol 47:75–82
Reneman RS, van der Starre PJ (1990) Serotonin and acute cardiovascular disorders. Cardiovasc Drugs Ther 4(Suppl 1):19–25
Robertson JI (1990) Carcinoid syndrome and serotonin: therapeutic effects of ketanserin. Cardiovasc Drugs Ther 4(Suppl 1):53–58
Robertson JI (1991) Serotonergic type-2 (5-HT2) antagonists: a novel class of cardiovascular drugs. J Cardiovasc Pharmacol 17(Suppl 5):S48–S53
Rummo G, Bordoni L (1889) Toxicité du sérum du sang de l’homme et des animaux à l’état normal et dans les maladies par infection. Archiv De Biologie Italienne XII:XLVI–XLVII
Sánchez-López A, Centurión D, Vázquez E, Arulmani U, Saxena PR, Villalón CM (2003) Pharmacological profile of the 5-HT-induced inhibition of cardioaccelerator sympathetic outflow in pithed rats: correlation with 5-HT1 and putative 5-ht5A/5B receptors. Br J Pharmacol 140:725–735
Sánchez-López A, Centurión D, Vázquez E, Arulmani U, Saxena PR, Villalón CM (2004) Further characterization of the 5-HT1 receptors mediating cardiac sympatho-inhibition in pithed rats: pharmacological correlation with the 5-HT1B and 5-HT1D subtypes. Naunyn-Schmiedeberg’s Arch Pharmacol 369:220–227
Saxena PR (1972) The effects of antimigraine drugs on the vascular responses by 5-hydroxytryptamine and related biogenic substances on the external carotid bed of dogs: Possible pharmacological implications to their antimigraine action. Headache 12:44–54
Saxena PR (1995) Serotonin receptors: subtypes, functional responses and therapeutic relevance. Pharmacol Ther 66:339–368
Saxena PR, De Vries P, Villalón CM (1998) 5-HT1-like receptors: a time to bid goodbye. Trends Pharmacol Sci 19:311–316
Saxena PR, Ferrari MD (1996) Pharmacology of antimigraine 5-HT1D receptor agonists. Expert Opin Invest Drugs 5:581–593
Saxena PR, Lawang A (1985) A comparison of cardiovascular and smooth muscle effects of 5-hydroxytryptamine and 5-carboxamidotryptamine, a selective agonist of 5-HT1 receptors. Arch Int Pharmacodyn Ther 277:235–252
Saxena PR, Villalón CM (1990) Cardiovascular effects of serotonin agonists and antagonists. J Cardiovasc Pharmacol 15(Suppl 7):S17–S34
Schoeffter P, Hoyer D (1990) 5-Hydroxytryptamine (5-HT)-induced endothelium-dependent relaxation of pig coronary arteries is mediated by 5-HT receptors similar to the 5- HT1D receptor subtype. J Pharmacol Exp Ther 252:387–395
Schoemaker RG, Du XY, Bax WA, Bos E, Saxena PR (1993) 5-Hydroxytryptamine stimulates human isolated atrium but not ventricle. Eur J Pharmacol 230:103–105
Scrogin KE (2003) 5-HT1A receptor agonist 8-OH-DPAT acts in the hindbrain to reverse the sympatholytic response to severe hemorrhage. Am J Physiol Regul Integr Comp Physiol 284:R782–R791
Scrogin KE, Johnson AK, Brooks VL (2000) Methysergide delays the decompensatory responses to severe hemorrhage by activating 5-HT1A) receptors. Am J Physiol Regul Integr Comp Physiol 279:R1776–R1786
Shepheard SL, Jordan D, Ramage AG (1994) Comparison of the effects of IVth ventricular administration of some tryptamine analogues with those of 8-OH-DPAT on autonomic outflow in the anaesthetized cat. Br J Pharmacol 111:616–624
Shepheard SL, Williamson DJ, Williams J, Hill RG, Hargreaves RJ (1995) Comparison of the effects of sumatriptan and the NK1 antagonist CP-99,994 on plasma extravasation in Dura mater and c-fos mRNA expression in trigeminal nucleus caudalis of rats. Neuropharmacology 34:255–261
Silver B, Weber J, Fisher M (1996) Medical therapy for ischemic stroke. Clin Neuropharmacol 19:101–128
Skingle M, Beattie DT, Scopes DI, Starkey SJ, Connor HE, Feniuk W, Tyers MB (1996) GR127935: a potent and selective 5-HT1D receptor antagonist. Behav Brain Res 73:157–161
Sumner MJ (1991) Characterization of the 5-HT receptor mediating endothelium-dependent relaxation in porcine vena cava. Br J Pharmacol 102:938–942
Thomas DR (2006) 5-ht5A receptors as a therapeutic target. Pharmacol Ther 111:707–714
Thor KB, Nickolaus S, Helke CJ (1993) Autoradiographic localization of 5-hydroxytryptamine1A, 5-hydroxytryptamine1B and 5-hydroxytryptamine1C/2 binding sites in the rat spinal cord. Neuroscience 55:235–252
Tramontana M, Giuliani S, Del BE, Lecci A, Maggi CA, Evangelista S, Geppetti P (1993) Effects of capsaicin and 5-HT3 antagonists on 5-hydroxytryptamine-evoked release of calcitonin gene-related peptide in the guinea-pig heart. Br J Pharmacol 108:431–435
Valdivia LF, Centurión D, Arulmani U, Saxena PR, Villalón CM (2004) 5-HT1B receptors, α2A/2C- and, to a lesser extent, α1-adrenoceptors mediate the external carotid vasoconstriction to ergotamine in vagosympathectomized dogs. Naunyn-Schmiedeberg’s Arch Pharmacol 370:46–53
Van Gelderen EM, Saxena PR (1992) Effect of N(G)-nitro-L-arginine methyl ester on the hypotensive and hypertensive responses to 5-hydroxytryptamine in pithed rats. Eur J Pharmacol 222:185–191
Van Nueten JM (1983) 5-hydroxytryptamine and precapillary vessels. Fed Proc 42:223–227
Van Nueten JM, Janssen PA, Van Beek J, Xhonneux R, Verbeuren TJ, Vanhoutte PM (1981) Vascular effects of ketanserin (R 41 468), a novel antagonist of 5-HT2 serotonergic receptors. J Pharmacol Exp Ther 218:217–230
Vialli M, Erspamer V (1933) Cellule enterocromaffine e cellule basigranulose acidofile nei vertebrati. Ztschr Zellforsch 19:743–773
Villalón CM, Centurión D, Bravo G, De Vries P, Saxena PR, Ortiz MI (2000) Further pharmacological analysis of the orphan 5-HT receptors mediating feline vasodepressor responses: close resemblance to the 5-HT7 receptor. Naunyn-Schmiedeberg’s Arch Pharmacol 361:665–671
Villalón CM, Centurión D, Fernández MM, Morán A, Sánchez-López A (1999a) 5-Hydroxytryptamine inhibits the tachycardia induced by selective preganglionic sympathetic stimulation in pithed rats. Life Sci 64:1839–1847
Villalón CM, Centurión D, Luján-Estrada M, Terrón JA, Sánchez-López A (1997a) Mediation of 5-HT-induced external carotid vasodilatation in GR 127935- pretreated vagosympathectomized dogs by the putative 5-HT7 receptor. Br J Pharmacol 120:1319–1327
Villalón CM, Centurión D, Rabelo G, De Vries P, Saxena PR, Sánchez-López A (1998) The 5-HT1-like receptors mediating inhibition of sympathetic vasopressor outflow in the pithed rat: operational correlation with the 5-HT1A, 5-HT1B and 5-HT1D subtypes. Br J Pharmacol 124:1001–1011
Villalón CM, Centurión D, Valdivia LF, De Vries P, Saxena PR (2002) An introduction to migraine: from ancient treatment to functional pharmacology and antimigraine therapy. Proc West Pharmacol Soc 45:199–210
Villalón CM, Centurión D, Willems EW, Arulmani U, Saxena PR, Valdivia LF (2004) 5-HT1B receptors and α2A/2C. -adrenoceptors mediate external carotid vasoconstriction to dihydroergotamine. Eur J Pharmacol 484:287–290
Villalón CM, Contreras J, Ramírez-San Juan E, Castillo C, Perusquía M, López-Muñoz FJ, Terrón JA (1995a) 5-Hydroxytryptamine inhibits pressor responses to preganglionic sympathetic nerve stimulation in pithed rats. Life Sci 57:803–812
Villalón CM, Contreras J, Ramírez-San Juan E, Castillo C, Perusquía M, Terrón JA (1995b) Characterization of prejunctional 5-HT receptors mediating inhibition of sympathetic vasopressor responses in the pithed rat. Br J Pharmacol 116:3330–3336
Villalón CM, De Vries P, Rabelo G, Centurión D, Sánchez-López A, Saxena PR (1999b) Canine external carotid vasoconstriction to methysergide, ergotamine and dihydroergotamine: role of 5-HT1B/1D receptors and α2-adrenoceptors. Br J Pharmacol 126:585–594
Villalón CM, De Vries P, Saxena PR (1997b) Serotonin receptors as cardiovascular targets. Drug Discov Today 2:294–300
Villalón CM, Den Boer MO, Heiligers JP, Saxena PR (1990) Mediation of 5-hydroxytryptamine-induced tachycardia in the pig by the putative 5-HT4 receptor. Br J Pharmacol 100:665–667
Villalón CM, Den Boer MO, Heiligers JP, Saxena PR (1991) Further characterization, by use of tryptamine and benzamide derivatives, of the putative 5-HT4 receptor mediating tachycardia in the pig. Br J Pharmacol 102:107–112
Villalón CM, Heiligers JP, Centurión D, De Vries P, Saxena PR (1997c) Characterization of putative 5-HT7 receptors mediating tachycardia in the cat. Br J Pharmacol 121:1187–1195
Villalón CM, Sánchez-López A, Centurión D (1996) Operational characteristics of the 5-HT1-like receptors mediating external carotid vasoconstriction in vagosympathectomized dogs: close resemblance to the 5-HT1D receptor subtype. Naunyn-Schmiedeberg’s Arch Pharmacol 354:550–556
Villalón CM, Sánchez-López A, Centurión D, Saxena PR (2001) Unravelling the pharmacological profile of the canine external carotid vasodilator “5-HT1-like” receptors: coexistence of sympatho-inhibitory 5-HT1B and postjunctional 5-HT7 receptors. Naunyn-Schmiedeberg’s Arch Pharmacol 363:73–80
Villalón CM, Terrón JA, Hong E (1993) Role of 5-HT1-like receptors in the increase in external carotid blood flow induced by 5-hydroxytryptamine in the dog. Eur J Pharmacol 240:9–20
Watts SW, Fink GD (1999) 5-HT2B-receptor antagonist LY-272015 is antihypertensive in DOCA-salt-hypertensive rats. Am J Physiol 276:H944–H952
Weiss O (1896) Ueber die Wirkungen von Blutserum-Injectionen ins Blut. Archiv für die Gesamte Physiologie des Menschen und der Thiere LXV:215–230
Wilson H, Coffman WJ, Cohen ML (1990) 5-Hydroxytryptamine3 receptors mediate tachycardia in conscious instrumented dogs. J Pharmacol Exp Ther 252:683–688
Yu XJ, Cutrer FM, Moskowitz MA, Waeber C (1997) The 5-HT1D receptor antagonist GR-127,935 prevents inhibitory effects of sumatriptan but not CP-122,288 and 5-CT on neurogenic plasma extravasation within guinea pig dura mater. Neuropharmacology 36:83–91
Yusof AP, Coote JH (1988) Excitatory and inhibitory actions of intrathecally administered 5-hydroxytryptamine on sympathetic nerve activity in the rat. J Auton Nerv Syst 22:229–236
Acknowledgements
We are grateful to all our colleagues who collaborated in the studies cited in this review. We are also indebted to CONACyT (Mexico) for their financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Villalón, C.M., Centurión, D. Cardiovascular responses produced by 5-hydroxytriptamine:a pharmacological update on the receptors/mechanisms involved and therapeutic implications. Naunyn-Schmied Arch Pharmacol 376, 45–63 (2007). https://doi.org/10.1007/s00210-007-0179-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-007-0179-1