
Abstract
Cannabinoids exert complex effects on blood pressure related to their interference with cardiovascular centres in the central nervous system and to their direct influence on vascular muscle, vascular endothelium and heart. In view of the relative lack of information on the occurrence of CB1 receptors on the vascular postganglionic sympathetic nerve fibres, the aim of the present study was to examine whether cannabinoid receptor ligands affect the electrically evoked tritium overflow in superfused vessels (tissue pieces) from the guinea-pig, the rat and the mouse preincubated with 3H-noradrenaline. The cannabinoid receptor agonist WIN 55,212-2 (R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]-pyrrolo[1,2,3-de]1,4-benzoxazinyl](1-naphthalenyl) methanone) inhibited the evoked tritium overflow in the guinea-pig aorta, but not in that of the rat or mouse. The concentration–response curve of WIN 55,212-2 was shifted to the right by the CB1 receptor antagonist rimonabant, yielding an apparent pA2 value of 7.9. The most pronounced (near-maximum) inhibition obtained at the highest WIN 55,212-2 concentration applied (3.2 μM) amounted to 40%. WIN 55,212-2 also inhibited the evoked overflow in guinea-pig pulmonary artery, basilar artery and portal vein, again in a manner sensitive to antagonism by rimonabant. The latter did not affect the evoked overflow by itself in the four vessels, but did increase the electrically evoked tritium overflow from superfused guinea-pig hippocampal slices preincubated with 3H-choline and from superfused guinea-pig retina discs preincubated with 3H-noradrenaline (labelling dopaminergic cells in this tissue). The inhibitory effect of 3.2 μM WIN 55,212-2 on the evoked overflow from the guineapig aorta was comparable in size to that obtained with agonists at the histamine H3, κ opioid (KOP) and ORL1 (NOP) receptor (1 or 10 μM, producing the respective near-maximum effects) whereas prostaglandin E2 1 μM caused a higher near-maximum inhibition of 70%. Prostaglandin E2 also induced an inhibition by 65 and 80% in the rat and mouse aorta respectively, indicating that the present conditions are basically suitable for detecting presynaptic receptor-mediated inhibition of noradrenaline release. The results show that the postganglionic sympathetic nerve fibres in the guineapig aorta, but not in the rat or mouse aorta, are endowed with presynaptic inhibitory cannabinoid CB1 receptors; such receptors also occur in guineapig pulmonary artery, basilar artery and portal vein. These CB1 receptors are not subject to an endogenous tone and the extent of inhibition obtainable via these receptors is within the same range as that of several other presynaptic heteroreceptors, but markedly lower than that obtainable via receptors for prostaglandin E2.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cannabinoids can elicit a variety of cardiovascular effects. Acute administration of hashish or marijuana in humans leads to an increase in heart rate whereas the effects on blood pressure are more variable ranging from a slight increase to orthostatic hypotension (the latter occurring at higher doses; for review, see Hollister 1986; Kalant 2004). However, more serious cardiovascular events including cerebral ischaemia and infarction (for review, see Mousouttas 2004) and an increased rate of myocardial infarction shortly after ingestion of cannabis preparations (for review, see Kalant 2004) have been reported as well. There is increasing evidence from animal models that the recently identified endogenous cannabinoid system is implicated in hemorrhagic, endotoxic and cardiogenic shock (for review, see Wagner et al. 1998; Pacher et al. 2005a, b) and in the vasodilated state associated with advanced liver cirrhosis (Batkai et al. 2001; Ros et al. 2002). The endocannabinoid system may also be implicated as a compensatory mechanism in various types of arterial hypertension (for review, see Pacher et al. 2005a).
The targets for the cardiovascular effects of the cannabinoids comprise the central nervous system (Niederhoffer and Szabo 2000; Kwolek et al. 2005), the vascular muscle (Gebremedhin et al. 1999), the vascular endothelium (for review, see Pacher et al. 2005b) and the heart (Bonz et al. 2003; Pacher et al. 2004). Many but by far not all effects are mediated via cannabinoid CB1 receptors. One of the endocannabinoids, anandamide, also activates the TRPV1 receptor. Anandamide is capable of inducing vasodilation by releasing calcitonin-gene-related peptide (CGRP) from sensory neurones (Zygmunt et al. 1999) and, in addition, leads to a decrease in heart rate and blood pressure by activating the Bezold-Jarisch reflex (Varga et al. 1995; Malinowska et al. 2001). Anandamide also stimulates the so-called abnormal cannabidiol receptor, activation of which is associated with an endothelium-dependent vasodilatation, e.g. of the mesenteric arteries (for review, see Pacher et al. 2005b). At this receptor (the exact molecular biology of which has not yet been clarified), cannabidiol, which, like Δ9-tetrahydrocannabinol, occurs in high concentrations in cannabis preparations, acts as an antagonist.
Many cannabinoid CB1 receptors are located presynaptically and their activation causes inhibition of the release of the respective transmitter (Schlicker and Kathmann 2001; Szabo and Schlicker 2005). The possibility that cannabinoids act via CB1 receptors on vascular postganglionic sympathetic nerve fibres has been considered in only a few studies. Thus, a CB1 receptor-mediated inhibition of noradrenaline release (or of the endorgan response elicited by noradrenaline) has been shown for the pithed rat and rabbit (Malinowska et al. 1997; Niederhoffer and Szabo 1999; Niederhoffer et al. 2003; Pfitzer et al. 2005) and for isolated mesenteric (Ralevic and Kendall 2002) and renal arteries (Deutsch et al. 1997) from the rat.
The aim of the present study was to extend those previous investigations to other blood vessels of other species, in a first step, to the aorta from the rat, guinea-pig and mouse. In addition, we examined whether release-modulating CB1 receptors also occur in other vessels of the guinea-pig and, using the CB1 receptor antagonist rimonabant (former name: SR 141716), whether the vascular CB1 receptor is subject to an endogenous tone. The occurrence of an endogenous tone has been shown for many CB1 receptors (for review, see Pertwee 2005), including the CB1 receptors reducing acetylcholine release from the guineapig hippocampus (Schultheiß et al. 2004) and dopamine release from the guinea-pig retina (Schlicker et al. 1996). Furthermore, the extent of inhibition of noradrenaline release via CB1 receptors and via another four types of presynaptic receptors was compared in the guinea-pig aorta. Finally, radioligand binding studies with 3H-rimonabant were carried out in guinea-pig cortex membranes to exclude that the agonists at those presynaptic receptors might, in addition, have affinity for CB1 receptors.
Materials and methods
Superfusion studies
Aortae were prepared from male Dunkin–Hartley guineapigs, male Wistar rats or male NMRI mice. Pulmonary artery, basilar artery, portal vein, hippocampus and retina were prepared from male Dunkin–Hartley guinea-pigs (for details, see Table 1). Preparations were incubated (37°C) for 60 min with physiological salt solution (PSS; Ca2+ 1.3 mM) containing 3H-noradrenaline or 3H-choline (for details, see Table 1). Subsequently, the preparations were transferred to superfusion chambers and superfused (0.5 ml/min) with PSS (37°C) (for Ca2+ concentration and auxiliary drugs, see Table 1). The superfusate was collected in 5-min samples; experiments lasted for 60 or 110 min (see Table 1). Tritium overflow was evoked by one or two 2-min period(s) of electrical field stimulation (for stimulation parameters, see Table 1) after 40 and/or 90 min of superfusion (S1 and S2). The drugs under study were present in the medium either throughout superfusion or from 62 min of superfusion onward, as indicated in the Results section. The PSS was composed as follows (mM): NaCl 118, KCl 4.8, CaCl2 1.3 or 3.25 (as indicated in Table 1), KH2PO4 1.2, MgSO4 1.2, NaHCO3 25, ascorbic acid 0.06, disodium EDTA 0.03, glucose 10, and the solution was aerated with 95% O2 and 5% CO2 (pH 7.4).
Tritium efflux was calculated as the fraction of the tritium content in the tissues at the beginning of the respective collection period (fractional rate of tritium efflux). To quantify effects of drugs on basal efflux, the ratio of the fractional rates in the 5-min period prior to S2 (t2) to those in the 5-min period 15–20 min after the onset of S1 (t1) was determined (for drugs added to the PSS from 62 min of superfusion onward) or the t1 values obtained in the absence or presence of a given drug were directly compared with each other (for drugs present in the PSS throughout superfusion). Stimulation-evoked tritium overflow was calculated by subtraction of the basal from the total efflux during stimulation and the subsequent 13 min and expressed as a percentage of the tritium present in the tissue at the onset of stimulation (basal efflux was assumed to decline linearly from the 5-min period before to that 15–20 min after the onset of stimulation). To quantify drug-induced effects on the stimulated tritium overflow, the ratio of the overflow evoked by S2 over that evoked by S1 was determined (S2/S1; for drugs added to the PSS from 62 min of superfusion) or the S1 values obtained in the absence or presence of a given drug were directly compared with each other (for drugs present throughout superfusion). The apparent pA2 value for rimonabant was calculated according to the formula\({\text{pA}}_{{\text{2}}} = \log {\left( {{{\left[ {{\text{A}}^{\prime } } \right]}} \mathord{\left/ {\vphantom {{{\left[ {{\text{A}}^{\prime } } \right]}} {{\left[ {\text{A}} \right]}}}} \right. \kern-\nulldelimiterspace} {{\left[ {\text{A}} \right]}} - 1} \right)} - \log {\left[ {\text{B}} \right]}\), where [A′] and [A] are the IC20% values for WIN 55,212-2 obtained in the presence and absence of rimonabant and [B] is the concentration of rimonabant.
Binding studies
Cerebral cortex membranes from male Dunkin–Hartley guinea-pigs were homogenised (Potter–Elvehjem) in 25 vol of ice-cold Tris-HCl buffer (Tris 50 mM, pH 7.5; EDTA 5 mM; sucrose 10.27%) and centrifuged at 1,500×g for 10 min (4°C). The supernatant was centrifuged at 39,000×g for 20 min and the pellet was resuspended in buffer (Tris 50 mM, pH 7.5; EDTA 5 mM) and frozen at −80°C.
For binding experiments, membranes were incubated with sucrose-free buffer in a final volume of 0.5 ml containing 40–70 μg protein. 3H-rimonabant was used at eight concentrations ranging from 0.05 to 8 nM for saturation experiments and at 0.5 nM for competition experiments. The incubation (25°C) was terminated after 60 min by filtration through Whatman GF/C filters. CP-55,940 (3 μM) was used to determine non-specific binding. Data were analysed using the programme GraphPadPrism (Prism; GraphPad Software, San Diego, CA, USA).
Statistics
Results are given as means±SEM of n experiments (superfusion) and of n experiments in duplicate or triplicate (saturation and competition experiments with 3H-rimonabant respectively). For comparison of mean values, the t test for unpaired data was used (superfusion studies); the Bonferroni correction was used when two or more values were compared with the same control. The F test was applied in order to evaluate whether the inhibition of 3H-rimonabant binding by drugs is better fitted by a one- or a two-site model.
Drugs used
[Methyl-3H]-choline chloride (specific activity 86 Ci/mmol), (R)-(−)-[ring-2,5,6-3H]-noradrenaline (spec. act. 53 Ci/mmol; NEN, Zaventem, Belgium); 3H-SR141716A (3H-rimonabant; spec. act. 44 Ci/mmol; Amersham, Braunschweig, Germany); AF-DX 384 (5,11-dihydro-11-{[(2-{2-[(dipropylamino)methyl]-1-piperidinyl}ethyl)amino]carbonyl}-6H-pyrido(2,3-β)(1,4)benzodiazepine-6-one; Boehringer–Ingelheim, Biberach an der Riss, Germany); CP-55,940 ((−)-cis-3-[2[hydroxy-4-(1,1-dimethylheptyl)phenyl]trans-4-(3-hydroxypropyl)-cyclohexanol; Biotrend, Cologne, Germany); desipramine hydrochloride (Novartis, Wehr, Germany); hemicholinium-3, prostaglandin E2, sulpiride, WIN 55,212-2 (R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]-pyrrolo[1,2,3-de]1,4-benzoxazinyl](1-naphthalenyl) methanone mesylate; Sigma, Munich, Germany); R-α-methylhistamine dihydrogenmaleate (Professor W. Schunack, Institut für Pharmazie, Freie Universität Berlin); nociceptin (Bachem, Bubendorf, Switzerland); nomifensine (Sanofi-Aventis, Frankfurt, Germany); rauwolscine hydrochloride (Roth, Karlsruhe, Germany); rimonabant hydrochloride (SR 141716A; Sanofi-Aventis, Montpellier, France); U-69,593 ((5α,7α,8β)-(+)-N-methyl-N-(7-[1-pyrrolidinyl]-1-oxaspiro[4.5]dec-8-yl)-benzeneacetamide; MP Biomedicals, Eschwege, Germany). Stock solutions of the drugs were prepared with ethanol (prostaglandin E2), dimethylsulfoxide (CP-55,940, rimonabant, WIN 55,212-2) or water and diluted with PSS (superfusion experiments) or water (binding experiments) to the concentration required.
Results
Transmitter release studies
Basal tritium efflux was not affected by any of the drugs or their organic solvents (DMSO up to 0.03% and ethanol 0.01%) in any of the preparations investigated (results not shown).
To investigate the effects of the drugs on the electrically evoked tritium overflow the drug under study was present in the medium before and during S2 (the second period of electrical stimulation); its effect on the evoked overflow was calculated as the ratio of the overflow evoked by S2 over that evoked by S1 and compared with the corresponding control S2/S1 ratio in the absence of the test drug. To study the interaction of WIN 55,212-2 with rimonabant, WIN 55,212-2 was present in the medium before and during S2 whereas rimonabant was present throughout superfusion. In the experiments designed to examine whether the CB1 receptor is subject to an endogenous tone (Fig. 4), rimonabant was present throughout superfusion of shorter experiments, which allowed one electrical stimulation only. The control S1 values are shown in Fig. 4 and Table 2, and the control S2/S1 ratios are depicted in Figs. 2 and 3. S1 and S2/S1 values were not affected by DMSO (up to 0.03%) or ethanol (0.01%).
The cannabinoid receptor agonist WIN 55,212-2 inhibited the electrically evoked tritium overflow (S2/S1) from guinea-pig aorta pieces preincubated with 3H-noradrenaline in a concentration-dependent manner (Fig. 1). Due to its limited solubility concentrations of WIN 55,212-2 higher than 3.2 μM could not be examined and its maximum inhibitory effect could not be determined precisely. If one assumes that WIN 55,212-2 inhibits the evoked overflow by a maximum of 40%, the concentration causing the half-maximum effect (i.e. an inhibition by 20%) will be 0.7 μM. The concentration–response curve of WIN 55,212-2 was shifted to the right by rimonabant 0.032 μM (Fig. 1), yielding an apparent pA2 value of 7.9. Rimonabant 0.032 μM by itself did not affect the evoked overflow (S1; Table 2).

Effect of WIN 55,212-2 on the electrically (3 Hz) evoked tritium overflow from guinea-pig aorta pieces preincubated with 3H-noradrenaline, and interaction of WIN 55,212-2 with rimonabant. Following incubation, the tissues were transferred to superfusion chambers and superfused at a flow rate of 0.5 ml/min. The superfusion medium contained WIN 55,212-2 from 62 min of superfusion onward and rimonabant as well as two auxiliary drugs (1 μM desipramine plus 1 μM rauwolscine) throughout superfusion (110 min). Tritium overflow was evoked twice, after 40 and 90 min of superfusion, and the ratio of the overflow evoked by S2 over that evoked by S1 was determined. Tritium overflow was expressed as a percentage of the S2/S1 value in controls (not shown). Means±SEM of three to five experiments
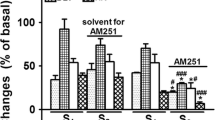
WIN 55,212-2 (1 μM) inhibited the electrically evoked tritium overflow (S2/S1) not only in the guinea-pig aorta (by 23%), but also in the pulmonary artery, basilar artery and portal vein from this species (by 26, 33 and 26% respectively; Fig. 2). The inhibitory effect was counteracted by rimonabant 0.32 μM (Fig. 2). Rimonabant by itself did not have a statistically significant effect, but tended to increase the evoked overflow in each of the eight experimental series (Fig. 2 [S2/S1]; Table 2 [S1]).

Effect of WIN 55,212-2 and rimonabant on the electrically (3 Hz) evoked tritium overflow from superfused vascular tissue pieces from guineapig (GP) preincubated with 3H-noradrenaline, and interaction of WIN 55,212-2 with rimonabant. White columns: the superfusion medium contained WIN 55,212-2 (WIN) or rimonabant (SR) from 62 min of superfusion onward. Dark columns: the superfusion medium contained WIN 55,212-2 from 62 min of superfusion onward and rimonabant throughout superfusion (110 min). Tritium overflow was evoked twice, after 40 and 90 min of superfusion, and the ratio of the overflow evoked by S2 over that evoked by S1 was determined (S2/S1). Means±SEM of three to eight experiments. *P<0.02, **P<0.005, compared with the corresponding control
WIN 55,212-2 (1 μM) failed to inhibit the electrically evoked tritium overflow (S2/S1) in the rat and mouse aorta (Fig. 3B, C). In the aorta from the two rodent species and from the guinea-pig, prostaglandin E2 (1 μM) inhibited the evoked overflow by more than 60% (Fig. 3A–C). The evoked overflow from the guinea-pig aorta was also inhibited by the histamine H3 receptor agonist R-α-methylhistamine, by nociceptin and by the κ opioid receptor agonist U-69,593; the extent of inhibition, representing the near-maximum effects of the respective drugs, was comparable to that obtained with WIN 55,212-2 (3.2 μM; Fig. 3A).

Effect of WIN 55,212-2 (WIN), prostaglandin E2 (PGE 2 ), R-α-methylhistamine (RαMH), nociceptin (Noci) and U-69,593 (U-69) on the electrically (3 Hz) evoked tritium overflow from superfused a guinea-pig, b rat and c mouse aorta pieces preincubated with 3H-noradrenaline. The superfusion medium contained the drug under study from 62 min of superfusion onward (total duration of superfusion 110 min). Tritium overflow was evoked twice, after 40 and 90 min of superfusion, and the ratio of the overflow evoked by S2 over that evoked by S1 was determined (S2/S1). Means±SEM of four to nine experiments. *P<0.05, **P<0.01
Finally, the effect of rimonabant on the electrically evoked tritium overflow in the guinea-pig aorta preincubated with 3H-noradrenaline was compared with its effect on the electrically evoked tritium overflow from guinea-pig hippocampal slices preincubated with 3H-choline and guinea-pig retina discs preincubated with 3H-noradrenaline (Fig. 4). In the latter two tissues, rimonabant 0.32 μM, which was ineffective in the aorta (see also above), facilitated the evoked tritium overflow (S1) by 70 and 125% respectively (Fig. 4).

Effect of rimonabant (SR) on the electrically evoked tritium overflow from superfused guinea-pig aorta pieces (stimulation frequency 3 Hz) and retina discs preincubated with 3H-noradrenaline (1 Hz), and from guinea-pig hippocampal slices preincubated with 3H-choline (3 Hz). The superfusion medium contained rimonabant throughout superfusion (60 min). Tritium overflow was evoked once, after 40 min of superfusion (S 1 ), and is expressed as a percentage of tissue tritium. Means±SEM of three to nine experiments. *P<0.05, **P<0.01
Binding studies
In saturation binding experiments on guinea-pig brain cortex membranes, using 3H-rimonabant at eight concentrations, a KD value of 2.12±0.56 nM with a maximum number of binding sites (Bmax) of 2,340±420 fmol/mg protein was determined (Fig. 5A). Scatchard analysis revealed a straight line with a Hill coefficient (nH) of unity (not shown). Unspecific binding (determined with 3 μM CP-55,940) was 27% for 0.5 nM 3H-rimonabant. In competition binding experiments, binding of 0.5 nM 3H-rimonabant was inhibited monophasically (nH near unity) by (unlabelled) rimonabant and the cannabinoid receptor agonists CP-55,940 and WIN 55,212-2, yielding pKi (±SEM) values of 8.72±0.06, 7.65±0.09 and 6.48±0.11 respectively (Fig. 5B). In the concentrations used for the experiments in Fig. 3, R-α-methylhistamine, nociceptin and prostaglandin E2 (1 μM each) and U-69,593 (10 μM) did not affect binding (Fig. 5B).

Specific 3H-rimonabant binding to guinea-pig cortex membranes. Membranes were incubated (25°C) for 60 min with 3H-rimonabant. Specific binding was defined as that inhibited by CP-55,940 (3 μM). A Saturation binding experiments. The effect of cannabinoid receptor ligands and of agonists at other presynaptic inhibitory receptors on the binding of 3H-rimonabant 0.5 nM is shown in B. Means±SEM of four experiments in A duplicate or B triplicate (for some data points, SEM is smaller than the symbols)
Discussion
The aim of the present study was to identify release-inhibiting cannabinoid CB1 receptors in superfused vascular tissue labelled by 3H-noradrenaline. In some experiments, we also examined guinea-pig hippocampal slices preincubated with 3H-choline, which in turn is metabolised to 3H-acetylcholine. Furthermore, retinal discs were preincubated with 3H-noradrenaline, which, however, is accumulated in dopaminergic amacrine cells (false-labelling; Schlicker et al. 1996). 3H-noradrenaline rather than 3H-dopamine was used since the scatter of variation of the results is lower when the former tracer is used (Schlicker et al. 1996). In the vascular and in the two non-vascular tissues electrical stimulation was used to induce quasi-physiological exocytotic release of the respective 3H-tracer molecule (or of 3H-acetylcholine in the case of 3H-choline) and the corresponding endogenous unlabelled neurotransmitter.
Superfusion of vascular pieces, hippocampal slices and retinal discs was routinely performed in the presence of an inhibitor of the high-affinity noradrenaline, choline and dopamine transporter, i.e. desipramine, hemicholinium and nomifensine respectively, to avoid interactions of the drugs under study with the respective transporters and to increase the amount of tritium overflow. In addition, an antagonist of the respective presynaptic inhibitory autoreceptors, i.e. rauwolscine, AF-DX 384 and sulpiride, was present in the medium to avoid interactions of the drugs under study with the respective autoreceptors and again to increase the amount of tritium overflow. There was, however, an additional reason to use the α2-adrenoceptor antagonist rauwolscine for the superfusion experiments on vessels. The extent of inhibition of noradrenaline release following activation of presynaptic heteroreceptors (e.g. opioid receptors) is usually increased in noradrenergically innervated tissues, including vessels, when the presynaptic α2-autoreceptor is blocked simultaneously (for review, see Schlicker and Göthert 1998). Such an interaction (which might take place at the level of G proteins; Schlicker and Göthert 1998) has also been shown for the presynaptic CB1 receptor causing inhibition of noradrenaline release in guinea-pig hippocampal slices (Schlicker and Göthert 1998) and may occur in guinea-pig vessels as well, although this has not been studied systematically so far. A direct effect of rauwolscine (1 μM) on CB1 receptors can be excluded since the drug did not exhibit an affinity for 3H-rimonabant binding in rat brain cortex membranes (Kathmann et al. 1999).
Noradrenaline release from the guinea-pig aorta was inhibited by the cannabinoid receptor agonist WIN 55,212-2 and its concentration–response curve was shifted to the right by the CB1 receptor antagonist rimonabant, suggesting that the effect is mediated via the CB1 receptor. Due to the limited solubility of WIN 55,212-2 full concentration–response curves could not be obtained and the maximum effect could not be determined. The apparent pA2 of 7.9 is identical to the reference value given in the review by Alexander et al. (2004); it is also close to the values obtained for the presynaptic CB1 receptors causing inhibition of noradrenaline release in the guinea-pig hippocampus (8.2; Schlicker et al. 1997) and of dopamine release in guinea-pig retina (8.3; Schlicker et al. 1996). The present study shows that presynaptic inhibitory CB1 receptors, in addition, occur in guinea-pig pulmonary artery, basilar artery and portal vein.
Presynaptic inhibitory CB1 receptors could not be identified in the aorta of the rat and the mouse. The possibility that the conditions used for electrical stimulation in the rodent aorta in the present study were not suitable to detect presynaptic modulation can be excluded since prostaglandin E2 inhibited noradrenaline release by at least 60%. The difference in the aorta of the various species is reminiscent of the lack of CB1 receptor-mediated modulation of noradrenaline release in the rodent hippocampus, whereas such a mechanism is operative in the guinea-pig hippocampus (Schlicker et al. 1997). On the other hand, the sympathetic nerve fibres innervating the resistance vessels and the mesenteric and renal arteries in the rat are endowed with presynaptic CB1 receptors (Deutsch et al. 1997; Malinowska et al. 1997; Ralevic and Kendall 2002; Niederhoffer et al. 2003; Pfitzer et al. 2005).
Rimonabant did not affect noradrenaline release in the four vessels of the guinea-pig under study (although there was a tendency towards an increase). In this respect, the vessels differ from guinea-pig hippocampus and retina in which the CB1 receptors involved in the inhibition of acetylcholine and dopamine release respectively are subject to an endogenous tone, i.e. rimonabant facilitates transmitter release (Schlicker et al. 1996; Schultheiß et al. 2004). To confirm the organ specificity of a tonical activation of CB1 receptors, we repeated the experiments on hippocampal slices and retina discs under the conditions of the present study, i.e. using a shorter exposure to rimonabant (40 vs. 60 min) and superfusion chambers with a lower volume. Again, rimonabant markedly increased acetylcholine and dopamine release respectively, demonstrating that a facilitation of transmitter release is possible under the experimental conditions of the present study. The possibility has to be considered anyway that the four guinea-pig vessels are subject to an endogenous tone in vivo (i.e. when they are in their natural surroundings, possibly providing endocannabinoids). On the other hand, rimonabant also failed to increase noradrenaline release in the resistance vessels of the pithed rat (Malinowska et al. 1997; Pfitzer et al. 2005), i.e. in an experimental model more closely resembling the in vivo situation compared with the in vitro experiments of the present study.
The maximum inhibitory effect of WIN 55,212-2 on noradrenaline release in the guinea-pig aorta was 40% or slightly higher. Since an inhibition of 40% appears to be relatively small, the effects of agonists at other presynaptic inhibitory heteroreceptors in the guinea-pig aorta, namely the histamine H3, ORL1, κ opioid and prostaglandin EP3 receptors (at concentrations causing the maximum or near-maximum effect at the respective receptors; Exner and Schlicker 1995; Schlicker et al. 1998; Timm et al. 1998; Bauer et al. 1999), were examined in this study as well. The inhibitory effects were 26% via H3, 33% via CB1, 36% via ORL1, 40% via κ and 66% via EP3 receptors. The effects of the same agonists (at identical concentrations) on noradrenaline release from guinea-pig hippocampal slices have been studied by us previously (Szabo and Schlicker 2005 and unpublished), yielding an extent of inhibition of 27% via H3, 49% via CB1, 82% via ORL1, 94% via κ and 21% via EP3 receptors. The comparisons show that in both experimental models the extent of CB1 receptor-mediated inhibition of noradrenaline release is moderate, but within the range mediated by other presynaptic inhibitory heteroreceptors. The inhibitory effect of R-α-methylhistamine, nociceptin, U-69,593 and prostaglandin E2 cannot be attributed to a putative agonistic property at CB1 receptors in addition to their respective specific properties since the four agonists at the concentrations under study did not have an affinity for CB1 receptor binding sites determined with 3H-rimonabant in guinea-pig cortex membranes. The lack of affinity of prostaglandin E2, which like some of the endocannabinoids is derived from arachidonic acid, had also been shown by Howlett et al. (1992) on rat brain membranes, using the radioligand 3H-CP-55,940.
In conclusion, the postganglionic sympathetic nerve fibres innervating guinea-pig aorta, pulmonary artery, basilar artery and portal vein are endowed with presynaptic inhibitory cannabinoid CB1 receptors, whereas such receptors could not be identified in rat and mouse aorta. The CB1 receptors in the guinea-pig vessels are not subject to an endogenous tone and the maximum extent of CB1 receptor-mediated inhibition is within the same range as that of several other presynaptic inhibitory heteroreceptors, except for EP3 receptors, which mediate a more pronounced inhibition.
References
Alexander SPH, Mathie A, Peters JA (2004) Guide to receptors and channels. Br J Pharmacol 141:S1–S126
Batkai S, Jarai Z, Wagner JA, Goparaju SK, Varga K, Liu J, Wang L, Mirshahi F, Khanolkar AD, Makriyannis A, Urbaschek R, Garcia N Jr, Sanyal AJ, Kunos G (2001) Endocannabinoids acting at vascular CB1 receptors mediate the vasodilated state in advanced liver cirrhosis. Nat Med 7:827–832
Bauer U, Nakazi M, Kathmann M, Göthert M, Schlicker E (1999) The stereoselective κ opioid receptor antagonist Mr 2266 does not exhibit stereoselectivity as an antagonist at the orphan opioid (ORL1) receptor. Naunyn-Schmiedebergs Arch Pharmacol 359:17–20
Bonz A, Laser M, Kullmer S, Kniesch S, Babin-Ebell J, Popp V, Ertl G, Wagner JA (2003) Cannabinoids acting on CB1 receptors decrease contractile performance in human atrial muscle. J Cardiovasc Pharmacol 41:657–664
Deutsch DG, Goligorsky MS, Schmid PC, Krebsbach RJ, Schmid HH, Das SK, Dey SK, Arreaza G, Thorup C, Stefano G, Moore LC (1997) Production and physiological actions of anandamide in the vasculature of the rat kidney. J Clin Invest 100:1538–1546
Exner HJ, Schlicker E (1995) Prostanoid receptors of the EP3 subtype mediate the inhibitory effect of prostaglandin E2 on noradrenaline release in the mouse brain cortex. Naunyn-Schmiedebergs Arch Pharmacol 351:46–52
Gebremedhin D, Lange AR, Campbell WB, Hillard CJ, Harder DR (1999) Cannabinoid CB1 receptor of cat cerebral arterial muscle functions to inhibit L-type Ca2+ channel current. Am J Physiol 276:H2085–H2093
Hollister LE (1986) Health aspects of cannabis. Pharmacol Rev 38:1–20
Howlett AC, Evans DM, Houston DB (1992) The cannabinoid receptor. In: Murphy L, Bartke A (eds) Marijuana/cannabinoids: neurobiology and neurophysiology. CRC, Boca Raton, pp 35–72
Kalant H (2004) Adverse effects of cannabis on health: an update of the literature since 1996. Prog Neuropsychopharmacol Biol Psychiatry 28:849–863
Kathmann M, Bauer U, Schlicker E, Göthert M (1999) Cannabinoid CB1 receptor-mediated inhibition of NMDA- and kainate-stimulated noradrenaline and dopamine release in the brain. Naunyn-Schmiedebergs Arch Pharmacol 359:466–470
Kwolek G, Zakrzeska A, Schlicker E, Göthert M, Godlewski G, Malinowska B (2005) Central and peripheral components of the pressor effect of anandamide in urethane-anaesthetized rats. Br J Pharmacol 145:567–575
Malinowska B, Godlewski G, Bucher B, Schlicker E (1997) Cannabinoid CB1 receptor-mediated inhibition of the neurogenic vasopressor response in the pithed rat. Naunyn-Schmiedebergs Arch Pharmacol 356:197–202
Malinowska B, Kwolek G, Göthert M (2001) Anandamide and methanandamide induce both vanilloid VR1- and cannabinoid CB1 receptor-mediated changes in heart rate and blood pressure in anaesthetized rats. Naunyn-Schmiedebergs Arch Pharmacol 364:562–569
Mousouttas M (2004) Cannabis use and cerebrovascular disease. Neurologist 10:47–53
Niederhoffer N, Szabo B (1999) Effect of the cannabinoid receptor agonist WIN55212-2 on sympathetic cardiovascular regulation. Br J Pharmacol 126:457–466
Niederhoffer N, Szabo B (2000) Cannabinoids cause central sympathoexcitation and bradycardia in rabbits. J Pharmacol Exp Ther 294:707–713
Niederhoffer N, Schmid K, Szabo B (2003) The peripheral sympathetic nervous system is the major target of cannabinoids in eliciting cardiovascular depression. Naunyn-Schmiedebergs Arch Pharmacol 367:434–443
Pacher P, Batkai S, Kunos G (2004) Haemodynamic profile and responsiveness to anandamide of TRPV1 receptor knock-out mice. J Physiol (Lond) 558:647–657
Pacher P, Batkai S, Kunos G (2005a) Blood pressure regulation by endocannabinoids and their receptors. Neuropharmacology 48:1130–1138
Pacher P, Batkai S, Kunos G (2005b) Cardiovascular pharmacology of cannabinoids. In: Pertwee RG (ed) Cannabinoids (handbook of experimental pharmacology, vol 168). Springer, Berlin Heidelberg New York, pp 599–625
Pertwee RG (2005) Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci 76:1307–1324
Pfitzer T, Niederhoffer N, Szabo B (2005) Search for an endogenous cannabinoid-mediated effect in the sympathetic nervous system. Naunyn-Schmiedebergs Arch Pharmacol 371:9–17
Ralevic V, Kendall DA (2002) Cannabinoids inhibit pre- and postjunctionally sympathetic neurotransmission in rat mesenteric arteries. Eur J Pharmacol 444:171–181
Ros J, Claria J, To-Figueras J, Planaguma A, Cejudo-Martin P, Fernandez-Varo G, Martin-Ruiz R, Arroyo V, Rivera F, Rodes J, Jimenez W (2002) Endogenous cannabinoids: a new system involved in the homeostasis of arterial pressure in experimental cirrhosis in the rat. Gastroenterology 122:85–93
Schlicker E, Göthert M (1998) Interactions between the presynaptic α2-autoreceptor and presynaptic inhibitory heteroreceptors on noradrenergic neurones. Brain Res Bull 47:129–132
Schlicker E, Kathmann M (2001) Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci 22:565–572
Schlicker E, Timm J, Göthert M (1996) Cannabinoid receptor-mediated inhibition of dopamine release in the retina. Naunyn-Schmiedebergs Arch Pharmacol 354:791–795
Schlicker E, Timm J, Zentner J, Göthert M (1997) Cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in the human and guinea-pig hippocampus. Naunyn-Schmiedebergs Arch Pharmacol 356:583–589
Schlicker E, Werthwein S, Kathmann M, Bauer U (1998) Nociceptin inhibits noradrenaline release in the mouse brain cortex via presynaptic ORL1 receptors. Naunyn-Schmiedebergs Arch Pharmacol 358:418–422
Schultheiß T, Flau K, Redmer A, Kathmann M, Reggio P, Seltzman HH, Schlicker E (2004) The facilitatory effect of SR141716 on transmitter release in guinea-pig hippocampus is due to its inverse agonist activity at cannabinoid CB1 receptors. Naunyn-Schmiedebergs Arch Pharmacol 369(Suppl 1):R84
Szabo B, Schlicker E (2005) Effects of cannabinoids on neurotransmission. In: Pertwee RG (ed) Cannabinoids (handbook of experimental pharmacology, vol 168). Springer, Berlin Heidelberg New York, pp 327–365
Timm J, Marr I, Werthwein S, Elz S, Schunack W, Schlicker E (1998) H2 receptor-mediated facilitation and H3 receptor-mediated inhibition of noradrenaline release in the guinea-pig brain. Naunyn-Schmiedebergs Arch Pharmacol 357:232–239
Varga K, Lake K, Martin BL, Kunos G (1995) Novel antagonist implicates the CB1 cannabinoid receptor in the hypotensive action of anandamide. Eur J Pharmacol 278:279–283
Wagner JA, Varga K, Kunos G (1998) Cardiovascular actions of cannabinoids and their generation during shock. J Mol Med 76:824–836
Zygmunt PM, Petersson J, Andersson DA, Chuang HH, Sorgard M, Di Marzo V, Julius D, Högestätt ED (1999) Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400:452–457
Acknowledgements
The financial support by the Deutsche Forschungsgemeinschaft (Graduiertenkolleg 246 and grant “Schl 266/5-5”) and by the faculty programme “BONFOR” is gratefully acknowledged. We would also like to thank Mrs. D. Petri and Mrs. P. Zeidler for their technical assistance and Professor Schunack and the companies Boehringer-Ingelheim, Novartis and Sanofi-Aventis for gifts of drugs.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Schultheiß, T., Flau, K., Kathmann, M. et al. Cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in guinea-pig vessels, but not in rat and mouse aorta. Naunyn Schmied Arch Pharmacol 372, 139–146 (2005). https://doi.org/10.1007/s00210-005-0007-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-005-0007-4