
Abstract
In this study we describe the activity of two cyclic nociceptin/orphanin FQ (N/OFQ) peptides; c[Cys10,14]N/OFQ(1–14)NH2 (c[Cys10,14]) and its [Nphe1] derivative c[Nphe1,Cys10,14]N/OFQ(1–14)NH2 (c[Nphe1,Cys10,14]) in native rat and mouse and recombinant human N/OFQ receptors (NOP). Cyclisation may protect the peptide from metabolic degradation.
In competition binding studies of rat, mouse and human NOP the following rank order pKi was obtained: N/OFQ(1–13)NH2(reference agonist)>N/OFQ=c[Cys10,14]>>c[Nphe1Cys10,14]. In GTPγ35S studies of Chinese hamster ovary cells expressing human NOP (CHOhNOP) c[Cys10,14] (pEC50 8.29) and N/OFQ(1–13)NH2 (pEC50 8.57) were full agonists whilst c[Nphe1Cys10,14] alone was inactive. Following 30 min pre-incubation c[Nphe1Cys10,14] competitively antagonised the effects of N/OFQ(1–13)NH2 with a pA2 and slope factor of 6.92 and 1.01 respectively. In cAMP assays c[Cys10,14] (pEC50 9.29, Emax 102% inhibition of the forskolin stimulated response), N/OFQ(1–13)NH2 (pEC50 10.16, Emax 103% inhibition) and c[Nphe1Cys10,14] (~80% inhibition at 10 μM) displayed agonist activity. In the mouse vas deferens c[Cys10,14] (pEC50 6.82, Emax 89% inhibition of electrically evoked contractions) and N/OFQ(1–13)NH2 (pEC50 7.47, Emax 93% inhibition) were full agonists whilst c[Nphe1Cys10,14] alone was inactive. c[Nphe1Cys10,14] (10 μM) competitively antagonised the effects of N/OFQ(1–13)NH2 with a pKB of 5.66. In a crude attempt to assess metabolic stability, c[Cys10,14] was incubated with rat brain membranes and then the supernatant assayed for remaining peptide. Following 60 min incubation 64% of the 1 nM added peptide was metabolised (compared with 54% for N/OFQ-NH2).
In summary, we report that c[Cys10,14] is a full agonist with a small reduction in potency but no improvement in stability whilst c[Nphe1Cys10,14] displays tissue (antagonist in the vas deferens) and assay (antagonist in the GTPγ35S assay and agonist in cAMP assay) dependent activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since its formal identification in 1995 (Meunier et al. 1995; Reinscheid et al. 1995), nociceptin/orphanin FQ (N/OFQ) has been the subject of intense study (Mogil and Pasternak 2001; Calo et al. 2000c) with particular emphasis on the design and evaluation of high affinity selective agonists and antagonists for the N/OFQ peptide receptor (NOP; Calo et al. 2000a). Activation of NOP by N/OFQ reduces cAMP formation, enhances an outward K+ conductance and closes voltage sensitive Ca2+ channels (Knoflach et al. 1996; Meunier et al. 1995; Reinscheid et al. 1995; Vaughan and Christie 1996). These actions decrease neurotransmission leading to a variety of organ/whole animal effects including; inhibition of neurogenic contractions in several tissues, antinociception/hyperalgesia, food intake stimulation, locomotor activity modulation, anxiolysis, diuresis and antinaturesis.
There are several novel peptide ligands currently available for the NOP receptor with varying degrees of efficacy, potency and selectivity and we and others have described these in some detail elsewhere (Hashiba et al. 2001; Calo et al. 2002; Guerrini et al. 2000). The major drawback with these peptide ligands is they are less stable due to their susceptibility to enzymatic cleavage (Terenius et al. 2000). In an attempt to limit metabolism we routinely use amidated and truncated forms of the native peptide such as N/OFQ(1–13)NH2 (Calo et al. 1996; Guerrini et al. 1997) and where practical high concentrations of peptidase inhibitors are also used (Terenius et al. 2000). One approach that has been used in the opioid peptide field involves peptide cyclisation to produce compounds such as [Dpen2,5]enkephalin (Kramer et al. 1991).
Addition of cysteine residues to peptides makes them suitable for cyclisation via disulphide bridge formation. Indeed, Ambo et al. (2001) reported a structure-activity relationship series of six cyclic N/OFQ analogues and compared these to their linear counterparts. One of these cyclic peptides c[Cys10,14]N/OFQ(1–14)NH2 displayed a small reduction in potency but retained full agonist activity and represents the starting point for this study. A full characterisation was not performed.
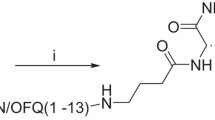
We have synthesised c[Cys10,14]N/OFQ(1–14)NH2 (c[Cys10,14]) and its [Nphe1] derivative c[Nphe1,Cys10,14]N/OFQ(1–14)NH2 (c[Nphe1,Cys10,14]; see structures in Fig. 1) with the former representing a cyclic agonist and the latter a putative cyclic antagonist. The latter [Nphe1] substitution is based on our previous observations that modification of phenylalanine at position 1 of both N/OFQ(1–13)NH2 and [Arg14,Lys15]N/OFQ-NH2 created two selective antagonists (Calo et al. 2000b, 2002). In this study we have performed a detailed characterisation of the pharmacological profile of these two cyclic peptides in:

Structures of A c[Cys10,14]N/OFQ(1–14)NH2 and B c[Nphe1, Cys10,14]N/OFQ(1–14)NH2. In B the C to N shift to create an antagonist is highlighted
-
1.
Radioligand binding studies to rat and mouse native NOP and to recombinant human NOP, MOP, DOP and KOP receptors expressed in Chinese hamster ovary (CHO) cells
-
2.
GTPγ35S and cAMP functional assays in CHOhNOP cells
-
3.
Electrically stimulated mouse vas deferens
In addition we have performed some crude studies to assess the metabolic stability of the peptides.
Materials and methods
Sources of reagents
All peptides (N/OFQ, N/OFQ(1–17)NH2, N/OFQ(1–13)NH2, UFP-101, c[Cys10,14] and c[Nphe1, Cys10,14]) were synthesised at one of our institutes using standard solid-phase synthesis techniques according to previously published methods (Guerrini et al. 1997). For the synthesis of the cyclic derivatives c[Cys10,14] and c[Nphe1, Cys10,14], the corresponding linear purified analogue was dissolved in a mixture of H2O/DMSO/TFA (75:25:0.1 v/v) at a concentration of 1 mg/ml (Annis et al. 1997). The cyclisation reaction completed within one day and was monitored by analytical HPLC and Ellman test (Ellman 1959). After partial evaporation of the solvent, the product was purified by preparative HPLC to yield the desired cyclic compound after lyophilisation. Peptide stocks were dissolved and frozen at −70°C in distilled water at 2 mM. All tissue culture media and supplements were from Invitrogen (Paisley, Scotland). [leucyl-3H]N/OFQ ([3H]-N/OFQ, 150 Ci/mmol) was from Amersham Pharmacia Biotech (Buckinghamshire, UK), GTPγ35S (1,250 Ci/mmol), [3H]diprenorphine ([3H]DPN, 50 Ci/mmol) and [3H]cAMP (32 Ci/mmol) were from Perkin Elmer Life Sciences (Boston, MA, USA). All receptor ligands, bacitracin, bovine serum albumin, GDP, unlabelled GTPγS, 3-isobutyl methylxanthine, peptidase inhibitors and forskolin were from Sigma (Poole, UK). All other consumables and reagents were of the highest purity available.
Cell culture and membrane preparation
Stock cultures of CHOhNOP cells (a gift from Dr F. Marshall and Mrs N. Bevan, GSK, Stevenage, UK) were maintained in DMEM: Ham F12(50:50) supplemented with 5% FCS, 100 IU/ml penicillin (P), 100 μg/ml streptomycin (S), 2.5 μg/ml fungizone, 200 μg/ml hygromycin B and 200 μg/ml G418 selection media. CHOhMOP/DOP/KOP cell stocks (gift from L. Toll, SRI International, Menlo Park, CA, US) were maintained in Ham F12 containing 10% FCS, 100 IU/ml P, 100 μg/ml S and 400 μg/ml G418, for CHO non-transfected cells G-418 and hygromycin B were omitted. Cell cultures were kept at 37°C in 5% CO2/humidified air. In all cases experimental cultures were free from selection agents (hygromycin B, G418). When confluence was reached (3–4 days), cells were harvested for use by the addition of HEPES (10 mM) buffered saline (0.9%) containing EDTA (0.05%). For native NOP preparations, female Wistar rats (250–300 g) or mice (25–30g) were decapitated following cervical dislocation. The brain was removed and rapidly transferred to ice-cold buffer (Tris-HCl 50 mM, pH 7.4) and the cerebrocortex dissected. All CHO and animal tissues were homogenized (Ultra Turrax, 30 s) on ice in; Tris (50 mM), EGTA (0.2 mM), pH 7.4 (CHOhNOP, GTPγ35S), Tris (50 mM), MgSO4 (5 mM), pH 7.4 for CHOhNOP, rat and mouse cerebrocortex (saturation/competition binding) or 50 mM Tris, pH 7.4 for CHOMOP/DOP/KOP cells. The homogenate was centrifuged at 20,374 g for 10 min at 4°C. This procedure was repeated twice more with re-suspension in fresh buffer each time. Rat and mouse tissue was frozen in small aliquots at −70°C until use. CHO membranes were prepared fresh each day.
Radioligand binding assay
[3H]-N/OFQ
Approximately 10 μg of CHOhNOP or 100–150 μg rat or mouse cerebrocortical membrane protein were incubated in 0.5 ml of homogenisation buffer containing 10 μM of peptidase inhibitors (captopril, amastatin, bestatin and phosphoramidon) and 0.5% BSA for 60 min at room temperature. In saturation experiments, various concentrations of [3H]-N/OFQ (0.001–1 nM) were used. In competition experiments, a single fixed concentration of 0.2 nM [3H]-N/OFQ was used. In all studies non-specific binding (NSB) was defined in the presence of 1 μM unlabelled N/OFQ. N/OFQ, N/OFQ(1–13)NH2, c[Cys10,14] and c[Nphe1Cys10,14] were included in various concentrations and combinations. Following incubation bound and free radioactivity was separated by vacuum filtration using a Brandel cell harvester through Whatman GF/B filters. Filters were soaked in polyethyleminine (PEI, 0.5%) to reduce NSB and loaded onto the harvester wet.
[3H]-diprenorphine
This is essentially as described above. Twenty-five to fifty micrograms CHOhMOP/DOP/KOP membrane protein was incubated in 0.5 ml of homogenisation buffer containing a cocktail of peptidase inhibitors (10 μM as above) and BSA for 60 min at room temperature. Competition binding studies were performed using 0.3–0.5 nM [3H]-DPN and N/OFQ(1–13)NH2, c[Cys10,14] and c[Nphe1Cys10,14] in various concentrations and combinations. In saturation studies up to 4 nM [3H]-DPN was used. NSB was defined in the presence of 10 μM naloxone. As reference compounds, naltrindole was included for CHOhDOP, norbinaltorphimine for CHOhKOP and fentanyl for CHOhMOP cells. Bound and free radioactivity was separated as described above.
GTPγ35S binding assay
Assays were performed essentially as described by (Albrecht et al. 1998; McDonald et al. 2002). Twenty micrograms of CHOhNOP membranes were incubated in 0.5 ml buffer containing Tris (50 mM), EGTA (0.2 mM), MgCl2 (1 mM), NaCl (100 mM), bacitracin (0.15 μM), amastatin (10 μM), bestatin (10 μM), captopril (10 μM), phosphoramidon (10 μM), GDP (100 μM) and ~150 pM GTPγ35S. Non-specific binding was determined in the presence of 10 μM unlabelled GTPγS. N/OFQ(1–13)NH2, c[Cys10,14] and c[Nphe1Cys10,14] were included in various concentrations to determine primary agonist actions. In order to probe any antagonist actions of c[Nphe1,Cys10,14] concentration response curves to N/OFQ(1–13)NH2 were constructed in the absence and presence of this peptide (0.1, 1, 10 μM added simultaneously with the agonist). In some of these antagonist experiments CHOhNOP membranes were pre-incubated for 30 min with c[Nphe1,Cys10,14] prior to N/OFQ(1–13)NH2 addition. Reactions were allowed to proceed for 60 min at 30°C with gentle shaking. Bound and free radiolabel was separated by vacuum filtration onto Whatman GF/B filters. PEI was not used.
Cyclic AMP accumulation assay
For measurement of cyclic AMP, whole CHOhNOP cells were incubated in 0.3 ml volumes of Krebs-HEPES buffer supplemented with BSA (0.5%), pH 7.4. cAMP formation was measured in the presence of isobutyl-methylxanthine (1 mM) and forskolin (1 μM). N/OFQ(1–13)NH2, c[Cys10,14] and c[Nphe1,Cys10,14] were included in various concentrations and combinations. After 15 min incubation at 37°C, reactions were terminated and cAMP extracted by addition of HCl (10 M)/NaOH (10 M)/Tris (1 M) and cyclic AMP was measured using a protein binding assay as described by Okawa et al. (1999).
Electrically stimulated mouse vas deferens
Vas deferens tissues was taken from male Swiss mice weighing 25–30 g; tissues prepared as previously described (Bigoni et al. 1999) and suspended in 5 ml organ baths containing Krebs buffer oxygenated with 95% O2 and 5% CO2. The temperature was set at 33°C and a resting tension of 0.3 g was applied to the tissues. Tissues were continuously stimulated through two platinum ring electrodes with supramaximal voltage rectangular pulses of 1 ms duration and 0.05 Hz frequency. Electrically evoked contractions were measured isotonically with a strain gauge transducer (Basile 7006) and recorded with the PC based acquisition system Autotrace 2.2 (RCS, Florence, Italy). Following an equilibration period of about 60 min, the contractions induced by electrical field stimulation (twitches) were stable. Measurements of twitch size were made. At this time, cumulative concentration response curves to agonists were constructed (0.5 log unit steps) in the absence and presence of antagonists. Antagonists were added to the bath 15 min before performing cumulative concentration response curves to agonists. In our experience with mVD, 15 min equilibration is more than ample for complete equilibration.
Crude metabolism experiments
N/OFQ, N/OFQ(1–17)NH2 and c[Cys10,14] (1 nM added from frozen stocks) were pre-incubated in 1,000 μl of saturation/competition homogenisation buffer containing 1 mg rat cerebrocortex membranes at 37°C for 15 min, 60 min and 120 min. Incubations were centrifuged at 16,000 g for 3 min at room temperature, then the supernatant was removed and kept on ice. [3H]-N/OFQ competition binding assays were performed with these and non-incubated stock samples as described above. At same time, a full competition curve for each peptide was constructed for subsequent use as a “standard curve”. In order to inactivate all peptidase activity some reactions (with c[Cys10,14]) were taken to pH 3.0 with 0.1 M trifluoroacetic (TFA) acid and incubated in parallel with an identical reaction at pH 7.4 for 1 h at 37°C. At the end of this period TFA was added to reaction at pH 7.4 so that both conditions contained identical quantities of TFA. Both reactions were then taken back to pH 7.4 using 2 M Tris (pH 7.4).
Data analysis
All data are expressed as mean ± SEM from ≥3 separate experiments. All curve fitting was performed using GraphPad PRISM V3.0 (San Diego, USA). In competition binding studies, the concentration of ligand producing 50% displacement of specific binding (IC50) was corrected for the competing mass of [3H]-N/OFQ or [3H]-DPN using KD values determined in this study or that of Hashiba et al. (2002b) to yield pKi. Agonist potencies were measured as pEC50, which is the negative logarithm to base 10 of the agonist molar concentration that produces 50% of the maximal possible effect of that agonist. In GTPγ35S binding experiments antagonist potencies were expressed as pA2 values from Schild regression plots, which is the negative logarithm to base 10 of the antagonist molar concentration at which the agonist concentration must be doubled to restore its original response, while in bioassays studies antagonist potencies were calculated with the Gaddum-Schild equation pKb = (CR-1)/[antagonist], assuming a slope equal to one. In metabolism experiments peptide concentrations were estimated from constructed standard curves using GraphPad PRISM. Statistical analysis has been performed using the Student’s t-test or one-way ANOVA followed by Bonferroni correction as appropriate with p values less than 0.05 considered significant.
Results
Radioligand binding
The binding of [3H]-N/OFQ to human recombinant NOP expressed in CHO cells (CHOhNOP) and native rat (Hashiba et al. 2002b) and mouse NOP expressed in cerebrocortical membranes was concentration dependent and saturable with pKD and Bmax values shown in Table 1. N/OFQ, N/OFQ(1–13)NH2, c[Cys10,14] and c[Nphe1Cys10,14] displaced the binding of [3H]-N/OFQ to all tissues in a concentration dependent manner (Fig. 2) with pKi values shown in Table 1. When compared to N/OFQ(1–13)NH2, c[Cys10,14] modification per se resulted in a modest (approximately 3-fold) loss of binding affinity whereas the effects of further [Nphe1] modification produced a more substantial (about 2 orders of magnitude) loss of affinity. In all three species of NOP the following rank order pKi was obtained: N/OFQ(1–13)NH2 >N/OFQ = c[Cys10,14] >>c[Nphe1Cys10,14].

Displacement of [3H]-N/OFQ binding by N/OFQ, N/OFQ(1–13)NH2, c[Cys10,14] and c[Nphe1Cys10,14] to human NOP expressed in A CHO cells, B native NOP expressed in rat cerebrocortical membranes and C native NOP expressed in mouse cerebrocortical membranes. All data are mean ± SEM for n=4. pKi values are shown in Table 1
In order to determine selectivity of c[Cys10,14] and c[Nphe1Cys10,14] for NOP receptor over classical MOP/DOP/KOP receptor a series of [3H]-DPN competition binding studies were performed using CHO cells expressing recombinant human MOP/DOP/KOP. The binding of [3H]-DPN was concentration dependent and saturable, pKD and Bmax values are shown in Table 1. It can be seen that CHOhDOP cells expressed the highest receptor density although the affinity of the N/OFQ analogues was weakest at this receptor More importantly, c[Cys10,14] and c[Nphe1Cys10,14] showed at least 2 orders of magnitude selectivity for NOP.
GTPγ35S binding
N/OFQ(1–13)NH2 and c[Cys10,14] stimulated the binding of GTPγ35S to CHOhNOP membranes in a concentration dependent and saturable manner (Fig. 3). c[Cys10,14] modification resulted in a modest loss of functional potency (~2 fold) without major modification of efficacy (1 μM). However, in all experiments the highest c[Cys10,14] concentration used 10 μM (and achievable based on the stocks in use) produced a response some 22% lower than that achieved at 1 μM (p<0.05). c[Nphe1Cys10,14] alone was inactive in the GTPγ35S binding assay. Full concentration response curves to N/OFQ(1–13)NH2, c[Cys10,14] and c[Nphe1Cys10,14] were constructed in non-transfected CHO cells in which all peptides failed to increase GTPγ35S binding (n=3 data not shown). Moreover, no differences in GTPγ35S binding at 10 μM compared to 1 μM c[Cys10,14] were observed indicating that the difference obtained in CHOhNOP membranes required the presence of the receptor.

N/OFQ(1–13)NH2 and c[Cys10,14] but not c[Nphe1Cys10,14] stimulate the binding of GTPγ35S to CHOhNOP membranes in a concentration dependent and saturable manner. Data are expressed as stimulation factor (ratio specific stimulated to specific basal binding) and represent mean ± SEM for n=7. pEC50 and Emax values are shown in Table 2. *Smaller than the response at 1 μM
c[Nphe1Cys10,14] was then tested as an antagonist where it produced a concentration dependent rightward shift in the concentration response curve to N/OFQ(1–13)NH2 without modification of the maximum response obtained (Fig. 4A). Schild analysis of these data yielded a pA2 and slope of 7.35 and 0.87 respectively. As this slope was less than unity it was suspected that the antagonist was not at equilibrium and hence these studies were repeated with a 30 min pre-incubation of c[Nphe1Cys10,14], Fig. 4B. Under these experimental conditions the pA2 decreased to 6.92 with a slope of 1.01.

Effects of c[Nphe1Cys10,14] on N/OFQ(1–13)NH2 stimulated GTPγ35S binding to CHOhNOP membranes (stimulation factor expressed as a % of the maximum N/ OFQ(1–13)NH2 response) either A without pre-incubation (i.e. added simultaneously with the agonist) or B with 30 min pre-incubation. In both experiments, c[Nphe1Cys10,14] produced a parallel rightward shift, which was used to generate the Schild plot shown as an insert in each panel. Data are mean ± SEM from n=5
Inhibition of cAMP formation
N/OFQ, N/OFQ(1–13)NH2 and c[Cys10,14] produced a concentration dependent inhibition of forskolin stimulated cAMP formation (Fig. 5). In this assay all peptides behaved as agonists with the following rank order potency (pEC50) N/OFQ(1–13)NH2 >N/OFQ >c[Cys10,14], Table 2. In contrast to the GTPγ35S studies described above, c[Nphe1Cys10,14] also produced a marked inhibition of cAMP formation (Fig. 5). However, due to the low potency of the peptide it was not possible to complete the concentration response curve to this peptide, which induced a 80% inhibition at 10 μM.

N/OFQ, N/OFQ(1–13)NH2, c[Cys10,14] and c[Nphe1Cys10,14] produce a concentration dependent inhibition of forskolin stimulated cAMP formation in whole CHOhNOP cells. pEC50 and Emax values are shown in Table 2. Data are means ± SEM for n=3–7
Mouse vas deferens
In the mouse vas deferens (Fig. 6A) N/OFQ(1–13)NH2 produced a concentration dependent inhibition of electrically evoked twitches with a pEC50 and Emax of 7.47 and −93%. c[Cys10,14] mimicked the inhibitory effects of the linear peptide with reduced potency but similar efficacy (Table 3). The kinetics of action of the cyclic compound were similar to those of N/OFQ(1–13)NH2. Indeed, the action of this peptide occurred immediately after addition to the bath, was rapidly reversible after washing, and could be repeated in the same tissue (data not shown).

A Comparison of N/OFQ(1–13)NH2, c[Cys10,14] actions in isolated mVD. B Antagonism of N/OFQ(1–13)NH2 inhibition of the electrically evoked contractile response by c[Nphe1, Cys10,14]N/OFQ(1–14)NH2 (10 μM). pEC50, Emax and pKB values are shown in Tables 3 and 4. Data are inhibition of twitch size (% twitches) and mean ± SEM for n≥4
The effects of N/OFQ(1–13)NH2 and c[Cys10,14] were reassessed in the presence of the universal opioid receptor antagonist naloxone and of the selective NOP antagonist UFP-101 (Fig. 7). Naloxone (1 μM) did not modify the concentration response curve to N/OFQ(1–13)NH2 or c[Cys10,14], while the selective NOP receptor antagonist, UFP-101 (1 μM) clearly shifted to the right the concentration response curve to both agonists without modifying their maximal effects. pKB values for UFP-101 vs. N/OFQ(1–13)NH2 and c[Cys10,14], respectively are shown in Table 4. c[Nphe1,Cys10,14]N/OFQ(1–14)NH2, up to 10 μM, did not significantly modify per se the control twitches (data not shown), but was able to shift to the right the concentration response curve to N/OFQ(1–13)NH2 without producing any change in maximal effects: a pKB value of 5.66 (Table 4) was derived from these experiments (Fig. 6B).

Effects of naloxone (1 μM) and UFP-101 (1 μM) on A N/OFQ(1–13)NH2 and B c[Cys10,14] inhibition of electrically evoked contractions of the mouse vas deferens. pKB values are summarised in Table 4. Data are mean ± SEM for n≥4
Crude metabolism experiments
In an attempt to determine any increased metabolic stability inferred by cyclisation N/OFQ, N/OFQ(1–17)NH2 and c[Cys10,14] were incubated with 1 mg of a crude rat cerebrocortical membrane preparation in the absence of any peptidase inhibitors. We used a standard competition binding assay (with peptidase inhibitors) with each peptide in order to determine actual tube concentrations. For all peptides 1 nM was added which we later confirmed to vary between 0.9–1.3 nM (Table 5). There was a time dependent loss of peptide such that following 2 h incubation ~88% of N/OFQ, 71% of N/OFQ-NH2 and 73% of c[Cys10,14] was lost. Amidation appeared to provide some protection when compared with N/OFQ while cyclisation did not appear to provide any additional increase in metabolic stability (Table 5). When the pH of the reaction was reduced to 3.0 all peptidase activity ceased such that there was no statistical difference between the quantity of c[Cys10,14] added and that remaining after 1 h incubation (Fig. 8).

Reducing buffer pH to 3.0 with trifluoroacetic acid (TFA) prevents metabolism of 1 nM (added) c[Cys10,14]. Data are mean ± SEM for n=4
Discussion
In this study we have shown that c[Cys10,14] and c[Nphe1Cys10,14] bind to native (mouse and rat) and recombinant NOP expressed in CHO cells. Whilst there was a dramatic loss of affinity on c[Nphe1Cys10,14] modification, both peptides displayed high selectivity over classical MOP/DOP/KOP. In GTPγ35S, cAMP inhibition and mouse vas deferens (mVD) assays c[Cys10,14] displayed agonist activity. In the GTPγ35S and mVD assays c[Nphe1Cys10,14] behaved as a competitive antagonist. However, in cAMP assays where signal amplification is apparent, this peptide behaved as an agonist (with reduced pEC50). Aware of these tissue and assay differences we conclude that c[Nphe1Cys10,14] is most likely to be a partial agonist. The initial premise of this study was that cyclisation would produce a peptide with improved metabolic stability and therefore be of more use in vivo. However, in a series of crude metabolic stability studies cyclisation provided no increased enzymatic protection when compared with its amidated equivalent, N/OFQ-NH2.
In radioligand binding studies c[Cys10,14] shows a high degree of selectivity over classical MOP/DOP/KOP. Indeed this is confirmed with isolated tissue studies. In the mVD the effects of c[Cys10,14] and N/OFQ(1–13)NH2 are unaffected by the classical opioid receptor antagonist naloxone but competitively antagonised by the selective NOP antagonist, UFP-101 with a pKB (7.3) value consistent to that reported previously by us (Calo et al. 2002) thus demonstrating that the actions of both peptides are exclusively due to NOP receptor activation.
c[Cys10,14] displays a small reduction in binding affinity that is likely to result from modification of the C-terminal address domain (responsible for receptor binding) of the peptide (Guerrini et al. 1997) where the cyclisation straddles Arg11 and Lys12. The importance of these C-terminally located basic residues is confirmed though studies demonstrating N/OFQ(1–13)NH2 as the shortest fragment that retains biological activity of the full length peptide and from creation of the first peptide with greater potency/affinity than N/OFQ, containing a triple Arg-Lys repeat motif (Okada et al. 2000). Indeed we have used this Arg-Lys triple repeat strategy to design UFP-101 (Calo et al. 2002). There was also a very small overall reduction in efficacy in the GTPγ35S but not the amplified cAMP assay. This discrepancy is a common feature of this system (Bigoni et al. 2002). These data are largely consistent with the paper of Ambo et al. where c[Cys10,14] was evaluated using NOP in HEK-293 cells (Ambo et al. 2001). In this brief article there was a small increase in binding affinity relative to N/OFQ(1–13)NH2 and [Cys10,14]N/OFQ(1–14)NH2, the linear version of this cyclic peptide. In GTPγ35S assays there was a small reduction in functional potency but all analogues were full agonists.
We have previously reported that [Nphe1] substitution in the N/OFQ(1–13)NH2 and [Arg14,Lys15]N/OFQ-NH2 sequences results in highly selective NOP antagonists with the latter molecule displaying improved affinity (Calo et al. 2000b, 2002; McDonald et al. 2003a). In this study we hoped to make a more metabolically stable cyclic antagonist, c[Nphe1Cys10,14]. In GTPγ35S and mVD studies clear competitive but relatively low affinity antagonism was observed. However, there appeared to be some equilibration problems encountered with c[Nphe1Cys10,14] since in initial studies a slope <1 was obtained which was increased to unity by pre-incubation. Whilst we have no evidence to support this notion these data may suggest that cyclisation and [Nphe1] substitution may modify the kinetics (on and/or off rates) of binding of c[Nphe1Cys10,14] to the receptor.
In GTPγ35S and mouse vas deferens assays there was no indication of residual agonist activity with c[Nphe1Cys10,14] but in cAMP studies this peptide was an agonist, such that at the highest concentration tested there was an 80% inhibition of the forskolin stimulated response with a crudely estimated agonist potency (based on a theoretical 100% maximum inhibition) of 5.76.
It is worthy of note that the [Nphe1] modification produces pure antagonists (Calo et al. 2000b) when applied to the linear templates N/OFQ(1–13)NH2 and [Arg14,Lys15]N/OFQ-NH2 (Calo et al. 2002) but when applied to the cyclic template c[Cys10,14] antagonism is observed in GTPγ35S and mVD assays but agonism is observed in (amplified) cAMP assays (present data). This difference is not easy to interpret. However, the C-terminal region of N/OFQ (i.e. the address domain) can assume an alpha helix structure (Zhang et al. 2002). This is likely to be important for NOP binding particularly with the second extra-cellular loop which contains several acidic residues (Topham et al. 1998), reminiscent to that of dynorphin binding to KOP receptor (Paterlini et al. 1997). Cyclisation of the 10–14 region of the address domain may interfere with the ability to assume the alpha helix conformation and this may affect the relative spatial orientation between the message (where Phe1 or Nphe1 modification is located) and the address domains. These changes may be negligible for agonist activity while they may limit the ability of the Nphe1 modification to reduce efficacy possibly generating a partial agonist.
For a partial agonist pEC50 should predict pA2/pKB and in general pKi should also predict pA2/pKB. In binding experiments c[Nphe1Cys10,14] displayed a pKi of ~8 in GTPγ35S a pA2 of ~7 and in cAMP studies a very rough estimate of pEC50 of ~5.8 can be obtained. This latter, pEC50 measured downstream in whole cells agrees well the pKB value obtained in mVD of 5.7. These discrepancies and differences with accepted theory are common to this system and are likely to result from buffer and assay sensitivity differences (Calo et al. 2000c). For example, Na+ would reduce binding affinity (Dooley and Houghten 2000) but this is necessary for GTPγ35S binding and cAMP/mVD assays use whole cells/tissues with relatively high concentrations of guanine nucleotides. Thus in comparison with the Na+ free receptor binding system a reduced pA2 in GTPγ35S might be predicted and further reductions in binding affinity for whole cells could also be anticipated. Indeed, we have discussed this previously (Calo et al. 2000c).
The high agonist activity of c[Nphe1Cys10,14] for inhibition of forskolin stimulated cAMP formation is worthy of a further contextual note. As cAMP formation is downstream in the signal transduction cascade this functional assay is liable to significant signal amplification. The net result of this being the creation of a receptor reserve, with partial agonists behaving as full agonists (Kenakin 2002; McDonald et al. 2003b). Indeed, we have previously reported that the partial agonists Ac-Arg-Tyr-Tyr-Arg-Trp-Lys-NH2 (Okawa et al. 1998) and [F/G]N/OFQ(1–13)NH2 (Okawa et al. 1999) behave as full agonists in this assay system. Moreover, using an ecdysone inducible expression system for hNOP we were able to vary intrinsic activity of [F/G]N/OFQ(1–13)NH2 relative to N/OFQ(1–13)NH2 by increasing receptor density in cAMP but not GTPγ35S assays (McDonald et al. 2003b). Again these studies underscore the need for caution in defining pharmacology based on a single functional end point.
The initial premise of this cyclisation study was to generate a peptide with improved metabolic stability. In a crude attempt to assess metabolic stability we exposed the agonist template c[Cys10,14] to rat cerebrocortical membranes as a potential source of peptidases for increasing times. Peptide remaining in the supernatant was assessed using a simple competition binding “bioassay”. There was a time dependent metabolism of N/OFQ, which could be reduced by amidation, but to our surprise cyclisation offered no additional protection. The drastic step of reducing buffer pH to 3.0 was able to prevent c[Cys10,14] metabolism. In essence, in this system the peptide did not have improved metabolic stability. There are several potential problems with this series of experiments in that we have not used N/OFQ(1–14)NH2 or a linear version of c[Cys10,14] as reference standards although we feel it is unlikely that this will make a significant difference. However, and potentially of more concern is the source of metabolic “activity” (i.e., rat brain homogenate); is this a representative example of in vivo peptide metabolism? Certainly most of our in vivo studies use direct i.c.v. injection so in this respect this is a representative model. However, it might be useful to perform future studies with plasma as a representation of i.v. administration. Moreover, the real test is simple in vivo administration at both i.c.v. and i.v. sites.
In summary, we have reported that cyclisation at positions 10 and 14 produced an agonist with slightly reduced potency (pEC50) in GTPγ35S and vas deferens assays and [Nphe1] substitution of that agonist, to yield c[Nphe1Cys10,14], produced an antagonist in GTPγ35S and vas deferens assays and an agonist in cAMP assays. With uncertainty regarding metabolic activity, in vivo studies are clearly warranted.
References
Albrecht E, Samovilova NN, Oswald S, Baeger I, Berger H (1998) Nociceptin (orphanin FQ): high-affinity and high-capacity binding site coupled to low-potency stimulation of guanylyl-5’-O-(gamma-thio)-triphosphate binding in rat brain membranes. J Pharmacol Exp Ther 286:896–902
Ambo A, Hamazaki N, Yamada Y, Nakata E, Sasaki Y (2001) Structure-activity studies on nociceptin analogues: ORL1 receptor binding and biological activity of cyclic disulfide-containing analogues of nociceptin peptides. J Med Chem 44:4015–4018
Annis I, Hargittai B, Barany G (1997) Disulfide bond formation in peptides. Methods Enzymol 289:198–221
Bigoni R, Giuliani S, Calo G, Rizzi A, Guerrini R, Salvadori S, Regoli D, Maggi CA (1999) Characterization of nociceptin receptors in the periphery: in vitro and in vivo studies. Naunyn-Schmiedebergs Arch Pharmacol 359:160–167
Bigoni R, Cao G, Rizzi A, Okawa H, Regoli D, Smart D, Lambert DG (2002) Effects of naloxone benzoylhydrazone on native and recombinant nociceptin/orphanin FQ receptors. Can J Physiol Pharmacol 80:407–412
Calo G, Rizzi A, Bogoni G, Neugebauer V, Salvadori S, Guerrini R, Bianchi C, Regoli D (1996) The mouse vas deferens: a pharmacological preparation sensitive to nociceptin. Eur J Pharmacol 311:R3–R5
Calo G, Bigoni R, Rizzi A, Guerrini R, Salvadori S, Regoli D (2000a) Nociceptin/orphanin FQ receptor ligands. Peptides 21:935–947
Calo G, Guerrini R, Bigoni R, Rizzi A, Marzola G, Okawa H, Bianchi C, Lambert DG, Salvadori S, Regoli D (2000b) Characterization of [Nphe1]nociceptin(1–13)NH2, a new selective nociceptin receptor antagonist. Br J Pharmacol 129:1183–1193
Calo G, Guerrini R, Rizzi A, Salvadori S, Regoli D (2000c) Pharmacology of nociceptin and its receptor: a novel therapeutic target. Br J Pharmacol 129:1261–1283
Calo G, Rizzi A, Rizzi D, Bigoni R, Guerrini R, Marzola G, Marti M, McDonald J, Morari M, Lambert DG, Salvadori S, Regoli D (2002) [Nphe1,Arg14,Lys15]Nociceptin-NH2, a novel potent and selective antagonist of the nociceptin/orphanin FQ receptor. Br J Pharmacol 136:303–311
Dooley CT, Houghten RA (2000) Orphanin FQ/nociceptin receptor binding studies. Peptides 21:949–960
Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82:70–77
Guerrini R, Calo G, Rizzi A, Bianchi C, Lazarus LH, Salvadori S, Temussi PA, Regoli D (1997) Address and message sequences for the nociceptin receptor: a structure-activity study of nociceptin-(1–13)-peptide amide. J Med Chem 40:1789–1793
Guerrini R, Calo G, Rizzi A, Bigoni R, Rizzi D, Regoli D, Salvadori S (2000) Structure-activity relationships of nociceptin and related peptides: comparison with dynorphin A. Peptides 21:923–933
Hashiba E, Harrison C, Calo G, Guerrini R, Rowbotham DJ, Smith G, Lambert DG (2001) Characterisation and comparison of novel ligands for the nociceptin/orphanin FQ receptor. Naunyn-Schmiedebergs Arch Pharmacol 363:28–33
Hashiba E, Lambert DG, Farkas J, Toth G, Smith G (2002a) Comparison of the binding of [(3)H]nociceptin/orphaninFQ(1–13)NH(2), [(3)H]nociceptin/orphaninFQ(1–17)OH and [(125)I]Tyr(14)nociceptin/orphaninFQ(1–17)OH to recombinant human and native rat cerebrocortical nociceptin/orphanin FQ receptors. Neurosci Lett 328:5–8
Hashiba E, Lambert DG, Jenck F, Wichmann J, Smith G (2002b) Characterisation of the non-peptide nociceptin receptor agonist, Ro64–6198 in Chinese hamster ovary cells expressing recombinant human nociceptin receptors. Life Sci 70:1719–1725
Kenakin T (2002) Drug efficacy at G protein-coupled receptors. Annu Rev Pharmacol Toxicol 42:349–379
Knoflach F, Reinscheid RK, Civelli O, Kemp JA (1996) Modulation of voltage-gated calcium channels by orphanin FQ in freshly dissociated hippocampal neurons. J Neurosci 16:6657–6664
Kramer TH, Toth G, Haaseth RC, Matsunaga TO, Davis P, Hruby VJ, Burks TF (1991) Influence of peptidase inhibitors on the apparent agonist potency of delta selective opioid peptides in vitro. Life Sci 48:881–886
McDonald J, Barnes TA, Calo G, Guerrini R, Rowbotham DJ, Lambert DG (2002) Effects of [(pF)Phe(4)]nociceptin/orphanin FQ-(1–13)NH(2) on GTPgamma35S binding and cAMP formation in Chinese hamster ovary cells expressing the human nociceptin/orphanin FQ receptor. Eur J Pharmacol 443:7–12
McDonald J, Calo G, Guerrini R, Lambert DG (2003a) UFP-101, a high affinity antagonist for the nociceptin/orphanin FQ receptor: radioligand and GTPgamma(35)S binding studies. Naunyn-Schmiedebergs Arch Pharmacol 367:183–187
McDonald J, Barnes TA, Okawa H, Williams J, Calo G, Rowbotham DJ, Lambert DG (2003b) Partial agonist behaviour depends upon the level of nociceptin/orphanin FQ receptor expression. Studies using the ecdysone inducible mammalian expression system. Br J Pharmacol 140:61–70
Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, Butour JL, Guillemot JC, Ferrara P, Monsarrat B et al (1995) Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature 377:532–535
Mogil JS, Pasternak GW (2001) The molecular and behavioral pharmacology of the orphanin FQ/nociceptin peptide and receptor family. Pharmacol Rev 53:381–415
Okada K, Sujaku T, Chuman Y, Nakashima R, Nose T, Costa T, Yamada Y, Yokoyama M, Nagahisa A, Shimohigashi Y (2000) Highly potent nociceptin analog containing the Arg-Lys triple repeat. Biochem Biophys Res Commun 278:493–498
Okawa H, Hirst RA, Smart D, McKnight AT, Lambert DG (1998) Rat central ORL-1 receptor uncouples from adenylyl cyclase during membrane preparation. Neurosci Lett 246:49–52
Okawa H, Nicol B, Bigoni R, Hirst RA, Calo G, Guerrini R, Rowbotham DJ, Smart D, McKnight AT, Lambert DG (1999) Comparison of the effects of [Phe1psi(CH2-NH)Gly2]nociceptin(1–13)NH2 in rat brain, rat vas deferens and CHO cells expressing recombinant human nociceptin receptors. Br J Pharmacol 127:123–130
Paterlini G, Portoghese PS, Ferguson DM (1997) Molecular simulation of dynorphin A-(1–10) binding to extracellular loop 2 of the kappa-opioid receptor. A model for receptor activation. J Med Chem 40:3254–3262
Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma Jr FJ, Civelli O (1995) Orphanin FQ: a neuropeptide that activates an opioid-like G protein-coupled receptor. Science 270:792–794
Terenius L, Sandin J, Sakurada T (2000) Nociceptin/orphanin FQ metabolism and bioactive metabolites. Peptides 21:919–922
Topham CM, Mouledous L, Poda G, Maigret B, Meunier JC (1998) Molecular modelling of the ORL1 receptor and its complex with nociceptin. Protein Eng 11:1163–1179
Vaughan CW, Christie MJ (1996) Increase by the ORL1 receptor (opioid receptor-like1) ligand, nociceptin, of inwardly rectifying K conductance in dorsal raphe nucleus neurones. Br J Pharmacol 117:1609–1611
Zhang C, Miller W, Valenzano KJ, Kyle DJ (2002) Novel, potent ORL-1 receptor agonist peptides containing alpha-Helix-promoting conformational constraints. J Med Chem 45:5280–5286
Acknowledgement
The authors would like to thank the International Association for the Study of Pain for provision of a collaborative travel grant between Leicester (UK) and Ferrara (Italy).
Author information
Authors and Affiliations
Corresponding author
Additional information
Presented in part to The British Pharmacological Society at the Brighton, UK Meeting January 2003
Rights and permissions
About this article
Cite this article
Kitayama, M., Barnes, T.A., Carra, G. et al. Pharmacological profile of the cyclic nociceptin/orphanin FQ analogues c[Cys10,14]N/OFQ(1–14)NH2 and c[Nphe1,Cys10,14]N/OFQ(1–14)NH2 . Naunyn-Schmiedeberg's Arch Pharmacol 368, 528–537 (2003). https://doi.org/10.1007/s00210-003-0821-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-003-0821-5