
Abstract
5F-MDMB-PINACA and 4F-MDMB-BINACA are synthetic cannabinoids (SCs) that elicit cannabinoid psychoactive effects. Defining pharmacokinetic–pharmacodynamic (PK–PD) relationships governing SCs and their metabolites are paramount to investigating their in vivo toxicological outcomes. However, the disposition kinetics and cannabinoid receptor (CB) activities of the primary metabolites of SCs are largely unknown. Additionally, reasons underlying the selection of ester hydrolysis metabolites (EHMs) as urinary biomarkers are often unclear. Here, metabolic reaction phenotyping was performed to identify key metabolizing enzymes of the parent SCs. Hepatic clearances of parent SCs and their EHMs were estimated from microsomal metabolic stability studies. Renal clearances were simulated using a mechanistic kidney model incorporating in vitro permeability and organic anionic transporter 3 (OAT3)-mediated uptake data. Overall clearances were considered in tandem with estimated volumes of distribution for in vivo biological half-lives (t1/2) predictions. Interactions of the compounds with CB1 and CB2 were investigated using a G-protein coupled receptor activation assay. We demonstrated that similar enzymatic isoforms were implicated in the metabolism of 5F-MDMB-PINACA and 4F-MDMB-BINACA. Our in vivo t1/2 determinations verified the rapid elimination of parent SCs and suggest prolonged circulation of their EHMs. The pronounced attenuation of the potencies and efficacies of the metabolites against CB1 and CB2 further suggests how toxic manifestations of SC abuse are likely precipitated by augmented exposure to parent SCs. Notably, basolateral OAT3-mediated uptake of the EHMs substantiates their higher urinary abundance. These novel insights underscore the importance of mechanistic, quantitative and systematic characterization of PK–PD relationships in rationalizing the toxicities of SCs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Between 2016 and 2018, synthetic cannabinoids (SCs) and synthetic opioids accounted for approximately half of the cases of new psychoactive substances abuse reported to the United Nations Office of Drug and Crime (United Nations Office on Drugs and Crime 2019). Notably, while a reduction in the prevalence of opioid use was detected, an upward trajectory was observed in the instances of SC usage (United Nations Office on Drugs and Crime 2019). Similar to Δ9‐tetrahydrocannabinol, the primary psychoactive compound in cannabis, SCs bind to the cannabinoid receptors (CB) of the endocannabinoid system, thereby inducing euphoric and psychogenic effects (Huestis et al. 2001; Gurney et al. 2014). However, the dramatically higher potency of SCs can result in mild to devastating consequences, ranging from vomiting, agitation and confusion to seizures, cardiac arrest and death (Gatch and Forster 2019).
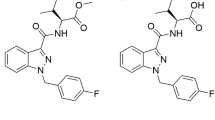
Since its emergence in September 2014, the most seized SC in the United States (US) has been 5F-MDMB-PINACA (methyl 2‐[1-(5‐fluoropentyl)‐1H‐indazole‐3‐carboxamido]‐3,3‐dimethylbutanoate, Fig. 1a) (European Monitoring Centre for Drugs and Drug Addiction 2017; Krotulski et al. 2018), surpassing the expected lifespan of SCs in the illicit drug market (Krotulski et al. 2019). Another dangerous SC gaining popularity is 4F-MDMB-BINACA (methyl 2‐[1‐(4‐fluorobutyl)‐1H‐indazole‐3‐carboxamido]‐3,3-dimethylbutanoate, Fig. 1b), which has been implicated in twenty fatalities in the US since December 2018 (Krotulski et al. 2019). These two indazole carboxamide SCs are structurally similar homologues that differ by one methylene group along their N-fluoroalkyl chain. Despite the increasing prevalence of abuse, severe toxicities associated with SC intake coupled with uncertainties in identifying the parent drug in biological samples remain perennial problems undermining clinical and forensic management of this global public health issue (Harris and Brown 2013).

Metabolic pathways of a 5F-MDMB-PINACA, b 4F-MDMB-BINACA and their urinary biomarker metabolites c 5F-MDMB-PINACA N-5-hydroxypentyl metabolite, d 5F-MDMB-PINACA EHM, e 4F-MDMB-BINACA EHM and f 4F-MDMB-BINACA N-4-hydroxybutyl metabolite. The question marks denote unknown elimination pathways and kinetics
SCs are known to be extensively metabolized, implicating cytochrome P450 enzymes (CYP450), carboxylesterases (CES) and UDP-glucuronosyltransferases (UGT) (Kong et al. 2018). Consequently, drug–drug interactions (DDIs) or genetic polymorphisms of xenobiotic metabolizing enzyme become pivotal factors governing toxic manifestations of SC abuse (Meyer et al. 2015; Holm et al. 2015a). For instance, the concomitant use of SCs with quetiapine had resulted in a case report of rhabdomyolysis, speculating its toxic potentiation via CYP3A4-mediated DDIs (Zhao et al. 2015). Nevertheless, the type and extent of metabolizing enzymes contributing to the metabolism of 5F-MDMB-PINACA and 4F-MDMB-BINACA are unknown. Based on the reported metabolic pathways of 5F-MDMB-PINACA and 4F-MDMB-BINACA as illustrated in Fig. 1 to form their respective ester hydrolysis metabolites (EHMs) and N-hydroxyalkyl metabolites (Haschimi et al. 2019; Yeter and Ozturk 2019), we hypothesized that CYP450 and CES are implicated in the metabolism of parent SCs while UGT are unlikely involved as glucuronic acid conjugates of the parent SCs have not been reported. Hence, our first aim was to perform metabolic phenotyping experiments to identify the specific isoforms of enzymes responsible for the metabolism of the two SCs.
However, the likelihood of toxicological outcomes stemming from altered metabolism and resultant variations in parent SC exposure must also be accompanied by careful consideration of the biological significance of metabolic intermediates. Notably, the N-4-hydroxypentyl metabolite of 5F-MDMB-PINACA has been demonstrated to retain full efficacy at CB1 and CB2, albeit with diminished affinity and potency (Gamage et al. 2019). Hence, active metabolites could potentially prolong psychotropic and physiological effects elicited by parent SCs, contributing to the in vivo toxicity profiles of SCs in an additive or synergistic manner. To date, however, there has been no systematic investigation on the relative CB1 and CB2 potencies (and efficacies) of 5F-MDMB-PINACA, 4F-MDMB-BINACA, their respective EHMs (Fig. 1d, e), and their respective N-5-hydroxypentyl and N-4-hydroxybutyl metabolites (Fig. 1c, f). Consequently, to gain further insight into the relative contributions of potential active metabolites to the toxicities observed with 5F-MDMB-PINACA and 4F-MDMB-BINACA, their in vitro biological activities at CB1 and CB2 were evaluated.
Additionally, rapid and reliable detection of SCs in biological matrices is also critical in clinical and forensic settings. Apart from associating adverse events with proof of consumption, evidence of widespread usage could also provide lawmakers with justification for future scheduling and control of the relevant SCs. Unfortunately, due to rapid hepatic metabolism, parent 5F-MDMB-PINACA and 4F-MDMB-BINACA are usually undetectable in urine samples. Castaneto et al. (2015) suggested that metabolites of SCs could represent potential biomarkers to document intake, but cautioned that such biomarkers have to be carefully curated. The metabolite profiles of 5F-MDMB-PINACA and 4F-MDMB-BINACA have been elucidated (Fig. 1) and their respective EHMs have been recommended as urinary biomarkers (Fig. 1d, e) to confirm the abuse of 5F-MDMB-PINACA and 4F-MDMB-BINACA (Haschimi et al. 2019; Krotulski et al. 2019; Yeter and Ozturk 2019).
Unambiguous identification of the EHMs as diagnostic markers for detecting consumption was guided by consistent outcomes obtained from both pooled human liver microsomes (HLM) and analyses of authentic urine samples (Haschimi et al. 2019; Yeter and Ozturk 2019). This two-pronged approach was adopted because discrepancies between the major metabolites detected via HLM studies and those found in urine following SC intake have been reported (Holm et al. 2015b; Diao and Huestis 2017). Nevertheless, this dependence on human urine specimens for verification of marker metabolites remains a major limitation impairing pre-emptive monitoring of SC intake. Hence, our third aim was to determine if systematic in vitro characterization of the disposition kinetics of the EHMs could enable rationalization of their preferential abundance in urine over other metabolites of 5F-MDMB-PINACA and 4F-MDMB-BINACA.
Materials and methods
Chemicals and reagents
5F-MDMB-PINACA (methyl (S)-2-(1-(5-fluoropentyl)-1H-indazole-3-carboxamido)-3,3-dimethylbutanoate), 5F-MDMB-PINACA EHM ((S)-2-(1-(5-fluoropentyl)-1H-indazole-3-carboxamido)-3,3-dimethylbutanoic acid), 5F-MDMB-PINACA N-5-hydroxypentyl metabolite (methyl (S)-2-(1-(5-hydroxypentyl)-1H-indazole-3-carboxamido)-3,3-dimethylbutanoate), 4F-MDMB-BINACA (methyl (S)-2-(1-(4-fluorobutyl)-1H-indazole-3-carboxamido)-3,3-dimethylbutanoate), 4F-MDMB-BINACA EHM ((S)-2-(1-(4-fluorobutyl)-1H-indazole-3-carboxamido)-3,3-dimethylbutanoic acid), 4F-MDMB-BINACA N-4-hydroxybutyl metabolite (methyl (S)-2-(1-(4-hydroxybutyl)-1H-indazole-3-carboxamido)-3,3-dimethylbutanoate), JWH-018 (1-naphthalenyl(1-pentyl-1H-indol-3-yl)-methanone) and D5-AM2201 N-4-hydroxypentyl metabolite (1-(5-fluoro-4-hydroxypentyl)-1H-indol-D5-3-yl](naphthalene-1-yl) methanone) were purchased from Cayman Chemical (Ann Arbor, MI, USA). Heroin was acquired from Cerilliant Corporation (Round Rock, Texas, USA). Bis(4-nitrophenyl) phosphate (BNPP), α-naphthoflavone, quercetin, sulfaphenazole, ticlopidine, S-( +)-N-3-benzylnirvanol, quinidine, ketoconazole, nitrendipine, loperamide, propranolol, estrone-3-sulfate and para-aminohippurate were purchased from Sigma-Aldrich (St. Louis, MO, USA), while atenolol was obtained from Tokyo Chemical Industry (Nihonbashihoncho, Tokyo, Japan). NADPH regenerating system solutions A (26 mM NADP+, 66 mM glucose-6-phosphate and 66 mM MgCl2 in water) and B (40 U/mL glucose-6-phosphate dehydrogenase in 5 mM sodium citrate), UGT reaction mix solutions A (250 mM Tris–HCl, 40 mM MgCl2, 0.125 mg/mL alamethicin in water) and B (25 mM uridine 5′-diphospho-glucuronic acid) were acquired from Corning Inc. (Corning, NY, USA). ACS-grade hydrochloric acid (HCl) and analytical-grade sodium hydroxide (NaOH) were obtained from Merck (Kenilworth, NJ, USA). All other commercially available chemicals were of analytical grade.
Dulbecco’s Modified Eagle’s Medium (DMEM) GlutaMAX™, DMEM high-glucose, Opti-MEM® I Reduced Serum Medium, penicillin–streptomycin, hygromycin B (50 mg/mL), amphotericin B (250 μg/mL), trypsin–EDTA (0.5%), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), Hank’s Balanced Salt Solution (HBSS), Dulbecco’s Phosphate-buffered Saline (DPBS), lucifer yellow dilithium CH salt and the Bicinchoninic acid (BCA) Protein Assay kit were purchased from Thermo Fischer Scientific Inc. (Waltham, MA, USA). Fetal bovine serum (FBS) and poly-d-lysine were supplied by Sigma-Aldrich (Overijse, Belgium). FBS was also alternatively of Hyclone™ brand purchased from GE Healthcare Life Sciences (Chicago, IL, USA). The Nano-Glo® Live Cell reagent, which was used for the readout of the bioassay, was procured from Promega (Madison, WI, USA).
Human liver microsomes and recombinant enzymes
150-donor pooled HLM (20 mg/mL protein in 250 mM sucrose) were purchased from Corning Inc. (Corning, NY, USA). Recombinant human CYP450 (rhCYP450) and recombinant human CES (rhCES) enzymes, which include CYP1A2 with oxidoreductase (catalog no. 456203), CYP2C8 with oxidoreductase and cytochrome b5 (catalog no. 456252), CYP2C9*1 (Arg 144) with oxidoreductase (catalog no. 456218), CYP2C19 with oxidoreductase (catalog no. 456219), CYP2D6*1 (Val374) with oxidoreductase (456217), CYP3A4 with oxidoreductase and cytochrome b5 (catalog no. 456202), CYP3A5 with oxidoreductase and cytochrome b5 (catalog no. 456256), CES1b (catalog no. 453320) and CES2 (catalog no. 453322) were also purchased from Corning Inc. (Corning, NY, USA).
Reaction phenotyping
Determining the in vitro formation clearance of EHMs and main classes of metabolizing enzymes via HLM incubations
Experimental procedure
The in vitro intrinsic formation clearances (CLint,formation) of the EHMs were determined with reaction mixtures comprising potassium phosphate buffer (100 mM, pH 7.4), HLM (0.5 mg/mL) and substrate (0.5 μM, 5F-MDMB-PINACA or 4F-MDMB-BINACA), and varied as summarized in Table 1: (1) Uninhibited metabolism in the presence of NADPH A; (2) restricted metabolism in the absence of NADPH A; (3) completely inhibited metabolism in the presence of irreversible CES inhibitor, BNPP, and absence of NADPH A. The final volume of reaction mixture was 100 μL comprising 1% methanol (v/v). After pre-warming at 37 °C and mixing at 120 rpm for 5 min in a shaking incubator, the reaction was initiated by transferring the initiation mixture into the incubation mixture (Table 1). At stipulated time-points (Table 1), the reaction was terminated by transferring 80 μL of reaction mixture into an equal volume of ice-cold acetonitrile with 0.25 μM of internal standard (D5-AM2201 N-4-hydroxypentyl metabolite). Quenched samples were centrifuged at 2775 g, 4 °C for 30 min, before the supernatant was analyzed via liquid chromatography–tandem mass spectrometry (LC–MS/MS).
Data analysis
The peak area ratio of substrate to internal standard was converted to percentage of substrate remaining and plotted against time (Obach 1999). The curve was ln transformed and fitted with linear regression using GraphPad Prism software v7.04 (San Diego, CA, USA). Elimination rate constant, k, was derived from the gradient of the plot and in vitro half-life (t1/2) was calculated using Eq. 1. As presented in Eq. 2, in vitro CLint,formation of each EHM was determined in the absence of NADPH after accounting for any non-CES contribution, while in vitro total intrinsic clearance of the parent SC (CLint,total) was calculated from the incubations with uninhibited metabolism via Eq. 3. Human in vitro–in vivo scaling factors were applied to in vitro CLint,formation and in vitro CLint,total to derive CLint,formation (Eq. 4) and CLint,total (Eq. 5), respectively. The in vivo formation clearances of the EHMs (CLformation) and total hepatic clearance of the parent SCs (CLH,parent) were subsequently determined using the well-stirred model (Eqs. 6 and 7). The percentage of metabolism via CES-mediated ester hydrolysis was calculated with Eq. 8.
MPPGL, QH, fub, Cb/C represent microsomal protein per gram of liver, liver blood flow, fraction unbound of drug in blood and drug concentration in blood to plasma ratio, respectively. Liver weight and QH were assumed to be 1.648 kg and 90 L/h, respectively. MPPGL was 43 mg/g according to the HLM product application notes. fub was assumed to follow fu, the fraction unbound of drug in plasma, which was predicted by Simcyp® simulator (Simcyp Ltd, version 18, Sheffield, UK) to be 0.071 and 0.107 for 5F-MDMB-PINACA and 4F-MDMB-BINACA, respectively. Similarly, their respective Cb/C was predicted to be 1.26 and 1.23, respectively.
Identifying specific metabolizing enzyme isoforms with isoform-specific chemical inhibitors and recombinant enzymes
Experimental procedure
To obtain preliminary insights into the relative contributions of specific drug metabolizing enzymes to the elimination of 5F-MDMB-PINACA and 4F-MDMB-BINACA, examination of the metabolic reactions of interest was performed in the absence and presence of CYP450 and CES isoform-specific chemical inhibitors. They include α-naphthoflavone (CYP1A2), quercetin (CYP2C8), sulfaphenazole (CYP2C9), ticlopidine (CYP2B6 and CYP2C19), S-( +)-N-3-benzylnirvanol (CYP2C19), quinidine (CYP2D6), ketoconazole (CYP3A4 and CYP3A5), nitrendipine (CES1) and loperamide (CES2) (Yanjiao et al. 2013; Thomsen et al. 2015; U.S. Food and Drug Administration 2017). Concentrations of inhibitors used were at least tenfold higher than their reported inhibitory constant to ensure complete inhibition of the particular enzymatic isoform (Prakash et al. 2000; Yanjiao et al. 2013; Thomsen et al. 2015). Test inhibitors or methanol (0.5% v/v; vehicle) were co-incubated with the SC in the initiation mixture, apart from time-dependent inhibitor ticlopidine which required a pre-incubation of 30 min at 37 °C with HLM. Additionally, all incubations were conducted in the presence of BNPP or in the absence of NADPH to negate CES and CYP450 contribution, respectively, depending on whether CYP450 or CES pathways were being investigated (Table 1).
Given that a single approach in isolation may be inadequate in ascertaining the metabolic routes governing the clearances of 5F-MDMB-PINACA and 4F-MDMB-BINACA, we next determined if recombinant human enzymes would be able to catalyze the relevant biotransformation pathways identified using selective inhibitors in the previous section. Each reaction mixture incorporated relevant enzymatic isoforms (rhCYP450, 20 pmol/mL; or rhCES, 200 μg/mL) as highlighted in Table 1. United States Food and Drug Administration (USFDA)-recommended probe substrates for each rhCYP450 and heroin for rhCES were tested as positive controls (Table S1 in the Electronic Supplementary Material—ESM) (Meyer et al. 2015; U.S. Food and Drug Administration 2017).
Data analysis
Substrate depletion-time data obtained from recombinant assays were processed as described previously for the HLM incubations. Scaling of the in vitro intrinsic clearance attributed to the jth recombinant enzyme (CLint,rhj) (Eq. 9) to the corresponding in vitro intrinsic clearance (CLint,j) in HLM was performed via consideration of the intersystem extrapolation factor (ISEF) as well as CYP450/CES abundance in the liver (Eq. 10). Finally, the percentage contribution of each isoform to the hepatic intrinsic microsomal clearance of the parent SC was calculated by Eqs. 11 and 12 for CES and CYP450, respectively.
Determination of the relative in vitro biological activities of 5F-MDMB-PINACA, 4F-MDMB-BINACA, their EHMs and N-hydroxyalkyl metabolites at CB1 and CB2
Live cell-based in vitro reporter assays that monitor the activation of CB1 and CB2 via their interaction with β-arrestin 2 using the NanoLuc Binary Technology, were applied to assess the biological activity of the compounds. Details regarding the development of the stable cell lines used here have been reported elsewhere (Cannaert et al. 2016, 2017).
The modified human embryonic kidney (HEK) 293 T cells were routinely maintained at 37 °C, 5% CO2, under humidified atmosphere in DMEM GlutaMAX™ supplemented with 10% heat-inactivated FBS, 100 IU/mL of penicillin, 100 μg/mL of streptomycin and 0.25 μg/mL of amphotericin B. For experiments, cells were plated on poly-d-lysine coated 96-well plates at 5 × 104 cells/well and incubated overnight. Next, the cells were washed twice with Opti-MEM® I Reduced Serum Medium to remove any remaining FBS, and 100 μL Opti-MEM® I and 25 µL of the Nano-Glo® Live Cell reagent was added to each well. Subsequently, the plate was placed into a TriStar2 LB 942 multimode microplate reader (Berthold Technologies GmbH & Co., Germany). Luminescence was monitored during the equilibration period until the signal stabilized (15 min). Next, 10 μL was added per well of the test compounds, present as concentrated (13.5-fold, as 10 μL to generate a final volume of 135 μL) stock solutions in Opti-MEM® I. The luminescence was continuously monitored for 120 min. Solvent controls were included in all experiments.
The results are represented as mean area under the curve (AUC) ± standard error of mean (SEM), obtained in minimum three independent experiments, with duplicates run in every experiment. All results were normalized to the Emax of JWH-018 (= 100%), our reference compound. Curve fitting of concentration–response curves via non-linear regression (four–parameter logistic fit) was employed to determine EC50 (a measure of potency) and Emax (a measure of efficacy) values, using GraphPad Prism software v7.04 (San Diego, CA, USA).
Establishing the disposition kinetics of the EHMs
Determining metabolic stabilities of the EHMs via HLM incubations
To investigate the in vitro metabolic clearances (CLint,metabolite) of the EHMs, the metabolites were incubated with NADPH and UGT co-factors to determine their Phase I and II metabolic stabilities, respectively (Table 1). Equations 3, 5 and 7 were similarly utilized to derive the hepatic clearances of the EHMs (CLH,metabolite). The Cb/C of the metabolites was assumed to be 0.55, which is a common Cb/C for acidic drugs (Riley et al. 2005).
Predicting the renal clearances of the EHMs via a mechanistic kidney model
To predict the renal clearance of the EHMs, a dynamic, physiologically based kidney model developed and verified by Huang and Isoherranen (2018) was used. The 35-compartment model consists of equations describing the physiological and pharmacokinetic processes underlying unbound glomerular filtration, pH-dependent bidirectional passive diffusion and active tubular secretion, thus providing a mechanistic framework for bottom-up simulations of renal clearance via incorporation of the plasma unbound fraction (fu), acid dissociation constant (pKa), in vitro apparent permeability (Papp) and in vivo basolateral secretion clearance (CLbsl,scr) (Huang and Isoherranen 2018). pKa and fu of the EHMs were derived computationally using Chemicalize (ChemAxon, Budapest, Hungary) and the Simcyp® simulator (Simcyp Ltd, version 18, Sheffield, UK), respectively; while, Papp and CLbsl,scr were determined experimentally. Simulations were initiated via a continuous infusion to allow renal clearance to be predicted under conditions of distribution equilibrium.
Permeability assay in MDCK II cells to determine P app
Experimental procedure
Madin–Darby Canine Kidney II (MDCK II) cells were kindly provided by Prof Ho Chi Lui, Paul (National University of Singapore, Singapore) and was used to determine the Papp of the EHMs. Propranolol and atenolol were utilized as high and low permeability markers, respectively. Culture techniques and assay procedures are outlined in the ESM.
Data analysis
Metabolite Papp and absolute recovery were calculated using Eqs. 13 and 14, respectively (Tran et al. 2004).
where VD and VR are the volumes of the apical and basolateral compartments (cm3), A is the area of the insert available for permeation (cm2), t is the time of sampling (s), CD(t) and CR(t) are the concentrations of metabolite in the apical and basolateral compartments at time t (μM).
Transport uptake assay to determine CLbsl,scr
HEK 293 cells were obtained from Dr. Kathleen Giacomini (University of California, San Francisco, San Francisco, CA) in three variants: wild-type, overexpressing organic anionic transporter 1 (OAT1) and overexpressing OAT3. Culture techniques and assay preparation steps are described in the ESM.
OAT transporter functionality was first verified using 1 μM of probe substrate, para-aminohippuric acid (PAH) for OAT1 and estrone-3-sulfate (E3S) for OAT3, respectively (US FDA 2017). Investigation of the putative OAT transporters governing the uptake of the EHMs (1 µM) was subsequently done in the presence and absence of OAT1/3 inhibitor probenecid (50 µM) (US FDA 2017).
Data analysis
For verification of transporter functionality and investigation of potential OAT-mediated uptake of the EHMs, cellular uptake rates (pmol/min/mg) are presented after normalization for total protein content and incubation time per well. Subsequently, in vitro intrinsic passive (CLint,passive) and total uptake clearances (CLint,passive+active) were derived from time course experiments performed in HEK wild-type and OAT-transfected cells, respectively (Eq. 15) (Izumi et al. 2018).
where Xcell(t) (pmol/mg), Cbuffer(t) (pmol/µL), AUC0-t,buffer (pmol min/µL) and V0 represent the amount of test compound taken up into the cells at time t, the compound concentration in the incubation buffer at time t, the area under the compound concentration − time curve from zero to time t in the incubation buffer, and the initial distribution volume, respectively. CLint,passive+active and CLint,passive were derived via linear regression of the initial slopes of the plots of uptake volume against AUC0-t,buffer/Cbuffer(t) using GraphPad Prism software v7.04 (San Diego, CA, USA). Intrinsic transporter-mediated active uptake clearance (CLint,active) was calculated by subtracting CLint,passive from CLint,active+passive.
In vitro CLint,active (μL/min/mg protein) was first converted to a unit of μL/min/106 cells via consideration of the number of cells per mg of measured protein as reported by Mathialagan et al. (2017). This was followed by quantitative in vitro in vivo extrapolation (IVIVE) using Eq. 16 to obtain in vivo CLbsl,scr (L/h).
In Eq. 16, the relative expression factor (REF), informed by differential OAT abundance in the kidney and transfected HEK cells, the number of proximal tubular cells per gram kidney (PTCPGK) (60 million in a healthy population) and kidney weight (300 g) were used as IVIVE scaling factors.
Predicting the in vivo t 1/2 and relative exposure of 5F-MDMB-PINACA, 4F-MDMB-BINACA and their EHMs via static modeling
The in vivo volumes of distribution at steady state (Vss) of the parent SCs and metabolites were estimated in the Simcyp® simulator (Simcyp Ltd, version 18, Sheffield, UK), using methods devised by Poulin and Theil (2002) and Rodgers and Rowland (2006), respectively, given that the former is more suitable for neutral compounds, while the latter has been shown to provide better predictions for acids. In vivo t1/2 (in a 70 kg man) and relative area under the curve of each EHM to parent SC (AUCm/AUCp) were calculated using Eqs. 17 and 18, respectively, where Fm refers to the systemic availability of the EHM (Nguyen et al. 2017).
LC–MS/MS analysis
Samples were analyzed using a Waters (Milford, MA, USA) liquid chromatography–tandem mass spectrometer comprising an Ultra Performance Liquid Chromatograph (UPLC®) and Xevo TQD Tandem Mass Spectrometer. Data were acquired using MassLynx software 4.2 and processed using TargetLynx XS Application Manager. For LC, an ACQUITY UPLC® ethylene bridged hybrid (BEH) C18, 1.7 μM, 2.1 × 50 mm column was employed. The mobile phase consisted of 10 mM ammonium formate with 0.1% formic acid (A) and acetonitrile with 0.1% formic acid (B). The composition of A and B was as follows: isocratic at 30% B (0–0.2 min), linear gradient from 30 to 95% B (0.2–4.5 min), isocratic at 95% B (4.5–4.8 min), linear gradient from 95 to 30% B (4.8–4.81 min), isocratic at 30% B (4.81–6 min). Flow rate was 0.4 mL/min, injection volume was 2 μL and column and sample temperatures were maintained at 30 °C and 4 °C, respectively. MS analysis was performed in electrospray positive mode, with capillary voltage 0.50 kV, source temperature 150 °C, desolvation temperature 400 °C, desolvation gas flow 800 L/h, cone gas flow 20 L/h. The compound-specific multiple reaction monitoring transitions are presented in Table 2. The first transition was used as the target transition, while the second and third transitions were the qualifying transitions. Dwell time was 0.015 s for all compounds.
Estimation of EHMs and N-hydroxyalkyl metabolites of 5F-MDMB-PINACA and 4F-MDMB-BINACA in authentic urine samples
Drug abusers’ urine samples that were identified with the EHMs or N-hydroxyalkyl metabolites of 5F-MDMB-PINACA and/or 4F-MDMB-BINACA (n = 32; mean age = 40 years; 20 Malay, 6 Chinese, 6 Indian; 31 male) were acquired, processed and analyzed as described in the following sections.
An aliquot of 0.5 mL of urine sample was hydrolyzed with β-glucuronidase/sulfatase enzyme from Haliotis Rufescens (abalone), adjusted to pH 11–12, then subjected to Supported Liquid Extraction (SLE) on a Biotage Isolute cartridge and eluted with 2 × 2 mL isopropanol:ethyl acetate (5:95 v/v). The eluate was evaporated to dryness, and reconstituted in 500 µL water:acetonitrile (80:20 v/v) for instrumental analysis.
Metabolite detection was performed on an Agilent Infinity II Liquid Chromatograph and SCIEX X500R Quadrupole Time-of-Flight Mass Spectrometer. Chromatographic separation was achieved with a Kinetex Biphenyl (2.1 × 100 mm, 2.6 µm) column at 40 °C by gradient elution of 10 mM ammonium formate with 0.1% formic acid (A) and methanol with 0.1% formic acid (B), 10–95% B over 10 min. Mass spectrometric data were acquired in Information-Dependent Acquisition (IDA)-MS/MS mode using positive electrospray ionization. Collision energy of 35 V with a spread of 15 V was used for the MS/MS. The concentrations of the analytes were estimated from fortified standards of known concentrations (2 ng/mL for 5F-MDMB-PINACA EHM and 5F-MDMB-PINACA N-5-hydroxypentyl metabolite; 5 ng/mL of 4F-MDMB-BINACA EHM and 4F-MDMB-BINACA N-4-hydroxybutyl metabolite).
Results
Metabolism of structurally similar 5F-MDMB-PINACA and 4F-MDMB-BINACA implicated comparable enzymatic pathways
Without inhibiting any metabolic pathways, the in vitro t1/2 of 5F-MDMB-PINACA determined in HLM was 7.82 min (95% CI 7.04 to 8.80 min) and its elimination rate constant k was 0.089 min−1 which was scaled to yield an in vitro CLint,total of 0.18 mL/min/mg, CLint,total of 12,561 mL/min and CLH,parent of 705 mL/min. For 4F-MDMB-BINACA, its HLM t1/2, k, in vitro CLint,total, CLint,total and CLH,parent were 10.27 min (95% CI 9.00 to 11.94 min), 0.068 min−1, 0.14 mL/min/mg, 9571 mL/min and 751 mL/min, respectively. The metabolic stability profiles of 5F-MDMB-PINACA and 4F-MDMB-BINACA in HLM incubations are shown in Fig. 2a, b, respectively.

Investigating the metabolizing enzymes of 5F-MDMB-PINACA and 4F-MDMB-BINACA via a, b non-specific inhibitors, c–f isoform-specific inhibitors, g, h recombinant enzymes and i, j relative contributions, respectively. Two-way ANOVA with Dunnett’s multiple comparisons test revealed significant differences between the control and some inhibitors for (c–f) (*p < 0.05). Experiments were conducted in duplicates. Values are mean ± SD (n = 2)
In the reaction mixture containing BNPP and excluding NADPH, no significant turnover of 4F-MDMB-BINACA was observed (Fig. 2b), displaying a similar trend to the negative control that did not contain HLM. In the absence of NADPH, the kNo NADPH, in vitro CLint,formation, CLint,formation and CLformation values were 0.047 min−1, 0.093 mL/min/mg, 6607 mL/min and 593 mL/min, respectively. The percentage of metabolism attributed to CES-mediated ester hydrolysis was 69.0%, while the remaining 31.0% was CYP450 mediated.
In the presence of BNPP and the absence of NADPH, 5F-MDMB-PINACA declined marginally to give a kBNPP+No NADPH of 0.015 min−1 (Fig. 2a). Moreover, in the sole absence of NADPH, kNo NADPH was 0.050 min−1. After taking the difference of the k values (kNo NADPH − kBNPP+No NADPH), in vitro CLint,formation, CLint,formation and CLformation values of 0.071 mL/min/mg, 5038 mL/min and 364 mL/min, respectively, were calculated. Taken together, our results indicate a non-CYP450 and non-CES pathway mediating 16.6% of the overall metabolism of 5F-MDMB-PINACA, with the remaining 40.1% and 43.3% being mediated by CES and CYP450, respectively.
To first identify the key CYP450/CES isoforms responsible for the metabolism of the parent SCs, inhibition studies utilizing enzyme-specific chemical inhibitors were simultaneously conducted in HLM. When compared to the control incubation which represents the theoretical maximum extent of enzyme-mediated metabolism, the selective inhibitor ketoconazole (CYP3A4 and CYP3A5) almost completely inhibited the metabolism of 5F-MDMB-PINACA (Fig. 2c) and 4F-MDMB-PINACA (Fig. 2d) post-inhibition at 15 and 30 min. Additionally, there was a statistically greater percentage of 5F-MDMB-PINACA remaining post-inhibition at 15 min and 30 min compared to the vehicle control in the presence of the CYP2C8 inhibitor quercetin and the CYP2C9 inhibitor sulphaphenazole, and at 30 min in the presence of the CYP2D6 inhibitor quinidine (Fig. 2c). For 4F-MDMB-BINACA, CYP2C8, CYP2C9, CYP2C19 and CYP2B6 inhibition was further demonstrated at 30 min (Fig. 2d). In the presence of the CES1 inhibitor nitrendipine, there was a statistically greater percentage of 4F-MDMB-BINACA remaining for CES1 at 30 min (Fig. 2f). The percentages of 5F-MDMB-PINACA remaining at 15 min and 30 min were not statistically greater than the vehicle control with concomitant CES1 and CES2 inhibition (Fig. 2e).
Further screening of 5F-MDMB-PINACA and 4F-MDMB-BINACA with rhCYP450 and rhCES isoforms was performed. As illustrated in Fig. 2g, h, rapid metabolism of both SCs was observed in the rhCYP3A4 and rhCYP3A5 incubations (greatest k value). Minor contributions from rhCYP2C8 and rhCES1b for 5F-MDMB-PINACA (Fig. 2g) and from rhCYP2C19 and rhCES1b for 4F-MDMB-BINACA (Fig. 2h) were observed. All other isoforms of rhCYP450 and rhCES did not contribute significantly to the metabolism of both SCs.
Upon consideration of the relative abundance, ISEF and in vitro CLint,rhj of each isoform as summarized in Table S2, both 5F-MDMB-PINACA and 4F-MDMB-BINACA were established to be predominantly metabolized by CYP3A4 and CES1b (Fig. 2i, j).
EHMs and N-hydroxyalkyl metabolites exhibited inferior potencies, along with either poorer or comparable efficacies at CB1 and CB2 relative to 5F-MDMB-PINACA and 4F-MDMB-BINACA
In vitro CB1 and CB2 activation analyses established that both 4F-MDMB-BINACA and 5F-MDMB-PINACA are potent agonists, with EC50 values in the low- or even sub-nanomolar range (0.69–5.69 nM; Table 3). In contrast, both EHMs and N-hydroxyalkyl metabolites of 4F-MDMB-BINACA and 5F-MDMB-PINACA were less potent than their respective parent compounds.
At CB1 and CB2, the relative potencies of the parent SCs and their metabolites revealed similar patterns when comparing 5F-MDMB-BINACA (Fig. 3a, c) and 4F-MDMB-PINACA (Fig. 3b, d), with the rank orders presented as follows: parent compound > N-hydroxyalkyl metabolites > EHMs (Table 3). Receptor-dependent differences were identified with the N-hydroxyalkyl metabolites. While hydroxylation of the alkyl side chain resulted in a marked attenuation of CB1 activation, this only culminated in a slightly reduced potency at CB2 (Fig. 3, Table 3).

Concentration-dependent interaction of cannabinoid receptor (CB) 1 (upper panel) and CB2 (lower panel) with β-arrestin 2 upon stimulation with 5F-MDMB-PINACA (left) and 4F-MDMB-BINACA (right) and their respective N-hydroxy metabolites and EHMs. Data are given as mean receptor activation ± SEM (n = 3), normalized to the Emax of JWH-018 (= 100%). The data for 5F-MDMB-PINACA (dotted line) were derived from Antonides et al. (2019)
When efficacies (Emax) of the parent SCs and their metabolites at CB1 were assessed, the Emax values of all compounds greatly exceeded that of JWH-018, whereas this was less pronounced or not the case at CB2 (Table 3). Additionally, as presented in the upper panel of Fig. 3, measured efficacies at CB1 declined in order of the parent SCs, followed by the N-hydroxyalkyl metabolites and finally, the EHMs. In comparison, the efficacies of both metabolites remained comparable to that of the parent compounds in the CB2 activation assay (Fig. 3, lower panel).
Disposition kinetics of the EHMs of 5F-MDMB-PINACA and 4F-MDMB-BINACA
The metabolites were metabolically stable
The EHMs of 5F-MDMB-PINACA and 4F-MDMB-BINACA did not display appreciable decline in both phase I and II metabolic incubations (Fig. S1 in the ESM). Consequently, the CLH,metabolite of the EHMs of 5F-MDMB-PINACA and 4F-MDMB-BINACA were relatively low at 1.26 mL/min and 3.18 mL/min, respectively.
The metabolites exhibited poor permeability
The calculated Papp values of propranolol and atenolol were 120.43 and 6.91 nm/s, respectively. The EHMs of 5F-MDMB-PINACA and 4F-MDMB-BINACA yielded Papp values of 8.55 and 8.03 nm/s, respectively, which tended towards that of atenolol. The permeability of lucifer yellow was not greater than 20 nm/s across all experiments, except for the negative control, and all recoveries were close to 100%.
The metabolites were substrates of OAT3 but not OAT1
Upon verifying the functionality of the OAT1/OAT3-transfected HEK systems using PAH and E3S as probe substrates of OAT1 and OAT3, respectively (Fig. S2 in the ESM), preliminary investigation of potential OAT1/3-mediated uptake of 5F-MDMB-PINACA and 4F-MDMB-BINACA EHMs was performed.
The rates of transport of both metabolites did not differ significantly between wild-type and OAT1-expressing HEK cells (Fig. 4a, b). Moreover, in OAT1-transfected HEK cells, the extent of uptake in the presence and absence of the OAT1 inhibitor probenecid was comparable (Fig. 4a, b), further affirming that the EHMs were not substrates of OAT1. In contrast, the rates of uptake of the EHMs of 5F-MDMB-PINACA (Fig. 4c) and 4F-MDMB-BINACA (Fig. 4d) were 1.8- and 2.4-fold greater in OAT3-expressing HEK cells compared to wild-type, respectively. The presence of the OAT3 inhibitor probenecid attenuated the rate of transport to a similar value between the OAT-expressing cells and wild type.

Determining the rate of transport of 5F-MDMB-PINACA and 4F-MDMB-BINACA EHMs across a, b OAT1-transfected versus wild type cells and across c, d OAT3-transfected versus wild type cells, respectively, and e, f determining their CLint,active in OAT3-transfected versus wild type cells, respectively. Experiments were conducted in triplicates. Values are mean ± SD. [n = 1 for (a–d) and n = 2 for (e, f)]
Subsequent time-dependent uptake experiments revealed a linear increase in uptake volumes of the EHMs in both HEK wild-type cells and OAT3-transfected cells over the first 5 min of incubation (Fig. 4e, f). CLint,passive and CLint,passive+active were obtained from the initial slopes and the difference yielded CLint,active values of 2.14 and 4.42 μL/min/mg protein for the EHMs of 5F-MDMB-PINACA (Fig. 4e) and 4F-MDMB-BINACA, respectively (Fig. 4f), which were scaled using Eq. 16 to yield an in vivo CLbsl,scr of 9.61 and 19.87 mL/min, respectively (Table S3 in the ESM).
The pKa, fu, Papp and CLbsl,scr values were integrated into the mechanistic kidney model and CLR,metabolite of the EHMs of 5F-MDMB-PINACA and 4F-MDMB-BINACA were eventually determined to be 3.15 and 5.46 mL/min, respectively.
The in vivo t 1/2 of the EHMs of 5F-MDMB-PINACA and 4F-MDMB-BINACA was predicted
Using Eq. 17, the in vivo t1/2 of 5F-MDMB-PINACA and 4F-MDMB-BINACA was predicted to be 6.6 and 4.3 h, respectively; while, their EHMs had significantly longer estimated half-lives of 17.5 and 9.0 h, respectively (Table 4). The AUCm/AUCp of both SCs was greater than 1.
The EHMs were more abundant in urine compared to the N-hydroxyalkyl metabolites
From the urinary data, the EHMs of 5F-MDMB-PINACA and/or 4F-MDMB-BINACA were detected in all 32 samples, whereas 5F-MDMB-PINACA N-5-hydroxypentyl and 4F-MDMB-BINACA N-4-hydroxybutyl metabolites were not present in some samples. As illustrated in Fig. 5a, b, the median concentration of each EHM was higher than that of the N-hydroxyalkyl metabolite of each SC (*p < 0.05).

Estimated urinary concentrations of a 5F-MDMB-PINACA EHM and 5F-MDMB-PINACA N-5-hydroxypentyl metabolite (n = 26) and b 4F-MDMB-BINACA EHM and 4F-MDMB-BINACA N-4-hydroxybutyl metabolite (n = 20). A non-parametric Mann–Whitney test revealed significant difference between the median concentrations of 5F-MDMB-PINACA EHM and 5F-MDMB-PINACA N-5-hydroxypentyl metabolite, and between those of 4F-MDMB-BINACA EHM and 4F-MDMB-BINACA N-4-hydroxybutyl metabolite (****p < 0.0001)
Discussion
SCs were originally developed in the 2000s to be utilized as CB agonists for biomedical research (Presley et al. 2013). However, their ability to induce psychoactive effects comparable to or surpassing that of cannabis has unwittingly culminated in the rapid proliferation of SC abuse. Consequently, there is a particularly acute need for forensic and public health authorities to better characterize the disposition kinetics of SCs, which will enable rationalization of marked interindividual variabilities in toxic manifestations and inform the development of analytical methods for robust detection (Diao and Huestis 2019). Unfortunately, controlled SC administration studies are severely restricted due to the lack of preclinical acute and chronic drug toxicity data. Moreover, the rapid emergence of new SCs, structurally modified to circumvent prohibitive legislation, hampers the feasibility of performing dedicated trials for each entity.
In this study, we have demonstrated the utility of detailed in vitro investigations as viable alternatives to in vivo clinical studies in (1) delineating factors that could affect the exposure and, hence, exacerbate toxic adverse effects associated with 5F-MDMB-PINACA and 4F-MDMB-BINACA usage and (2) establishing the diagnostic relevance of the EHMs in verifying the abuse of 5F-MDMB-PINACA and 4F-MDMB-BINACA.
In most clinical cases, 5F-MDMB-PINACA and 4F-MDMB-BINACA were undetectable in urine samples, suggesting that renal clearance is possibly not a major contributing factor to their elimination (Haschimi et al. 2019; Krotulski et al. 2019; Yeter and Ozturk 2019). Corroboratively, we established the metabolic instability of these parent SCs (HLM t1/2 < 30 min) and confirmed hepatic metabolism as their main route of elimination.
Based on the reported metabolic pathways of 5F-MDMB-PINACA and 4F-MDMB-BINACA to form the EHMs as well as N-hydroxyalkyl metabolites as reported by Yeter et al. and Haschimi et al., we initially hypothesized CYP450 and CES to be the main classes of enzymes implicated in the hepatic elimination of our SCs of interest (Haschimi et al. 2019; Yeter and Ozturk 2019). Negligible 4F-MDMB-BINACA depletion in the presence of the CES inhibitor, BNPP and absence of NADPH confirmed our postulation. Ensuing experiments in the absence of NADPH circumvented CYP450-mediated reactions but did not inhibit CES. Subsequently, we established the percentage contribution of CES and CYP450 to the metabolism of 4F-MDMB-BINACA as 69.0% and 31.0%, respectively. In contrast, there was apparent metabolism of 5F-MDMB-PINACA in the reaction mixture with BNPP and in the absence of NADPH. Among SCs bearing fluoropentyl groups, a phenomenon of co-factor independent oxidative defluorination has been reported, which was reasoned to be a nucleophilic reaction with proteins within HLM or metabolism by an unknown non-CYP450 enzyme (Holm et al. 2015a; Diao and Huestis 2017). Given that metabolism of 5F-MDMB-PINACA was not observed in the presence of heat-inactivated HLM, the role of non-CYP450 enzymes might be significant but this needs to be further investigated. Consequently, we attributed 16.6% of the metabolism of 5F-MDMB-PINACA to the unknown route and assigned 40.1% and 43.3% to CES and CYP450 pathways, respectively.
Our subsequent reaction phenotyping results further established that the key metabolizing enzymes of 5F-MDMB-PINACA and 4F-MDMB-BINACA were similar. Given that both 5F-MDMB-PINACA and 4F-MDMB-BINACA were compounds with large acyl moieties and small alcohol parts, characteristic features typically observed among CES1 substrates, preferential CES1-mediated hydrolysis was expected (Wang et al. 2018). Aligned with this postulation, 40.1% and 69.0% of the metabolism of 5F-MDMB-PINACA and 4F-MDMB-BINACA, respectively, were revealed to be mediated by CES1b. Furthermore, CYP3A4 played an instrumental role in the metabolism of 5F-MDMB-PINACA (32.0%) and 4F-MDMB-BINACA (22.9%). Taken together, the identified metabolic pathways of these structurally analogous SCs could expound their susceptibilities to intrinsic and extrinsic variabilities, underscoring the critical role of reaction phenotyping experiments in aiding interpretation and clinical management of potential adverse outcomes arising from SC abuse.
Genetic polymorphisms and DDIs represent key perpetrators of intrinsic and extrinsic variabilities, respectively, which can result in severe toxicities among abusers. For instance, five loss-of-function non-synonymous single nucleotide polymorphisms (nsSNPs) of CES1 (L40Ter, E220G, A158V, R199H and T290M) were reported to culminate in the reduced hydrolysis of pharmaceutical drugs (Wang et al. 2017). Consequently, an abuser who possesses these nsSNPs of CES1 could potentially experience exacerbated psychoactive and toxic effects of 5F-MDMB-PINACA and 4F-MDMB-BINACA. Additionally, among reports of acute intoxications and deaths with confirmed exposure to 5F-MDMB-PINACA, analyses of both clinical and post-mortem samples uncovered the presence of substances such as other SCs, opiates as well as various central nervous system depressants (i.e., benzodiazepines) and stimulants (i.e., amphetamines) (European Monitoring Centre for Drugs and Drug Addiction 2017). With the prevalence of CYP3A4-mediated metabolism among these compounds, these could act as competing substrates, hindering the metabolism of 5F-MDMB-PINACA and 4F-MDMB-BINACA, hence potentiating toxic effects of these SCs (Meyer et al. 2015; Hussaarts et al. 2019).
However, besides focusing on factors which could augment the systemic exposure of 5F-MDMB-PINACA and 4F-MDMB-BINACA, considering the rapid elimination of these parent SCs, tandem investigation of the interplay between the parent SCs and their major metabolites in perpetrating toxic manifestations also becomes pivotal. Our in vitro metabolic assays have revealed that the EHMs are potentially the major systemic metabolites of both 5F-MDMB-PINACA and 4F-MDMB-BINACA. As presented in Fig. S3 in the ESM, when relative metabolite formation was quantified in metabolic depletion assays of 5F-MDMB-PINACA, there was a greater concentration of the EHM formed compared to that of the N-5-hydroxypentyl metabolite after 15 min till the end of the experiment. We posit that metabolism of 4F-MDMB-BINACA would produce similar findings. Nevertheless, functional evaluation of the EHMs in comparison to their parent SCs and the N-hydroxyalkyl metabolites revealed significantly attenuated potencies (EC50), with negligible CB1 and CB2 activation observed at concentrations up to 1 µM (Table 3). Notably, the findings at both receptors are aligned with observations made with several other SCs, where the amide moiety is composed of an amino acid derivative (Wouters et al. 2019; Noble et al. 2019). Despite the strong reduction in potencies, the efficacies (Emax) of the ester hydrolysis metabolites were shown to exceed that of reference compound JWH-018 across both CB1 and CB2 (Table 3), suggesting that these may still contribute to the overall in vivo toxicological response assuming their systemic exposure is sufficiently high. Yet, as reported by Seither et al., based on analyses of blood samples from 36 post-mortem cases, the average concentration of the EHM of 5F-MDMB-PINACA was determined to be 49 ng/mL (135 nM; log M = − 6.87) in central blood specimens and 21 ng/mL (79.8 nM; log M = − 7.10) in peripheral blood specimens (Reidy et al. 2018). Preliminary evaluation of these measured blood concentrations using the relevant sigmoidal concentration–response curves shown in Fig. 3a, c highlights how no detectable CB1 and CB2 activation would occur. While blood concentrations may not reflect the actual exposure of the ester hydrolysis metabolites at CB1 (ubiquitously expressed in the central nervous system) and CB2 (abundantly located on immune cells in peripheral regions) (Tai and Fantegrossi 2016), their hydrophilic nature is likely to hamper substantial tissue accumulation. Consequently, it becomes clear that holistic understanding of the role of the identified major systemic metabolites in mediating toxicities arising from SC abuse is contingent on additional mechanistic characterization of exposure–response relationships.
Interestingly, despite their limited contribution to observed toxicological outcomes, the EHMs continue to retain considerable utility as markers of SC consumption, with analyses of human urine specimens indicating that concentrations of the EHMs of both 5F-MDMB-PINACA and 4F-MDMB-BINACA significantly surpassed that of the corresponding N-hydroxyalkyl metabolites (Fig. 5). Yet, in the absence of authentic urine samples, it is unclear how optimal urinary marker metabolites for verifying SC intake could be determined or selected from characteristic phase I metabolites identified in HLM incubations. With the relentless and rapid emergence of new SCs, there remains a profound need to streamline processes for SC detection by focusing efforts on the synthesis of standards and the development of targeted analytical methods on the most appropriate marker metabolites (Diao and Huestis 2019). In this study, we have outlined how detailed in vitro investigations of the various processes governing renal elimination (i.e., glomerular filtration, tubular reabsorption and tubular secretion) sufficed to identify the EHMs over the N-hydroxyalkyl metabolites as the main urinary markers of both 5F-MDMB-PINACA and 4F-MDMB-BINACA consumption. Low measured in vitro Papp values in MDCK II cells first revealed the minor role of tubular reabsorption in the renal elimination of the EHMs. Critically, subsequent in vitro investigations of potential tubular secretion mediated by OAT1/3 transporters located at the basolateral side of the proximal tubular epithelium identified both EHMs as substrates of OAT3 (Fig. 4c–f) but not OAT1 (Fig. 4a, b). Expectedly, preliminary results indicated that the N-hydroxyalkyl metabolites are not substrates of OAT1 and OAT3 (data not shown), which could be attributed to the absence of anionic acidic groups in the N-hydroxyalkyl metabolites as compared to the EHMs. Therefore, the presence of basolateral active secretion is likely the primary factor governing the preferential renal elimination of the EHMs. Moreover, the reported downstream metabolism of the N-hydroxyalkyl metabolites further substantiates their comparatively lower abundance in the urine (Haschimi et al. 2019; Yeter and Ozturk 2019). Hence, beyond the current repertoire of in vitro methodologies largely aimed at identifying the full spectrum of SC metabolites via human hepatocyte or HLM incubations, we have demonstrated the complementary nature of bottom-up approaches in independently rationalizing the choice of characteristic urinary SC marker metabolites. We envision that our findings could eventually empower clinical and forensic laboratories to incorporate additional assays investigating the renal disposition kinetics of potential marker metabolites into their screening workflows.
Integrating both in vitro-derived CLH and in silico predictions of Vss into Eq. 17, the in vivo t1/2 of 5F-MDMB-PINACA and 4F-MDMB-BINACA were estimated to be 6.6 h and 4.3 h, respectively. While there was an approximate twofold difference between the in vitro CLint,total derived from HLM versus the summed in vitro CLint,j obtained from rhCYP450 and rhCES, the magnitude of discrepancy was deemed to be reasonably consistent due to the use of ISEF which could vary between laboratories depending on the source of enzymes and experimental conditions amongst other factors (Chen et al. 2011). For subsequent IVIVE to CLH, the CLint,total derived from HLM was selected as the enzymes in pooled HLM are more representative of a human physiological system. Nevertheless, the accuracy of in vivo t1/2 calculations presented in this study could be undermined by underlying assumptions. First, the tissue-to-plasma partition coefficients required to derive Vss values for the parent SCs were predicted based on mechanistic tissue composition-based equations proposed by Poulin and Theil (2002), where partitioning of a compound into tissue components is assumed to occur solely by passive permeation, governed by compound-specific properties such as lipophilicity and plasma protein binding. Although application of this mechanism-based method in predicting rat and human Vss for 123 structurally unrelated drugs demonstrated reasonable congruence with reported in vivo Vss values (average ratio of predicted to experimental Vss = 1.06), there remained a subset of compounds in which predicted and experimental Vss values differed by a factor larger than two (Poulin and Theil 2002). As such, if additional specific binding or uptake processes occur that could additionally facilitate the tissue accumulation of 5F-MDMB-PINACA and 4F-MDMB-BINACA, application of Poulin and Theil’s framework would result in potential underpredictions of Vss. Conversely, if distribution into peripheral tissues is limited, Vss would be overpredicted. Furthermore, another key premise governing the application of Eq. 17 in t1/2 calculations is that the identified metabolic elimination processes of the parent SCs adhere to linear, first-order kinetics. However, a typical characteristic of enzymatic reactions is capacity-limited metabolism and hence, for some compounds, it is observed that elimination appears to become zero order at high concentrations when saturation occurs. Often, SCs are sprayed on dried plant material and the end user smokes or vapes the product. Furthermore, rapid elimination of the parent SCs is likely to culminate in transient psychoactive effects, increasing the likelihood of repeated abuse. In such instances, if the administered dose or frequency of dosing results in concentrations or accumulation sufficient to saturate enzymatic pathways, ensuing reductions in hepatic clearances would manifest in the prolongation of apparent half-lives. Evidently, considering the abovementioned assumptions, increased confidence in the validity of t1/2 estimates for the SCs necessarily entails further verification with clinical data.
Similarly, while the t1/2 of the EHMs was predicted to be significantly longer, at 17.5 h and 9.0 h, respectively, implying that they could be present up to approximately 4 and 2 days after ingestion of 5F-MDMB-PINACA and 4F-MDMB-BINACA (~ 5 t1/2), these findings must also be carefully interpreted in light of gaps in the quantitative translation of CLint,active to CLbsl,scr as presented in Table S3. Specifically, the assumption of similar OAT3 abundance (REF = 1) between the in vitro experimental system utilized (i.e., OAT3-transfected HEK cells) and the kidney cortex must be justified with additional protein abundance data. Hence, at present, the t1/2 estimates displayed in Table 4 should only serve as preliminary references for forensic and toxicology laboratories to further investigate and define the detection windows of SCs and their metabolites within the systemic circulation. Nonetheless, positive trends in our attempts to link in vitro studies to in vivo outcomes were still observed. Using Eq. 18, our calculations demonstrated that the AUCm/AUCp of both SCs were greater than 1, indicating greater in vivo systemic exposure of the EHMs when compared to their respective parent SC. Consistently, Yeter and Öztürk (2019) reported that blood concentrations of 5F-MDMB-PINACA and its EHM ranged from 0.10 to 1.55 ng/mL (mean = 0.40 ng/mL) and 0.15 to 23.4 ng/mL (mean = 2.69 ng/mL), respectively, across 70 autopsy cases (mean Cm/Cp = 6.73). In a separate study by Seither et al. (2020), analyses of blood samples from 36 post-mortem cases also yielded average blood concentrations of 0.29 ng/mL and 49 ng/mL for 5F-MDMB-PINACA and its EHM, respectively (mean Cm/Cp = 169). This convergence of top-down and bottom-up outcomes continues to ratify the value of predicting in vivo SC disposition kinetics via scaling of biochemical data obtained from in vitro experimental systems.
Conclusion
Given the existing and rapidly expanding array of illicit drugs, for toxicology and forensic laboratories, besides providing insights into possible causative factors governing SC-associated toxicities, future-proofing capabilities in verifying intake of emerging SCs also become essential. In this study, utilizing case studies of 5F-MDMB-PINACA and 4F-MDMB-BINACA, we first underscored the utility of reaction phenotyping experiments in uncovering major biotransformation pathways of the putative SCs, thereby enabling informed rationalization and management of potential interindividual variabilities in toxic manifestations. We further established how accurate delineation of the relative contributions of parent SCs and their metabolites to in vivo toxicological outcomes is predicated on mechanistic PK-PD characterization, where in vitro investigations of the disposition kinetics of SCs and their metabolites must be performed in tandem with activity-based characterization at CB1 and CB2. Finally, we demonstrated how systematic in vitro characterization of renal disposition kinetics could present a viable alternative to using authentic urine samples to support the choice of the EHMs as the major urinary makers of 5F-MDMB-PINACA and 4F-MDMB-BINACA consumption.
Abbreviations
- AUCm/AUCp :
-
Relative area under the curve of metabolite to parent
- BNPP:
-
Bis(4-nitrophenyl) phosphate
- CB:
-
Cannabinoid receptor
- C b/C :
-
Ratio of drug concentration in blood to plasma
- CLbsl,scr :
-
In vivo basolateral secretion clearance
- CLformation :
-
In vivo formation clearance of the ester hydrolysis metabolite
- CLH :
-
In vivo hepatic clearance
- CLint,active :
-
In vitro intrinsic transporter-mediated active uptake
- CLint,passive :
-
In vitro intrinsic passive uptake
- CLint,formation :
-
In vitro intrinsic formation clearance of the ester hydrolysis metabolite
- CLint,metabolic :
-
In vitro intrinsic metabolic clearance of the ester hydrolysis metabolite
- CLint,total :
-
In vitro total intrinsic clearance of the parent synthetic cannabinoid
- CLR :
-
In vivo renal clearance
- CYP450:
-
Cytochrome P450 enzyme
- CES:
-
Carboxylesterase
- DDI:
-
Drug–drug interaction
- EC50 :
-
Concentration of agonist that provokes a response halfway between the baseline and maximum response
- EHM:
-
Ester hydrolysis metabolite
- E max :
-
The maximal limit of response to an agonist
- f u :
-
Fraction unbound of drug in plasma
- f ub :
-
Fraction unbound of drug in blood
- 4F-MDMB-BINACA:
-
Methyl 2‐[1-(4‐fluorobutyl)‐1H‐indazole‐3‐carboxamido]‐3,3-dimethylbutanoate
- 5F-MDMB-PINACA:
-
Methyl 2‐[1-(5‐fluoropentyl)‐1H‐indazole‐3‐carboxamido]‐3,3‐dimethylbutanoate
- HEK:
-
Human embryonic kidney 293
- HLM:
-
Human liver microsomes
- ISEF:
-
Intersystem extrapolation factor
- IVIVE:
-
In vitro in vivo extrapolation
- k :
-
Elimination rate constant
- MDCK II:
-
Madin–Darby Canine Kidney II
- nsSNP:
-
Non-synonymous small nucleotide polymorphism
- OAT:
-
Organic anion transporter
- P app :
-
Apparent permeability
- pK a :
-
Acid dissociation constant
- rhCYP450:
-
Recombinant human cytochrome P450 enzyme
- rhCES:
-
Recombinant human carboxylesterase
- SC:
-
Synthetic cannabinoid
- t 1/2 :
-
Half-life
- UGT:
-
Uridine 5′-diphospho-glucuronosyltransferase
- USFDA:
-
United States Food and Drug Administration
- V ss :
-
Volume of distribution at steady state
References
Antonides LH, Cannaert A, Norman C et al (2019) Enantiospecific synthesis, chiral separation, and biological activity of four indazole-3-carboxamide-type synthetic cannabinoid receptor agonists and their detection in seized drug samples. Front Chem 7:1–20
Cannaert A, Storme J, Franz F et al (2016) Detection and activity profiling of synthetic cannabinoids and metabolites with a newly developed bio-assay. Anal Chem 88:11476–11485
Cannaert A, Franz F, Auwärter V, Stove CP (2017) Activity-based detection of consumption of synthetic cannabinoids in authentic urine samples using a stable cannabinoid reporter system. Anal Chem 89:9527–9536
Castaneto MS, Wohlfarth A, Desrosiers NA et al (2015) Synthetic cannabinoids pharmacokinetics and detection methods in biological matrices. Drug Metab Rev 47:124–174
Chen Y, Liu L, Nguyen K, Fretland AJ (2011) Utility of intersystem extrapolation factors in early reaction phenotyping and the quantitative extrapolation of human liver microsomal intrinsic clearance using recombinant cytochromes P450. Drug Metab Dispos 39:373–382
Diao X, Huestis MA (2017) Approaches, challenges, and advances in metabolism of new synthetic cannabinoids and identification of optimal urinary marker metabolites. Clin Pharmacol Ther 101:239–253
Diao X, Huestis MA (2019) New synthetic cannabinoids metabolism and strategies to best identify optimal marker metabolites. Front Chem 7:109
European Monitoring Centre for Drugs and Drug Addiction (2017) 5F-MDMB-PINACA—Report on the risk assessment of methyl 2-{[1-(5-fluoropentyl)-1H-indazole-3-carbonyl]amino}-3,3-dimethylbutanoate in the framework of the Council Decision on new psychoactive substances
Gamage TF, Farquhar CE, McKinnie RJ et al (2019) Synthetic cannabinoid hydroxypentyl metabolites retain efficacy at human cannabinoid receptors. J Pharmacol Exp Ther 368:414–422
Gatch MB, Forster MJ (2019) Cannabinoid-like effects of five novel carboxamide synthetic cannabinoids. Neurotoxicology 70:72–79
Gurney SMR, Scott KS, Kacinko SL et al (2014) Pharmacology, toxicology, and adverse effects of synthetic cannabinoid drugs. Forensic Sci Rev 26:54–76
Harris CR, Brown A (2013) Synthetic cannabinoid intoxication: a case series and review. J Emerg Med 44:360–366
Haschimi B, Mogler L, Halter S et al (2019) Detection of the recently emerged synthetic cannabinoid 4F-MDMB-BINACA in ‘legal high’ products and human urine specimens. Drug Test Anal 11:1377–1386
Holm NB, Nielsen LM, Linnet K (2015a) CYP3A4 mediates oxidative metabolism of the synthetic cannabinoid AKB-48. AAPS J 17:1237–1245
Holm NB, Pedersen AJ, Dalsgaard PW, Linnet K (2015b) Metabolites of 5F-AKB-48, a synthetic cannabinoid receptor agonist, identified in human urine and liver microsomal preparations using liquid chromatography high-resolution mass spectrometry. Drug Test Anal 7:199–206
Huang W, Isoherranen N (2018) Development of a dynamic physiologically based mechanistic kidney model to predict renal clearance. CPT Pharmacometrics Syst Pharmacol 7:593–602
Huestis MA, Gorelick DA, Heishman SJ et al (2001) Blockade of effects of smoked marijuana by the CB1-selective cannabinoid receptor antagonist SR141716. Arch Gen Psychiatry 58:322–328
Hussaarts KGAM, Veerman GDM, Jansman FGA et al (2019) Clinically relevant drug interactions with multikinase inhibitors: a review. Ther Adv Med Oncol 11:1–34
Izumi S, Nozaki Y, Kusuhara H et al (2018) Relative activity factor (raf)-based scaling of uptake clearance mediated by organic anion transporting polypeptide (OATP) 1B1 and OATP1B3 in human hepatocytes. Mol Pharm 15:2277–2288
Kong TY, Kim JH, Kim DK, Lee HS (2018) Synthetic cannabinoids are substrates and inhibitors of multiple drug-metabolizing enzymes. Arch Pharm Res 41:691–710
Krotulski AJ, Mohr AL, Logan BK (2018) Trend Report: Q4 2018 Synthetic Cannabinoids in the United States
Krotulski AJ, Mohr AL, Kacinko SL et al (2019) 4F-MDMB-BINACA: a new synthetic cannabinoid widely implicated in forensic casework. J Forensic Sci 64:1451–1461
Mathialagan S, Piotrowski MA, Tess DA et al (2017) Quantitative prediction of human renal clearance and drug-drug interactions of organic anion transporter substrates using in vitro transport data: a relative activity factor approach. Drug Metab Dispos 45:409–417
Meyer MR, Schütz A, Maurer HH (2015) Contribution of human esterases to the metabolism of selected drugs of abuse. Toxicol Lett 232:159–166
Nguyen HQ, Lin J, Kimoto E et al (2017) Prediction of losartan-active carboxylic acid metabolite exposure following losartan administration using static and physiologically based pharmacokinetic models. J Pharm Sci 106:2758–2770
Noble C, Cannaert A, Linnet K, Stove CP (2019) Application of an activity-based receptor bioassay to investigate the in vitro activity of selected indole- and indazole-3-carboxamide-based synthetic cannabinoids at CB1 and CB2 receptors. Drug Test Anal 11:501–511
Obach RS (1999) Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos 27:1350–1359
Poulin P, Theil FP (2002) Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J Pharm Sci 91:129–156
Prakash C, Kamel A, Cui D et al (2000) Identification of the major human liver cytochrome P450 isoform(s) responsible for the formation of the primary metabolites of ziprasidone and prediction of possible drug interactions. Br J Clin Pharmacol 49:35–42
Presley BC, Jansen-Varnum SA, Logan BK (2013) Analysis of synthetic cannabinoids in botanical material: a review of analytical methods and findings. Forensic Sci Rev 25:27–46
Reidy L, Seither J, Boland D (2018) Identification of synthetic cannabinoid 5-fluoro-ADB in human performance and postmortem samples: a case series. J Forensic Toxicol Pharmacol 7:1–9
Riley RJ, McGinnity DF, Austin RP (2005) A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug Metab Dispos 33:1304–1311
Rodgers T, Rowland M (2006) Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci 95:1238–1257. https://doi.org/10.1002/jps.20502
Seither JZ, Reidy LJ, Boland DM (2020) Identification and quantification of 5-fluoro ADB and the 5-fluoro ADB ester hydrolysis metabolite in postmortem blood samples by LC-MS/MS. J Anal Toxicol 44:133–139
Tai S, Fantegrossi WE (2016) Pharmacological and toxicological effects of synthetic cannabinoids and their metabolites. In: Current topics in behavioral neurosciences. Springer Verlag, pp 249–262
Thomsen R, Nielsen LM, Holm NB et al (2015) Synthetic cannabimimetic agents metabolized by carboxylesterases. Drug Test Anal 7:565–576
Tran TT, Mittal A, Gales T et al (2004) Exact kinetic analysis of passive transport across a polarized confluent MDCK cell monolayer modeled as a single barrier. J Pharm Sci 93:2108–2123
United Nations Office on Drugs and Crime (2019) Current NPS Threats, vol 1
U.S. Food and Drug Administration (2017) Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers. Accessed 18 Aug 2019
US FDA (2017) Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers
Wang X, Rida N, Shi J et al (2017) A comprehensive functional assessment of carboxylesterase 1 nonsynonymous polymorphisms. Drug Metab Dispos 45:1149–1155
Wang D, Zou L, Jin Q et al (2018) Human carboxylesterases: a comprehensive review. Acta Pharm Sin B 8:699–712
Wouters E, Mogler L, Cannaert A et al (2019) Functional evaluation of carboxy metabolites of synthetic cannabinoid receptor agonists featuring scaffolds based on L-valine or L-tert-leucine. Drug Test Anal 11:1183–1191
Yanjiao X, Chengliang Z, Xiping L et al (2013) Evaluation of the inhibitory effects of antihypertensive drugs on human carboxylesterase in vitro. Drug Metab Pharmacokinet 28:468–474
Yeter O, Öztürk YE (2019) Detection and quantification of 5F-ADB and its methyl ester hydrolysis metabolite in fatal intoxication cases by liquid chromatography–high resolution mass spectrometry. Forensic Sci Int 302:109866
Yeter O, Ozturk YE (2019) Metabolic profiling of synthetic cannabinoid 5F-ADB by human liver microsome incubations and urine samples using high-resolution mass spectrometry. Drug Test Anal 11:847–858
Zhao A, Tan M, Maung A et al (2015) Rhabdomyolysis and acute kidney injury requiring dialysis as a result of concomitant use of atypical neuroleptics and synthetic cannabinoids. Case Reports Nephrol 2015:1–4
Acknowledgements
The authors thank Jye Ing Soah, Yen Li Tan and Ching Yee Fong for their assistance in the LC-MS/MS instrumentation. The authors also thank Chi Pang Lui for his feedback. The project is supported by the Singapore Health Sciences Authority (HSA) and the National University of Singapore (NUS) Department of Pharmacy’s Final Year Project (FYP) funding provided to E.C.Y.C. A. C. acknowledges funding as a postdoctoral research fellow from the Research Foundation-Flanders (FWO; 12Y9520N). C. S. acknowledges funding from the Ghent University—Special Research Fund (Grants no. 01N00814 and no. 01J15517).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lie, W., Cheong, E.J.Y., Goh, E.M.L. et al. Diagnosing intake and rationalizing toxicities associated with 5F-MDMB-PINACA and 4F-MDMB-BINACA abuse. Arch Toxicol 95, 489–508 (2021). https://doi.org/10.1007/s00204-020-02948-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-020-02948-3