
Abstract
1,4-Dioxane is a widely used synthetic industrial chemical and its contamination of drinking water and food is a potential health concern. It induces liver tumors when administered in the drinking water to rats and mice. However, the mode of action (MOA) of the hepatocarcinogenicity of 1,4-dioxane remains unclear. Importantly, it is unknown if 1,4-dioxane is genotoxic, a key consideration for risk assessment. To determine the in vivo mutagenicity of 1,4-dioxane, gpt delta transgenic F344 rats were administered 1,4-dioxane at various doses in the drinking water for 16 weeks. The overall mutation frequency (MF) and A:T- to -G:C transitions and A:T- to -T:A transversions in the gpt transgene were significantly increased by administration of 5000 ppm 1,4-dioxane. A:T- to -T:A transversions were also significantly increased by administration of 1000 ppm 1,4-dioxane. Furthermore, the DNA repair enzyme MGMT was significantly induced at 5000 ppm 1,4-dioxane, implying that extensive genetic damage exceeded the repair capacity of the cells in the liver and consequently led to liver carcinogenesis. No evidence supporting other MOAs, including induction of oxidative stress, cytotoxicity, or nuclear receptor activation, that could contribute to the carcinogenic effects of 1,4-dioxane were found. These findings demonstrate that 1,4-dioxane is a genotoxic hepatocarcinogen and induces hepatocarcinogenesis through a mutagenic MOA in rats. Because our data indicate that 1,4-dioxane is a genotoxic carcinogen, we estimated the point of departure of the mutagenicity and carcinogenicity of 1,4-dioxane using the no-observed effect-level approach and the Benchmark dose approach to characterize its dose–response relationship at low doses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
1,4-Dioxane is a synthetic industrial chemical and is used as a stabilizer for chlorinated solvents such as 1,1,1-trichloroethane (TCA), and it is also widely used as a solvent in a wide variety of organic products such as paint strippers, dyes, greases, varnishes, waxes, detergents, cosmetics, and deodorants (EPA 2013, 2017; IARC 1999; Wilbur et al. 2012). 1,4-Dioxane is released into the environment during its production, the processing of other chemicals, and its use during the manufacture of several consumer products. It migrates rapidly into the ground water and is resistant to hydrolysis by water and to microbial degradation. The largest sources of 1,4-dioxane in the drinking water are wastewater discharge, unintended spills and leaks, and historical disposal practices of its host solvent, e.g., TCA. Conventional water treatment practices have proven to be ineffective at removing 1,4-dioxane from water. 1,4-Dioxane is also found as an impurity in some consumer products such as deodorants, shampoos, and cosmetics (EPA 2013, 2017; Wilbur et al. 2012). Traces of 1,4-dioxane may be present in some manufactured food additives, food containing residues from packaging material, or on food crops treated with pesticides that contain 1,4-dioxane as a solvent (EPA 2013, 2017; Wilbur et al. 2012). Consequently, its contamination of drinking water and food is of potential health concern.
An occupational cohort study of workers exposed to 1,4-dioxane did not reveal any elevation in the incidence of death by cancer (Buffler et al. 1978), but a significant increase in liver cancer was found in a comparative mortality study (Hansen et al. 1993). These studies, however, are considered inadequate for human carcinogenicity assessment because of the small sample size in the former study and lack of exposure data in the later study. Meanwhile, carcinogenicity bioassays have shown that 1,4-dioxane is carcinogenic to multiple sites in rats and mice. Exposure to 1,4-dioxane in drinking water induced hepatocellular and nasal tumors in rats and mice, and also induced mammary tumors and peritoneal mesotheliomas in rats but not in mice (Argus et al. 1965; Kano et al. 2009; Kociba et al. 1974; NTP 1978; Stickney et al. 2003). Inhalation exposure to 1,4-dioxane also induced hepatocellular and nasal tumors and peritoneal mesotheliomas in rats (Kasai et al. 2009). Based on sufficient evidence of carcinogenicity in experimental animals, the International Agency for Research on Cancer has classified 1,4-dioxane as Group 2B compound (possibly carcinogenic to humans) (IARC 1999). 1,4-Dioxane has been found in surface and groundwater in the United States, Japan, and Europe (EC 2002; EPA 2017; Wilbur et al. 2012). Therefore, although the concentrations of 1,4-dioxane in drinking water are orders of magnitude lower than the doses used in animal studies, it constitutes a potential hazard, and there is concern regarding the carcinogenic effects of low doses of this chemical.
The human relevance assessment framework for carcinogen risk is based on the mode of action (MOA) of the carcinogen (Bolt 2016; Boobis et al. 2006; Cohen et al. 2004; EPA 2005; Kobert and Willams 2016). Genotoxicity is a key event for MOA-based extrapolation of risk and clarifying how risk should be estimated for low-dose exposure. Linear non-threshold extrapolation is generally accepted for carcinogens that act by a genotoxic MOA. We assert, however, that genotoxic carcinogens have a practical threshold below which carcinogenicity is not distinguishable from background rates (Fukushima et al. 2002, 2010, 2016a), and the non-threshold approach has been challenged by quantification approaches using point-of-departure (PoD) metrics in defining acceptable exposure limits and assessing human risk (EPA 2005; Fukushima et al. 2016b; Gollapudi et al. 2013; MacGregor et al. 2015a, b; Slob 2014a; Thomas and Johnson 2016). For non-genotoxic carcinogens, in contrast, a threshold dose below which no carcinogenic effect is produced is presumed to exist.
Currently, available evidence is inadequate to establish an MOA by which 1,4-dioxane induces liver tumors in rats and mice (EPA 2013). Of particular importance, the genotoxicity of 1,4-dioxane is uncertain. 1,4-Dioxane was negative in the large majority of in vitro genotoxicity studies including mutagenicity assays with prokaryotic organisms and cytogenetic assays with non-mammalian eukaryotic organisms and mammalian cells, but it caused DNA strand breaks in rat hepatocytes and was positive in a cell transformation assay with BALB/3T3 cells at cytotoxic concentrations (EPA 2013; IARC 1999). Conflicting results were obtained in the in vivo micronucleus assays used to assess the induction of chromosomal aberrations in mice. Of five studies on the induction of bone-marrow micronuclei or peripheral blood micronuclei, three were negative (McFee et al. 1994; Morita and Hayashi 1998; Tinwell and Ashby 1994) and two were positive (Mirkova 1994; Roy et al. 2005). Notably, 1,4-dioxane was positive in a liver micronucleus assay in mice (Morita and Hayashi 1998). Moreover, 1,4-dioxane-induced hepatic DNA damage in rats (Kitchin and Brown 1990). Overall, the available literature indicates that 1,4-dioxane is non-genotoxic or weakly genotoxic (EPA 2013). However, there are no data available on the in vivo mutagenicity of 1,4-dioxane, especially for specific target organs, in rats.
In the present study, we determined the in vivo mutagenicity of 1,4-dioxane in the liver of gpt delta transgenic F344 rats. This rat model can provide crucial information for understanding the in vivo mechanism of organ-specific mutagenesis induced by various carcinogens (Hayashi et al. 2003; Nohmi 2016; Nohmi et al. 2017). In addition, we investigated other carcinogenic MOAs that could contribute to the carcinogenicity of 1,4-dioxane by evaluating oxidative DNA damage, expression levels of genes involved in metabolic activation of 1,4-dioxane, cell proliferation, and DNA damage repair in the liver of gpt delta transgenic F344 rats. To better understand the low-dose hepatocarcinogenicity of 1,4-dioxane in gpt delta transgenic F344 and wild-type F344 rats, we also examined induction of glutathione S-transferase placental form (GST-P) positive foci, which is a well-established preneoplastic lesion of the liver in rats and has been accepted as a useful end-point marker in assessment of carcinogenic effects of environmentally relevant concentrations of carcinogens as induction of GST-P positive foci can extend the range of observable effect levels of liver carcinogens (Fukushima et al. 2010, 2016a). Furthermore, to estimate exposure-related risk, we determined the PoDs for the mutagenicity and hepatocarcinogenicity endpoints by two quantitative approaches: the no-observed-effect level (NOEL) and the benchmark dose (BMD) approaches.
Materials and methods
Chemical
1,4-dioxane was purchased from Wako Pure Chemical Industries, Ltd., Osaka, Japan (purity > 99.9%). Bromodeoxyuridine (BrdU) was obtained from Sigma-Aldrich (St. Louis, MO, USA).
Animals
Five-week-old male gpt delta transgenic F344 rats were supplied by Japan SLC Inc. (Shizuoka, Japan). Five-week-old male wild-type F344 rats were supplied by Charles River Japan, Inc. (Hino, Shiga, Japan). Animal studies were approved by the Institutional Animal Care and Use Committee of Osaka City University Graduate School of Medicine and conducted in accordance with the Guidelines for Proper Conduct of Animal Experiments (Science Council of Japan, 2006). Animals were housed in polycarbonate cages in experimental animal rooms with a targeted temperature of 22 ± 3 °C, relative humidity of 55 ± 5%, and a 12-h light/dark cycle. Diet (pelleted CE-2, Clea Japan, Inc., Tokyo, Japan) and drinking water were available ad libitum throughout the study. Fresh distilled water containing 1,4-dioxane was changed three times weekly. Body weights, food consumption, and water intake were measured weekly.
Experiment 1
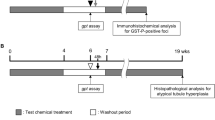
A total of 30 male gpt delta transgenic F344 rats, 6 weeks of age at the commencement of the experiment, were divided into 4 groups and administered 1,4-dioxane at doses of 0 (8 rats), 200 (7 rats), 1000 (7 rats), or 5000 ppm (8 rats) in their drinking water for 16 weeks. At the end of week 16, rats were fasted overnight and then euthanized by exsanguination via transection of the abdominal aorta under deep anesthesia (50 mg/kg sodium pentobarbital intraperitoneally). At necropsy, livers were excised and weighed. A total of 3 sections of liver tissue (one section each from the left lateral lobe, right middle lobe, and caudate lobe) were fixed in phosphate buffered formalin, embedded in paraffin, and processed for hematoxylin/eosin and immunohistochemical staining. The remaining liver tissues were snap frozen with liquid nitrogen and stored at − 80 °C for mutation assays, oxidative DNA damage, and gene expression analyses.
Experiment 2
A total of 20 male gpt delta transgenic F344 rats, 6 weeks of age at the commencement of the experiment, were divided into 4 groups (5 rats/group) and administered 1,4-dioxane at doses of 0, 0.2, 2, or 20 ppm in their drinking water for 16 weeks. At the end of week 16, rats were euthanized and liver tissues were processed as described in Experiment 1.
Experiment 3
A total of 180 male wild-type F344 rats, 3 weeks of age at the commencement of the experiment, were divided into 6 groups (30 rats/group) and administered 1,4-dioxane at doses of 0, 2, 20, 200, 2000, or 5000 ppm in their drinking water for 16 weeks. At the end of week 16, rats were euthanized and liver tissues were processed, as described in Experiment 1. One hour prior to sacrifice, ten rats from each group were injected i.p. with 100 mg BrdU/kg body weight for evaluation of cell proliferative activity in their livers.
In vivo mutation assays
The gpt and Spi− assays were conducted according to the published protocols by Nohmi et al. (2000). Genomic DNA was extracted from frozen liver tissue using the RecoverEase DNA Isolation kit according to the manufacturer’s protocol (Agilent Technologies, Santa Clara, CA). Lambda EG10 DNA in the genomic DNA was rescued as the lambda phage using the Transpack packaging extract (Agilent Technologies).
In the gpt assay, packaged phages were transfected into Escherichia coli YG6020 expressing Cre recombinase and cultured on selection plates containing chloramphenicol and 6-thioguanine (6-TG) for mutant selection. The gpt mutants [6-TG-resistant (6-TGR) phenotype] were isolated and re-streaked on selection plates to confirm the 6-TGR phenotype. All the confirmed gpt mutants were recovered and sequenced; identical mutations from the same rat were counted as one mutant. The mutation frequency (MF) of the gpt gene in the liver was calculated by dividing the number of confirmed 6-TGR colonies by the number of rescued plasmids [chloramphenicol-resistant (CmR) colonies]. DNA sequencing of the gpt gene was performed with the BigDye Terminater Cycle Sequencing Ready Reaction (Applied Biosystems, Inc., Carlsbad, CA, USA) on an Applied Biosystems PRISM 310 Genetic Analyzer. A:T-to-T:A transversions in the 299th bp of the gpt gene is a germline mutation in gpt delta F344 rat regardless of the experimental treatment (Masumura and Nohmi 2009; Masumura et al. 2015); therefore, A:T-to-T:A transversions in the 299th bp of the gpt gene were excluded from the MF and mutation spectra.
In the Spi−assay, packaged phages were incubated with E. coli XL-1 Blue MRA for survival titration and E. coli XL-1 Blue MRA P2 for mutant selection. Infected cells were mixed with molten lambda-trypticase agar and poured onto lambda-trypticase agar plates. The next day, plaques (Spi− candidates) were punched out with sterilized glass pipettes and the agar plugs were suspended in SM buffer. The Spi− phenotype was confirmed by spotting the suspensions on three types of plates in which XL-1 Blue MRA, XL-1 Blue MRA P2, or WL95 P2 strains were spread with lambda-trypticase soft agar. True Spi− mutants, which made clear plaques on all of the plates, were counted.
Immunohistochemical analysis
Paraffin sections of the liver of all animals in Experiments 1, 2, and 3 were examined for GST-P positive foci, and paraffin sections of the liver of ten rats from each group in Experiment 3 were examined for incorporation of BrdU by immunohistochemical staining using the avidin–biotin–peroxidase complex (ABC) method. For BrdU staining, antigen retrieval was performed by microwaving at 98 °C for 20 min in 0.01 M citrate buffer (pH 6.0). Endogenous peroxidase activity was blocked with 0.3% H2O2 in distilled water for 5 min. After blocking non-specific binding with goat serum at 37 °C for 30 min, sections were incubated with rabbit polyclonal anti-GST-P antibody (#311, Medical and Biological Laboratories Co., Ltd., Nagoya, Japan) diluted 1:1000 or mouse monoclonal anti-BrdU antibody (M0744, Dako, Tokyo, Japan) diluted 1:1000 overnight at 4 °C. Immunoreactivity was detected using a Rabbit IgG VECSTAIN ABC Kit (PK-4001, Vector Laboratories, Burlingame, CA, USA) for GST-P and a Mouse IgG VECSTAIN ABC Kit (PK-4002, Vector Laboratories, Burlingame, CA, USA) for BrdU and 3,3′-diaminobenzidine hydrochloride (Sigma-Aldrich Co., St Louis, MO, USA).
Evaluation of GST-P positive foci in the liver
GST-P positive hepatocellular foci composed of two or more cells were counted under a light microscope (Fukushima et al. 2002, 2010; Hoshi et al. 2004; Kang et al. 2006; Wei et al. 2006, 2011). Total areas of livers were measured using a color image processor IPAP (Sumica Technos, Osaka, Japan) and the number of GST-P positive foci per square centimeters of liver tissue was calculated.
Detection of oxidative damage in the liver DNA
Formation of 8-hydroxy-2′-deoxyguanosine (8-OHdG) in liver DNA was determined by high-performance liquid chromatography with electrochemical detection as described previously (Fukushima et al. 2002).
mRNA expression analysis in rat livers
The mRNA expression levels of the genes of interest were evaluated in five rats from each group in Experiment 1 by TaqMan real-time quantitative PCR. Total RNAs were isolated from the frozen liver tissue using TRIzol® Reagent (Life Technologies, Gaithersburg, MD, USA) according to the manufacturer’s instructions. PCR reagents and sequence-specific primers and probes for each gene (Taqman Gene Expression Assay) were purchased from Applied Biosystems, Inc., CA, USA. mRNA expression assays were performed using a 7500 Fast Real-Time PCR System (Applied Biosystems, Inc., CA, USA). ß-actin was employed as an internal control. Serially diluted standard cDNAs were included in each Taqman PCR reaction to create standard curves. The amounts of gene products in the test samples were estimated relative to the respective standard curves. Values for target genes were normalized to those for ß-actin.
Derivation of PoD
The point of departure (PoD) marks the beginning of low-dose extrapolation. In the NOEL approach, the PoD is the highest dose at which no statistically significant differences in response are observed compared with the background response. For the BMD approach, continuous models were used to fit dose–response data for MF and GST-P positive foci. Dose–response modeling was performed using two BMD software packages, the Benchmark Dose Software (BMDS, version 2.7) developed by the US EPA, and the PROAST software developed by the National Institute for Public Health and the Environment of Netherland (version 64.14). In the analyses using BMDS, a dose–response model was selected using methods consistent with US EPA guidelines (EPA 2012), and the benchmark response (BMR) was defined as an increase in the mean response equivalent to one standard deviation (BMD1SD) in control animals and its 95% lower confidence limit (BMDL1SD) was used as the PoD. In the analyses using PROAST, a dose–response model was selected using methods consistent with EFSA guidelines (Hardy et al. 2017), and the BMR was defined as the dose corresponding to a 10% extra risk (BMD10) and its 95% lower confidence limit (BMDL10) was used as the PoD.
Quantal models were used to fit dose–response data for liver tumor incidence in the 2-year carcinogenicity study in wild-type male F344 rats reported by Kano et al. (2009). For this study, the BMDL10 was used as the PoD in the analyses using BMDS and PROAST.
Statistical analysis
All mean values are reported as mean ± SD. Statistical analyses were performed using Prism 6 for Mac OS X (GraphPad Software, Inc., San Diego, CA, USA). Homogeneity of variance was tested by the Bartlett test. Differences in mean values between control (0 ppm) and treatment groups were analyzed by Dunnett’s Multiple Comparison Test when variance was homogeneous and Dunn’s test when variance was heterogeneous. p values less than 0.05 were considered significant.
Results
Mutagenicity and GST-P positive foci induction at high doses of 1,4-dioxane in gpt delta transgenic F344 rats (Experiment 1)
General findings and liver histopathology in gpt delta transgenic F344 rats
Data on final body weights, liver weights, food consumption and water intake, and 1,4-dioxane intake are summarized in Table 1. Final body weights were significantly decreased in the 5000 ppm group compared to the control group, along with slight suppression of food consumption. The intake of 1,4-dioxane was approximately proportional to the doses administered in the drinking water, although water intake showed a tendency to decrease in the 5000 ppm group. A slight but statistically significant increase in relative but not absolute liver weight was observed in the 5000 ppm group compared to the control group, possibly related to the lower body weights in this group. 1,4-dioxane at doses of 200 and 1000 ppm had no apparent effects on the body or liver weights, water intake, or food consumption.
No 1,4-dioxane treatment-related histopathological changes including hypertrophy, swelling, necrosis, apoptosis, or fatty changes were observed in the liver of any of the 1,4-dioxane treatment groups.
The gpt and Spi− MFs in the livers of F344 gpt delta transgenic rats
Data on MFs and mutation spectrum of the gpt transgene in the liver are summarized in Tables 2 and 3, respectively. The gpt MF was significantly increased in the 5000 ppm group compared to the control group (Table 2). A:T- to -G:C transition and A:T- to -T:A transversion frequencies were also significantly increased in this group (Table 3). In the 1000 ppm group, A:T- to -T:A transversion frequency was significantly increased, although the overall gpt MF did not significantly differ from the value of the control group. There were no significant differences in MF or mutation spectrum in the gpt transgene between the 200 ppm and control groups.
There were no significant differences in the frequency of Spi− MF in the liver between any of the groups administered 1,4-dioxane and the control group (Supplemental Table 1).
GST-P-positive foci induction in the livers of gpt delta transgenic F344 rats
As summarized in Table 2, the number of GST-P-positive foci per unit area of the liver was not significantly increased in the 200 or 1000 ppm groups, but a significant increase was seen in the 5000 ppm group.
Gene expression changes in the livers of gpt delta transgenic F344 rats
The relative mRNA expression of genes involved in cell proliferation [proliferating cell nuclear antigen (PCNA)], cell cycle regulation (p53 and p21Cip/WAF1), DNA damage repair [O-6-methylguanine–DNA methyltransferase (MGMT)], growth arrest and DNA damage-inducible protein 45 (GADD45), apurinic/apyrimidinic endodeoxyribonuclease 1 (APEX1), excision Repair Cross-Complementation Group 5 (ERCC5), MSH2, and MSH3 is shown in Fig. 1. A significant increase in PCNA was observed in the 5000 ppm group, but not in the groups administered lower doses of 1,4-dioxane (Fig. 1A). Furthermore, MGMT was significantly induced in the 5000 ppm group, but not in the groups administered lower doses of 1,4-dioxane (Fig. 1B). There was no significant change in expression levels of the other DNA damage repair genes, p53, or p21Cip/WAF1 between any of the groups administered 1,4-dioxane and the control group (Fig. 1C–I).

Relative mRNA expression levels of genes involved in cell proliferation, DNA repair, and cell cycle regulation in the livers of gpt delta transgenic rats at week 16 (Experiment 1). Significantly different from the non-treatment control group (0 ppm) at *p < 0.05; **p < 0.01
The relative mRNA expression of 12 CYP enzymes in families 1–4 (CYP1A1, CYP1B1, CYP2A1, CYP2A2, CYP2B1, CYP2B2, CYP2C11, CYP2E1, CYP3A1, CYP3A2, CYP4A1, and CYP4A2) is shown in Fig. 2. None of these CYP enzymes was affected by administration of 1,4-dioxane.

Relative mRNA expression levels of CYPs of families 1–4 in the livers of gpt delta transgenic rats at week 16 (Experiment 1)
8-OHdG formation in liver DNA
There were no significant differences in 8-OHdG levels in the liver DNA between any of the groups administered 1,4-dioxane and the control group (Supplemental Fig. 1).
Mutagenicity and GST-P positive foci induction at low doses of 1,4-dioxane in gpt delta transgenic F344 rats (Experiment 2)
General findings and liver histopathology in gpt delta transgenic F344 rats
As summarized in Table 1, administration of 1,4-dioxane at doses of 0.2 to 20 ppm had no apparent effects on the body or liver weights, water intake, or food consumption. The intake of 1,4-dioxane was approximately proportional to the doses administered in the drinking water. There were no treatment-related histopathological changes in the livers in any of the 1,4-dioxane treatment groups.
gpt mutations in the livers of gpt delta transgenic F344 rats
Data on MFs and mutation spectra of the gpt transgene the livers are summarized in Table 4 and Supplemental Table 2, respectively. There were no significant differences in gpt MF or mutation spectra between groups administered 0.2–20 ppm 1,4-dioxane, and the control group. Furthermore, there were no significant differences in Spi− MF between any of the groups administered 1,4-dioxane and the control group (Supplemental Table 3).
GST-P-positive foci induction in the liver of gpt delta transgenic F344 rats
As summarized in Table 4, 1,4-dioxane at doses of 0.2–20 ppm had no significant effect on the induction of GST-P-positive foci.
Induction of GST-P-positive foci by 1,4-dioxane in wild-type F344 rats (Experiment 3)
General findings and liver histopathology in wild-type F344 rats
Data on final body weights, liver weights, water intake and food consumption, and 1,4-dioxane intake are summarized in Table 1. Final body weights were significantly decreased in the wild-type F344 rats administered 5000 ppm 1,4-dioxane compared to the control group. 1,4-dioxane at doses of 2–2000 ppm had no effect on the body weight. Water intake and food consumption were similar between the 1,4-dioxane treatment groups and the control group. The intake of 1,4-dioxane was approximately proportional to the doses administered in the drinking water. There was no significant difference in absolute or relative liver weights between the 1,4-dioxane treatment groups and the control group. No treatment-related histopathological changes were observed in the livers in any of the treatment groups.
GST-P-positive foci induction and cell proliferation activity in the liver of wild-type F344 rats
The numbers of GST-P-positive foci per unit area of the liver were not significantly increased in the 2–200 ppm groups, but significant increases were detected in the 2000 and 5000 ppm groups (Table 5).
A significant increase in the BrdU labeling index was observed in the 5000 ppm group (Table 5). There was no significant difference in BrdU labeling index between groups administered 2–2000 ppm 1,4-dioxane, and the control group.
Derivation of PoD
Since 1,4-dioxane is a genotoxic carcinogen, as demonstrated above, to determine the PoDs, we analyzed the data on mutagenicity and GST-P positive foci induction from the present study and the data on liver tumors in a previous drinking water carcinogenicity study reported by Kano et al. (2009) by both the NOEL approach and the BMD approach using the BMDS and PROAST software packages. The derived PoDs are summarized in Table 6. The PoD values determined by the NOEL approach were 200 ppm for both mutations and GST-P positive foci (the lower value in Experiments 1 and 3) and 1000 ppm for liver tumors. In contrast, the PoD values derived by the BMD approach using PROAST (BMDL10) were 0.98 ppm for mutations, 1.66 ppm (lower values in Experiments 1 and 3) for GST-P positive foci, and 513 ppm for liver tumors. The PoD values derived by the BMD approach using BMDS were 576 ppm (BMDL1SD) for mutations, 1018 ppm (BMDL1SD) (lower values in Experiments 1 and 3) for GST-P positive foci, and 513 ppm (BMDL10) for liver tumors.
Discussion
There are no current data with which to establish an MOA by which 1,4-dioxane induces liver tumors in experiment animals (EPA 2013). In the present study, we clearly demonstrated for the first time that 1,4-dioxane is mutagenic to the liver, which is its target organ in rats. As 1,4-dioxane was negative in in vitro mutation assays, the results of the present study indicate that the in vitro mutagenicity of 1,4-dioxane does not reflect its in vivo mutagenicity in rodents. The discrepancy between in vivo and in vitro mutagenicity tests may result from organ-specific pathways of xenobiotic metabolism and DNA repair in vivo (Kirkland et al. 2005; Morita et al. 2016; Walmsley and Billinton 2011). Notably, administration of both 1000 and 5000 ppm 1,4-dioxane in the drinking water resulted in significantly increased DNA mutation frequency, while the DNA repair enzyme MGMT and cell proliferation were induced only by the mutagenic and carcinogenic dose of 5000 ppm 1,4-dioxane. This implies that the genetic damage caused by the 5000 ppm dose exceeded the repair capacity of liver cells and consequently led to the development of preneoplastic lesions, GST-P positive foci. These findings demonstrate that 1,4-dioxane is a genotoxic hepatocarcinogen and induces hepatocarcinogenesis through a mutagenic MOA in rats.
DNA-adduct formation is generally believed to play a crucial role in the mutagenicity and consequent carcinogenicity of genotoxic carcinogens. Although the precise mechanism underlying the mutagenicity of 1,4-dioxane in rat livers obtained in the present study remains to be elucidated, the altered mutation spectra of 1,4-dioxane exposed rats compared to control rats, shown in Table 3, implies the formation of 1,4-dioxane–DNA adducts. Exposure to 1000 ppm 1,4-dioxane resulted in a significant increase in A:T-to-T:A transversions, and this transversion is considered to be due to formation of adenosine adducts (Levine et al. 2000; Poon et al. 2014), suggesting that formation of adenosine adducts is a primary consequence of exposure to 1,4-dioxane. Exposure to 5000 ppm 1,4-dioxane resulted in significant increases in A:T-to-T:A transversions, A:T-to-G:C transitions, and expression of MGMT. MGMT is a DNA repair enzyme: the primary substrate of MGMT is the promutagenic O6-methylguanine (m6G) adduct that results in G:C-to-A:T mutations during DNA replication; MGMT also repairs O4-methylthymine (m4T) adducts that result in A:T–to-G:C mutations (Chatterjee and Walker 2017; Jin and Robertson 2013; Kaina et al. 2007; Sharma et al. 2009). Whether generation of A:T-to-G:C transitions without generation of G:C-to-A:T transitions is due to preferential thymidine adduct formation by exposure to 1,4-dioxane or preferential repair of guanosine adducts—m4T is a more persistent adduct than m6G (Encell and Loeb 2000; O’Toole et al. 1993; Singer 1986)—by MGMT is not known at this time. A collaborative study with researchers in the National Cancer Center Research Institute (Tokyo, Japan) is underway to determine 1,4-dioxane–DNA-adduct formation in the livers of rats administered various doses of 1,4-dioxane by DNA-adductomics analysis using LC–MS/MS, and principal component analysis has revealed that the DNA-adduct profile of rats administered 5000 ppm 1,4-dioxane is distinctly different from that of control rats (unpublished data). Intriguingly, these researchers have recently shown that DNA-adductomics analysis is useful in determining the carcinogenic potential of a test compound and risk assessment of carcinogens (Akiba et al. 2017; Ishino et al. 2015).
Several MOAs have been proposed for hepatocarcinogens in rodents including induction of oxidative stress, cytotoxicity followed by regenerative cell proliferation, CYP enzyme induction, and nuclear receptor activation (Cohen 2010). In the present study, neither significant changes in 8-OHdG levels nor necrosis were observed in rats treated with mutagenic or/and carcinogenic doses of 1,4-dioxane. This suggests that oxidative DNA damage and necrosis followed by regeneration do not appear to play a role in 1,4-dioxane-induced mutagenesis and carcinogenesis.
CYP enzymes are responsible for metabolizing an enormous number of compounds into intermediates that can be further degraded or excreted and are the primary enzymes responsible for the degradation and elimination of xenobiotics. However, a small percentage of CYP-generated metabolites are carcinogenic, and the MOA of a number of hepatocarcinogens is metabolic activation by CYP enzymes into intermediates that are mutagenic and carcinogenic (Nebert and Dalton 2006; Rodriguez-Antona and Ingelman-Sundberg 2006). Consequently, it is possible that aberrant activation of CYP enzymes can lead to generation of carcinogenic compounds. A well-known pathway of CYP-mediated metabolism of their substrates is induction of expression by nuclear receptors. The finding that 1,4-dioxane had no effect on the mRNA expression of 12 CYP enzymes in families CYP1-4 that are induced by the activation of nuclear transcription factors such as aryl hydrocarbon receptor, pregnane X receptor, constitutive androstane receptor, and peroxisome proliferator-activated receptor α (Gibson et al. 2002; Luch 2005; Nebert and Dalton 2006; Tompkins and Wallace 2007), suggesting that 1,4-dioxane is not likely to be a nuclear receptor activator and is not likely to induce aberrant activation of CYP enzymes.
Overall, there is no evidence for alternative non-genotoxic mechanisms that could contribute to the carcinogenic effects of 1,4-dioxane. Lack of non-genotoxic mechanisms provides indirect evidence supporting a mutagenic MOA of 1,4-dioxane.
Dose–response relationships for genotoxic carcinogens have been a topic of intense scientific and public debate. It is gradually shifting away from a linear non-threshold approach towards quantification approaches using PoD metrics in defining acceptable exposure limits and assessing the human risk of genotoxic carcinogens. The findings of the present study argue for the presence of no-effect levels of 1,4-dioxane for mutagenicity and hepatocarcinogenicity: (1) none of the measured parameters was statistically altered by administration of 1,4-dioxane at doses of 200 ppm and lower; (2) only in the groups administered higher doses of 1,4-dioxane were increases in mutation and/or altered mutation spectrum (at doses of 1000 ppm and higher) and GST-P positive foci (at doses of 2000 ppm and higher) observed; (3) in spite of induction of mutations by administration of 1000 ppm 1,4-dioxane, no significant increase in GST-P positive foci was observed in these rats; (4) cell proliferation activity was significantly increased only at the highest dose of 5000 ppm; and (5) significant induction of the DNA repair enzyme MGMT was observed only at the highest dose of 5000 ppm, indicating that the 1,4-dioxane-induced DNA damage response is dose-dependent. Therefore, these results indicate that 1,4-dioxane only has mutagenic and carcinogenic effects above a PoD.
Currently, there are two statistical approaches available for deriving a PoD, the NOEL approach and the BMD approach. In the present study on 1,4-dioxane, the PoD values derived from these different approaches are markedly different. It is well known that PoD values derived from the NOEL approach is dependent on dose selection, since the PoD is limited to one of the doses included in study, and the PoD is also highly dependent on sample size (and thus the statistical sensitivity of the study). Limitations of the NOEL approach include: it does not allow estimation of the probability of response for dose levels not included in the study, making it harder to compare separate studies, and the PoD will tend to be higher in studies with smaller numbers of animals per group (EPA 2012; Haber et al. 2018; Hardy et al. 2017; Slob 2014a, b). In the present study, for example, the PoD value derived by the NOEL approach for GST-P positive foci was 1000 ppm in Experiment 1, where the sample size was 7–8 rats per group, but was 200 ppm in Experiment 3, where the sample size was 30 rats per group. The advantages of the BMD approach using BMDS or PROAST have been well documented (EPA 2012; Haber et al. 2018; Hardy et al. 2017; Slob 2014a, b) and include the points that (1) when using the BMD approach, the PoD is not constrained to a dose used in the study; (2) the BMD approach takes the full shape of dose–response curve into account, thereby incorporating more dose–response information into the determination of the PoD; (3) because it incorporates all of the study data, it is better at taking into account statistical uncertainties, for example, those associated with inter-animal differences and study size; (4) it allows for cross-study comparison; and (5) one consequence of these points is that the BMD approach also makes more efficient use of animals. Differences in modeling that can lead to differences in the final PoD have also been extensively discussed (Haber et al. 2018; Hardy et al. 2017), including the mathematical models used, the choice of the benchmark response (BMR) for continuous data, and the use of unrestricted models. In addition, the use the PROAST and BMDS software packages can also lead to different PoD values. For example, the PROAST guideline recommends the BMR for continuous endpoints be defined as a percent change in the mean response (10% in the present study; BMD10), while the BMDS guideline recommends a BMR corresponding to a change in the control mean response of one control SD (BMD1SD). While determination of the best approach is beyond the scope of the present study, the PoDs (BMDL10) derived by the BMD approach using PROAST are conservative and in a logical ranking: Mutation < GST-P-positive foci < Liver tumor.
Several mechanisms for non-linear genotoxic and carcinogenic responses of genotoxic carcinogens have been suggested, including induction of detoxification processes, cell cycle delay, DNA repair, apoptosis, and the suppression of neoplastically transformed cells by the immune system (Jenkins et al. 2010; Johnson et al. 2012; Sofuni et al. 2000; Thomas and Johnson 2016). However, little in vivo evidence is available in the literature. The fact that exposure to 5000 ppm 1,4-dioxane resulted in an increase in A:T-to-G:C mutations suggests that 1,4-dioxane may cause O4-methylthymine (m4T) adduct formation, which as noted above can cause A:T-to-G:C transitions if not repaired. These data coupled with the result that in the lower dose groups, including the mutagenic dose group of 1000 ppm, there was no increase in A:T-to-G:C mutations suggest that normal physiological levels of MGMT are sufficient to repair the m4T adducts generated by 1000 ppm 1,4-dioxane, and that DNA repair is responsible, at least in part, for the observed no effect of low doses of 1,4-dioxane. However, as A:T-to-T:A mutations were significantly increased in both the 1000 and 5000 ppm groups, it is reasonable to presume that DNA repair by genes besides MGMT is responsible for the observed no effect of low doses of 1,4-dioxane in the present study. We previously demonstrated that suppression of cell cycle progression by p21Cip/WAF1 followed by DNA repair (induction of APEX1 and GADD45) is responsible for the observed no effect of low doses of 2-amino-3-methylimidazo[4,5-f]quinoline (IQ), a genotoxic hepatocarcinogen in rats (Wei et al. 2011). However, p53, p21Cip/WAF1, GADD45, APEX1, ERCC5, MSH2, and MSH3 gene expression was not induced by administration of 1,4-dioxane in the present study, suggesting that these genes do not have a significant impact on the mutagenicity and hepatocarcinogenicity of 1,4-dioxane in rats.
In conclusion, we demonstrated that 1,4-dioxane is a genotoxic carcinogen, even though it is negative in in vitro mutagenicity assays, and provide crucial mechanistic information for quantitative cancer risk assessment: 1,4-dioxane induces hepatocarcinogenesis in rats through a mutagenic MOA. The present study provides further evidence supporting the premise that the PoD should be considered when evaluating the risk of genotoxic carcinogens. To this end, further accumulation of data, especially mechanistic data, should be promoted to facilitate not only an understanding of the carcinogenic effects of low doses of genotoxic carcinogens but also to establish an accurate means of quantitative risk assessment.
References
Akiba N, Shiizaki K, Matsushima Y, Endo O, Inaba K, Totsuka Y (2017) Influence of GSH S-transferase on the mutagenicity induced by dichloromethane and 1,2-dichloropropane. Mutagenesis 32(4):455–462. https://doi.org/10.1093/mutage/gex014
Argus MF, Arcos JC, Hochligeti C (1965) Studies on the carcinogenic activity of protein-denaturing agents: hepatocarcinogenicity of dioxane. J Natl Cancer Inst 35(6):949–958
Bolt HM (2016) Practical thresholds in the derivation of occupational exposure limits (OELs) for carcinogens. In: Nohmi T, Fukushima S (eds) Thresholds of genotoxic carcinogens-from mechanisms to regulation. 1 Edn). Academic Press, Cambridge, p 117–128
Boobis AR, Cohen SM, Dellarco V, McGregor D, Meek ME, Vickers C, Willcocks D, Farland W (2006) IPCS framework for analyzing the relevance of a cancer mode of action for humans. Crit Rev Toxicol 36(10):781–792. https://doi.org/10.1080/10408440600977677
Buffler PA, Wood SM, Suarez L, Kilian DJ (1978) Mortality follow-up of workers exposed to 1,4-dioxane. J Occup Med 20(4):255–259
Chatterjee N, Walker GC (2017) Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 58(5):235–263. https://doi.org/10.1002/em.22087
Cohen SM (2010) Evaluation of possible carcinogenic risk to humans based on liver tumors in rodent assays: the two-year bioassay is no longer necessary. Toxicol Pathol 38(3):487–501. https://doi.org/10.1177/0192623310363813
Cohen SM, Klaunig J, Meek ME, Hill RN, Pastoor T, Lehman-McKeeman L, Bucher J, Longfellow DG, Seed J, Dellarco V, Fenner-Crisp P, Patton D (2004) Evaluating the human relevance of chemically induced animal tumors. Toxicol Sci 78(2):181–186. https://doi.org/10.1093/toxsci/kfh073
EC (2002) European Union risk assessment report: CAS No. 123-91-1: EINECS No. 204-661-8: 1,4-Dioxane. European Communities, Institute for Health and Consumer Protection, European Chemicals Bureau., Luxembourg
Encell LP, Loeb LA (2000) Enhanced in vivo repair of O(4)-methylthymine by a mutant human DNA alkyltransferase. Carcinogenesis 21(7):1397–1402
EPA (2005) United States Environmental Protection Agency’s guidelines for carcinogen risk assessment. https://www.epa.gov/sites/production/files/2013-09/documents/cancer_guidelines_final_3-25-05.pdf
EPA (2012) United States Environmental Protection Agency. Benchmark Dose Technical Guidance (100-R-12-001) https://www.epa.gov/sites/production/files/2015-01/documents/benchmark_dose_guidance.pdf
EPA (2013) United States Environmental Protection Agency, IRIS Toxicological Review of 1,4-Dioxane (with Inhalation Update) (EPA/635/R-11/003F). https://cfpub.epa.gov/si/si_public_record_report.cfm?dirEntryId=247852
EPA (2017) United States Environmental Protection Agency. Technical Fact Sheet–1,4-Dioxane (EPA 505-F-17-011) https://www.epa.gov/sites/production/files/2014-03/documents/ffrro_factsheet_contaminant_14-dioxane_january2014_final.pdf
Fukushima S, Wanibuchi H, Morimura K, Wei M, Nakae D, Konishi Y, Tsuda H, Uehara N, Imaida K, Shirai T, Tatematsu M, Tsukamoto T, Hirose M, Furukawa F, Wakabayashi K, Totsuka Y (2002) Lack of a dose-response relationship for carcinogenicity in the rat liver with low doses of 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline or N-nitrosodiethylamine. Jpn J Cancer Res 93(10):1076–1082
Fukushima S, Wei M, Kakehashi A, Wanibuchi H (2010) Thresholds for genotoxic carcinogens: evidence from mechanism-based carcinogenicity studies. In: Hsu C-H, Stedeford T (eds) Cancer risk assessment: chemical carcinogenesis, hazard evaluation, and risk quantification. John Wiley & Sons, New Jersey, pp 201–221
Fukushima S, Gi M, Kakehashi A, Wanibuchi H (2016a) Qualitative and quantitative assessment on low-dose carcinogenicity of genotoxic hepatocarcinogens: dose-response for key events in rat hepatocarcinogenesis. In: Takehiko N, Fukushima S (eds) Thresholds of genotoxic carcinogen. 1 Edn. Academic Press, Cambridge p 1–17
Fukushima S, Gi M, Kakehashi A, Wanibuchi H, Matsumoto M (2016b) Qualitative and quantitative approaches in the dose-response assessment of genotoxic carcinogens. Mutagenesis 31(3):341–346. https://doi.org/10.1093/mutage/gev049
Gibson GG, Plant NJ, Swales KE, Ayrton A, El-Sankary W (2002) Receptor-dependent transcriptional activation of cytochrome P4503A genes: induction mechanisms, species differences and interindividual variation in man. Xenobiotica 32(3):165–206. https://doi.org/10.1080/00498250110102674
Gollapudi BB, Johnson GE, Hernandez LG, Pottenger LH, Dearfield KL, Jeffrey AM, Julien E, Kim JH, Lovell DP, Macgregor JT, Moore MM, van Benthem J, White PA, Zeiger E, Thybaud V (2013) Quantitative approaches for assessing dose-response relationships in genetic toxicology studies. Environ Mol Mutagen 54(1):8–18. https://doi.org/10.1002/em.21727
Haber LT, Dourson ML, Allen BC, Hertzberg RC, Parker A, Vincent MJ, Maier A, Boobis AR (2018) Benchmark dose (BMD) modeling: current practice, issues, and challenges. Crit Rev Toxicol 48(5):387–415. https://doi.org/10.1080/10408444.2018.1430121
Hansen J, Schneider T, Olsen JH, Laursen B (1993) Availability of data on humans potentially exposed to suspected carcinogens in the Danish working environment. Pharmacol Toxicol 72(Suppl 1):77–85
Hardy A, Benford D, Halldorsson T, Jeger MJ, Knutsen KH, More S, Mortensen A, Naegeli H, Noteborn H, Ockleford C, Ricci A, Rychen G, Silano V, Solecki R, Turck D, Aerts M, Bodin L, Davis A, Edler L, Gundert-Remy U, Sand S, Slob W, Bottex B, Abrahantes JC, Marques DC, Kass G, Schlatter JR, Comm ES (2017) Update: use of the benchmark dose approach in risk assessment. Efsa J. https://doi.org/10.2903/j.efsa.2017.4658
Hayashi H, Kondo H, Masumura K, Shindo Y, Nohmi T (2003) Novel transgenic rat for in vivo genotoxicity assays using 6-thioguanine and Spi- selection. Environ Mol Mutagen 41(4):253–259
Hoshi M, Morimura K, Wanibuchi H, Wei M, Okochi E, Ushijima T, Takaoka K, Fukushima S (2004) No-observed effect levels for carcinogenicity and for in vivo mutagenicity of a genotoxic carcinogen. Toxicol Sci 81(2):273–279
IARC (1999) 1,4-Dioxane, in: re-evaluation of some organic chemicals, hydrazine and hydrogen peroxide. IARC Monogr Eval Carcinog Risks Hum. 71:589–602
Ishino K, Kato T, Kato M, Shibata T, Watanabe M, Wakabayashi K, Nakagama H, Totsuka Y (2015) Comprehensive DNA adduct analysis reveals pulmonary inflammatory response contributes to genotoxic action of magnetite nanoparticles. Int J Mol Sci 16(2):3474–3492. https://doi.org/10.3390/ijms16023474
Jenkins GJ, Zair Z, Johnson GE, Doak SH (2010) Genotoxic thresholds, DNA repair, and susceptibility in human populations. Toxicology 278(3):305–310. https://doi.org/10.1016/j.tox.2009.11.016
Jin B, Robertson KD (2013) DNA methyltransferases, DNA damage repair, and cancer. Adv Exp Med Biol 754:3–29. https://doi.org/10.1007/978-1-4419-9967-2_1
Johnson GE, Zoulikha MZ, Bodger OG, Lewis PD, Rees BJ, Verma JR, Thomas AD, Doak SH, Jenkins GJS (2012) Investigating mechanism for non-linear genotoxic responses, and analysing their effects in binary combination. Gene Environ 34(4):179–185. https://doi.org/10.1002/em.21870
Kaina B, Christmann M, Naumann S, Roos WP (2007) MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 6(8):1079–1099. https://doi.org/10.1016/j.dnarep.2007.03.008
Kang JS, Wanibuchi H, Morimura K, Totsuka Y, Yoshimura I, Fukushima S (2006) Existence of a no effect level for MeIQx hepatocarcinogenicity on a background of thioacetamide-induced liver damage in rats. Cancer Sci 97(6):453–458. https://doi.org/10.1111/j.1349-7006.2006.00201.x
Kano H, Umeda Y, Kasai T, Sasaki T, Matsumoto M, Yamazaki K, Nagano K, Arito H, Fukushima S (2009) Carcinogenicity studies of 1,4-dioxane administered in drinking-water to rats and mice for 2 years. Food Chem Toxicol 47(11):2776–2784. https://doi.org/10.1016/j.fct.2009.08.012
Kasai T, Kano H, Umeda Y, Sasaki T, Ikawa N, Nishizawa T, Nagano K, Arito H, Nagashima H, Fukushima S (2009) Two-year inhalation study of carcinogenicity and chronic toxicity of 1,4-dioxane in male rats. Inhal Toxicol 21(11):889–897. https://doi.org/10.1080/08958370802629610
Kirkland D, Aardema M, Henderson L, Muller L (2005) Evaluation of the ability of a battery of three in vitro genotoxicity tests to discriminate rodent carcinogens and non-carcinogens I. Sensitivity, specificity and relative predictivity. Mutat Res 584(1–2):1–256. https://doi.org/10.1016/j.mrgentox.2005.02.004
Kitchin KT, Brown JL (1990) Is 1,4-dioxane a genotoxic carcinogen? Cancer Lett 53(1):67–71
Kobert T, Willams GM (2016) Thresholds for hepatocarcinogenicity of DNA-reactive compounds. In: Nohmi T, Fukushima S (eds) Thresholds of genotoxic carcinogens-from mechanisms to regulation. 1 Edn. Academic Press, Cambridge, p 19–36
Kociba RJ, McCollister SB, Park C, Torkelson TR, Gehring PJ (1974) 1,4-dioxane. I. Results of a 2-year ingestion study in rats. Toxicol Appl Pharmacol 30:275–286
Levine RL, Yang IY, Hossain M, Pandya GA, Grollman AP, Moriya M (2000) Mutagenesis induced by a single 1,N6-ethenodeoxyadenosine adduct in human cells. Cancer Res 60(15):4098–4104
Luch A (2005) Nature and nurture—lessons from chemical carcinogenesis. Nat Rev Cancer 5(2):113–125. https://doi.org/10.1038/nrc1546
MacGregor JT, Frotschl R, White PA, Crump KS, Eastmond DA, Fukushima S, Guerard M, Hayashi M, Soeteman-Hernandez LG, Johnson GE, Kasamatsu T, Levy DD, Morita T, Muller L, Schoeny R, Schuler MJ, Thybaud V (2015a) IWGT report on quantitative approaches to genotoxicity risk assessment II. Use of point-of-departure (PoD) metrics in defining acceptable exposure limits and assessing human risk. Mutat Res Genet Toxicol Environ Mutagen 783:66–78. https://doi.org/10.1016/j.mrgentox.2014.10.008
MacGregor JT, Frotschl R, White PA, Crump KS, Eastmond DA, Fukushima S, Guerard M, Hayashi M, Soeteman-Hernandez LG, Kasamatsu T, Levy DD, Morita T, Muller L, Schoeny R, Schuler MJ, Thybaud V, Johnson GE (2015b) IWGT report on quantitative approaches to genotoxicity risk assessment I. Methods and metrics for defining exposure-response relationships and points of departure (PoDs). Mutat Res Genet Toxicol Environ Mutagen 783:55–65. https://doi.org/10.1016/j.mrgentox.2014.09.011
Masumura K, Nohmi T (2009) Spontaneous mutagenesis in rodents: spontaneous gene mutations identified by neutral reporter genes in gpt delta transgenic mice and rats. J Health Sci 55(1):40–49
Masumura K, Sakamoto Y, Kumita W, Honma M, Nishikawa A, Nohmi T (2015) Genomic integration of lambda EG10 transgene in gpt delta transgenic rodents. Genes Environ 37:24. https://doi.org/10.1186/s41021-015-0024-6
McFee AF, Abbott MG, Gulati DK, Shelby MD (1994) Results of mouse bone marrow micronucleus studies on 1,4-dioxane. Mutat Res 322(2):145–148
Mirkova ET (1994) Activity of the rodent carcinogen 1,4-dioxane in the mouse bone marrow micronucleus assay. Mutat Res 322(2):142–144
Morita T, Hayashi M (1998) 1,4-dioxane is not mutagenic in five in vitro assays and mouse peripheral blood micronucleus assay, but is in mouse liver micronucleus assay. Environ Mol Mutagen 32(3):269–280
Morita T, Hamada S, Masumura K, Wakata A, Maniwa J, Takasawa H, Yasunaga K, Hashizume T, Honma M (2016) Evaluation of the sensitivity and specificity of in vivo erythrocyte micronucleus and transgenic rodent gene mutation tests to detect rodent carcinogens. Mutat Res Genet Toxicol Environ Mutagen 802:1–29. https://doi.org/10.1016/j.mrgentox.2016.03.008
Nebert DW, Dalton TP (2006) The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat Rev Cancer 6(12):947–960. https://doi.org/10.1038/nrc2015
Nohmi T (2016) Past, Present and Future Directions of gpt delta Rodent Gene Mutation Assays. Food Saf 4(1):1–13
Nohmi T, Suzuki T, Masumura K (2000) Recent advances in the protocols of transgenic mouse mutation assays. Mutat Res 455(1–2):191–215
Nohmi T, Masumura K, Toyoda-Hokaiwado N (2017) Transgenic rat models for mutagenesis and carcinogenesis. Genes Environ 39:11. https://doi.org/10.1186/s41021-016-0072-6
NTP (1978) National Toxicology Program. Bioassay of 1,4-dioxane for possible carcinogenicity. Natl Cancer Inst Carcinog Tech Rep Ser 80:1–123
O’Toole SM, Pegg AE, Swenberg JA (1993) Repair of O6-methylguanine and O4-methylthymidine in F344 rat liver following treatment with 1,2-dimethylhydrazine and O6-benzylguanine. Cancer Res 53(17):3895–3898
Poon SL, McPherson JR, Tan P, Teh BT, Rozen SG (2014) Mutation signatures of carcinogen exposure: genome-wide detection and new opportunities for cancer prevention. Genome Med 6(3):24. https://doi.org/10.1186/gm541
Rodriguez-Antona C, Ingelman-Sundberg M (2006) Cytochrome P450 pharmacogenetics and cancer. Oncogene 25(11):1679–1691. https://doi.org/10.1038/sj.onc.1209377
Roy SK, Thilagar AK, Eastmond DA (2005) Chromosome breakage is primarily responsible for the micronuclei induced by 1,4-dioxane in the bone marrow and liver of young CD-1 mice. Mutat Res 586(1):28–37. https://doi.org/10.1016/j.mrgentox.2005.05.007
Sharma S, Salehi F, Scheithauer BW, Rotondo F, Syro LV, Kovacs K (2009) Role of MGMT in tumor development, progression, diagnosis, treatment and prognosis. Anticancer Res 29(10):3759–3768
Singer B (1986) O-alkyl pyrimidines in mutagenesis and carcinogenesis: occurrence and significance. Cancer Res 46(10):4879–4885
Slob W (2014a) Benchmark dose and the three Rs. Part I. Getting more information from the same number of animals. Crit Rev Toxicol 44(7):557–567. https://doi.org/10.3109/10408444.2014.925423
Slob W (2014b) Benchmark dose and the three Rs. Part II. Consequences for study design and animal use. Crit Rev Toxicol 44(7):568–580. https://doi.org/10.3109/10408444.2014.925424
Sofuni T, Hayashi M, Nohmi T, Matsuoka A, Yamada M, Kamata E (2000) Semi-quantitative evaluation of genotoxic activity of chemical substances and evidence for a biological threshold of genotoxic activity. Mutat Res 464(1):97–104
Stickney JA, Sager SL, Clarkson JR, Smith LA, Locey BJ, Bock MJ, Hartung R, Olp SF (2003) An updated evaluation of the carcinogenic potential of 1,4-dioxane. Regul Toxicol Pharmacol 38(2):183–195
Thomas AD, Johnson GE (2016) DNA repair and its influence on points of departure for alkylating agent genotoxicity. In: Nohmi T, Fukushima S (eds) Thresholds of genotoxic carcinogens-from mechanisms to regulation. 1 edn. Academic Press, Cambridge, p 67–82
Tinwell H, Ashby J (1994) Activity of 1,4-dioxane in mouse bone marrow micronucleus assays. Mutat Res 322(2):148–150
Tompkins LM, Wallace AD (2007) Mechanisms of cytochrome P450 induction. J Biochem Mol Toxicol 21(4):176–181
Walmsley RM, Billinton N (2011) How accurate is in vitro prediction of carcinogenicity? Br J Pharmacol 162(6):1250–1258. https://doi.org/10.1111/j.1476-5381.2010.01131.x
Wei M, Hori TA, Ichihara T, Wanibuchi H, Morimura K, Kang JS, Puatanachokchai R, Fukushima S (2006) Existence of no-observed effect levels for 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline on hepatic preneoplastic lesion development in BN rats. Cancer Lett 231(2):304–308. https://doi.org/10.1016/j.canlet.2005.02.029 pii]
Wei M, Wanibuchi H, Nakae D, Tsuda H, Takahashi S, Hirose M, Totsuka Y, Tatematsu M, Fukushima S (2011) Low-dose carcinogenicity of 2-amino-3-methylimidazo[4,5-f]quinoline in rats: evidence for the existence of no-effect levels and a mechanism involving p21(Cip / WAF1). Cancer Sci 102(1):88–94. https://doi.org/10.1111/j.1349-7006.2010.01761.x
Wilbur S, Jones D, Risher JF, Crawford J, Tencza B, Llados F, Diamond GL, Citra M, Osier MR, Lockwood LO (2012) Agency for toxic substances and disease registry (ATSDR) toxicological profiles toxicological profile for 1,4-dioxane. Agency for Toxic Substances and Disease Registry (US), Atlanta
Acknowledgements
This work was supported by Health Labor Sciences Research Grants from the Ministry of Health, Labor and Welfare of Japan, and a Grant from the Japan Food Chemical Research Foundation. The authors gratefully acknowledge the technical assistance of Rie Onodera, Kaori Nakakubo, Keiko Sakata, Yuko Hisabayashi, and Yukiko Iura (Department of Molecular Pathology, Osaka City University Graduate School of Medicine School), and Emi Donoue and Hideki Nakagawa (Research Support Platform, Osaka City University Graduate School of Medicine).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gi, M., Fujioka, M., Kakehashi, A. et al. In vivo positive mutagenicity of 1,4-dioxane and quantitative analysis of its mutagenicity and carcinogenicity in rats. Arch Toxicol 92, 3207–3221 (2018). https://doi.org/10.1007/s00204-018-2282-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-018-2282-0