
Abstract
Low-dose fluoride is an effective caries prophylactic, but high-dose fluoride is an environmental health hazard that causes skeletal and dental fluorosis. Treatments to prevent fluorosis and the molecular pathways responsive to fluoride exposure remain to be elucidated. Previously we showed that fluoride activates SIRT1 as an adaptive response to protect cells. Here, we demonstrate that fluoride induced p53 acetylation (Ac-p53) [Lys379], which is a SIRT1 deacetylation target, in ameloblast-derived LS8 cells in vitro and in enamel organ in vivo. Here we assessed SIRT1 function on fluoride-induced Ac-p53 formation using CRISPR/Cas9-mediated Sirt1 knockout (LS8Sirt/KO) cells or CRISPR/dCas9/SAM-mediated Sirt1 overexpressing (LS8Sirt1/over) cells. NaF (5 mM) induced Ac-p53 formation and increased cell cycle arrest via Cdkn1a/p21 expression in Wild-type (WT) cells. However, fluoride-induced Ac-p53 was suppressed by the SIRT1 activator resveratrol (50 µM). Without fluoride, Ac-p53 persisted in LS8Sirt/KO cells, whereas it decreased in LS8Sirt1/over. Fluoride-induced Ac-p53 formation was also suppressed in LS8Sirt1/over cells. Compared to WT cells, fluoride-induced Cdkn1a/p21 expression was elevated in LS8Sirt/KO and these cells were more susceptible to fluoride-induced growth inhibition. In contrast, LS8Sirt1/over cells were significantly more resistant. In addition, fluoride-induced cytochrome-c release and caspase-3 activation were suppressed in LS8Sirt1/over cells. Fluoride induced expression of the DNA double strand break marker γH2AX in WT cells and this was augmented in LS8Sirt1/KO cells, but was attenuated in LS8Sirt1/over cells. Our results suggest that SIRT1 deacetylates Ac-p53 to mitigate fluoride-induced cell growth inhibition, mitochondrial damage, DNA damage and apoptosis. This is the first report implicating Ac-p53 in fluoride toxicity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Dental caries is a largely preventable condition and fluoride has proven an effective caries prophylactic (Aoba and Fejerskov 2002). The US Public Health Service (PHS) recommends public water fluoridation at an optimal fluoride concentration of 0.7 ppm (corresponding to 0.04 mM NaF) (Health and Human Services Federal Panel on Community Water 2015). However, high-dose fluoride is an environmental health hazard. Acute or chronic fluoride over-exposure can result in enamel fluorosis (DenBesten 1999), skeletal fluorosis (Boivin et al. 1989), neurotoxicity (Adedara et al. 2016) and reproductive toxicity (Sm and Mahaboob Basha 2016). Dental fluorosis is a developmental disorder caused by fluoride over-exposure during enamel formation, resulting in hypomineralized, mottled, discolored, and porous enamel that is susceptible to decay. The prevalence of dental fluorosis among the USA population is increasing (Beltran-Aguilar et al. 2010). However, other than avoiding excessive fluoride intake during enamel development, treatments to prevent dental fluorosis remain unknown. Therefore, the validation of new therapeutic targets to prevent dental fluorosis is crucial.
Fluoride exerts diverse cellular effects in a dose, cell type, and tissue-dependent manner. We and others have shown in several tissues, including the enamel organ, that high-dose fluoride causes oxidative stress (Sun et al. 2011; Suzuki et al. 2015, 2014b), ER stress (Kubota et al. 2005; Sharma et al. 2008) and genotoxicity (Dutta et al. 2017). Fluoride reduces Klk4 and Tgf-β1 expression during enamel development (Suzuki et al. 2014a), increases mitochondrial damage, DNA damage and apoptosis in both ameloblast-derived LS8 cells in vitro and in ameloblasts in vivo (Suzuki et al. 2017). Fluoride exposure generates reactive oxygen species (ROS). ROS causes oxidative stress resulting in mitochondrial damage as can be observed by cytochrome-c release, up-regulation of UCP2, and attenuation of ATP synthesis. Elevated ROS can cause DNA damage. Fluoride treatment activated DNA damage pathways including H2AX phosphorylation [γH2AX], 8 oxoguanine and phosphorylation of ATM in rodent ameloblasts in vivo (Suzuki et al. 2015).
Previously we demonstrated that fluoride activates SIRT1 to ameliorate stress responses in LS8 cells. Sirtuins (SIRT1–SIRT7) are a family of highly conserved NAD+-dependent class III histone deacetylases (class III HDACs). SIRT1 is the mammalian homologue of yeast silent information regulator-2 (Sir2), which is the most widely studied of the sirtuins (Imai et al. 2000). SIRT1 expression increases in various physiological conditions, including nutrient starvation, aging and cell stress such as oxidative stress (Salminen et al. 2013). SIRT1 also deacetylates target histones H3 and H4 to repress gene transcription (Braunstein et al. 1996). SIRT1 deacetylates numerous non-histone protein substrates [Atgs, Foxo1, Foxo3, PGC-1α, NF-kB, E2F1 and p53] (Conrad et al. 2016) to play a key role in protecting against cell stress.
Posttranslational modification of the p53 peptide such as acetylation, phosphorylation, methylation and ubiquitination are key components of regulatory circuits that monitor signaling pathways including DNA damage, oncogenic events and normal cell processes (Reed and Quelle 2014). Acetylation increases p53 protein stability, binding to promoters, and association with signaling proteins. Ac-p53 is required for cell cycle checkpoint responses to DNA damage. p53 is acetylated at Lys379 following DNA damage (Sakaguchi et al. 1998). Additionally, Ac-p53 regulates cell cycle arrest via cyclin-dependent kinase inhibitor 1A/p21 (CDKN1A/p21) and regulates apoptosis (Bao et al. 2016). SIRT1 deacetylates p53 at Lys379 to inhibit p53-dependent apoptosis (Vaziri et al. 2001). As the number of reported SIRT1 targets increase (Poulose and Raju 2015), the critical target proteins and how SIRT1 regulates these target proteins are dependent on the kinds of stress and cell/tissue types. Under fluoride-induced stress, the SIRT1 targets and the molecular pathways of how SIRT1 protects cells from fluoride toxicity are not fully understood.
To assess SIRT1 function, we exploited CRISPR/Cas9 and CRISPR/dCas9/SAM systems to knockout or overexpress SIRT1 in LS8 cells. In early 2013, CRISPR/Cas9 [the clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR associated protein 9 (Cas9)] technology for mammalian genome editing was first reported (Cong et al. 2013). Recently, dCas9/SAM [the catalytically deficient Cas9 (dCas9) synergistic activation mediator (SAM)] system was developed to induce efficient transcriptional activation at endogenous genomic loci (Konermann et al. 2015). SAM comprises MS2-gRNA, dCas9–VP64 and the MS2–p65–HSF1 fusion protein that can specifically bind to any promoter region identified by a sgRNA. The transcription activator VP64 fused to dCas9 is capable of dramatically up-regulating transcription of targeted genes.
SIRT1 ablated cells derived from several tissues have been generated to delineate SIRT1 function (Bernier et al. 2011; He et al. 2016). However, this is the first report of ameloblast-derived cell line that constitutively overexpresses or ablates SIRT1 expression. Here we investigate SIRT1 function on fluoride-induced Ac-p53 formation and apoptosis using SIRT1 knockout (LS8Sirt/KO) and SIRT1 overexpressing (LS8Sirt1/over) cells.
Materials and methods
Cell culture
The mouse ameloblast-derived LS8 cells (Chen et al. 1992), WT, LS8Sirt1/KO, LS8Sirt1/over and negative controls; LS8Lacz/KO and LS8Lacz/over cells were maintained in alpha minimal essential medium (αMEM) with GlutaMAX (Thermo Fisher Scientific, Waltham, MA) supplemented with fetal bovine serum (10%) and sodium pyruvate (1 mM) without antibiotics. Reagents; sodium fluoride (NaF, Cat. S299-100, Fisher Scientific, Pittsburgh, PA). NaF 5 mM corresponds to 95 ppm F−. Resveratrol (RES, Cat.R5010-100MG, Sigma-Aldrich. St. Louis, MO).
Establishment of Sirt1 knockout (LS8Sirt1/KO) cells
LS8Sirt1/KO cells were established using the CRISPR/Cas9 system. The procedure was performed according to protocols by the Zhang Lab (Shalem et al. 2014). Details and sgRNA sequences are described in supplementary data.
Establishment of Sirt1 overexpressing (LS8Sirt1/over) cells
LS8Sirt1/over cells were established by the dCas9-SAM system (Konermann et al. 2015). The procedure was performed according to protocols by the Zhang Lab. Details and sgRNA sequences are described in supplementary data. All experiments using lentivirus vectors were performed in BSL2 environment approved by The Ohio State University IBC.
Animals
Sprague–Dawley rats (6-week-old) were purchased from Charles River Laboratories (Wilmington, MA) and were provided water containing 0 or 100 ppm fluoride as sodium fluoride ad libitum. After 6 weeks, the animals were euthanized and maxillary incisors were used for immunohistochemistry. All animals were treated humanely and all handling procedures were approved by the Institutional Animal Care Use Committee (IACUC) at The Forsyth Institute. The Forsyth Institute is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) and follows the Guide for the Care and Use of Laboratory Animals (NRC1996). Note that the first and senior authors were employed by The Forsyth Institute through October 2015 when the animal experiments were completed.
Immunohistochemistry
Immunohistochemistry was performed as described previously (Kubota et al. 2005). Briefly, rat maxillary incisors were extracted after fluoride treatment and fixed in paraformaldehyde, demineralized with EDTA for 2 weeks, and embedded in paraffin. Hematoxylin and eosin (H&E) staining and immunohistochemistry were performed. Sections were incubated with primary antibodies: rabbit anti-Acetyl-p53 [K370] (Abcam Inc., Cambridge, MA), followed by incubation with a peroxidase-conjugated secondary antibody, Vectastain Elite ABC Regent (Vector Labs, Burlingame, CA), and ImmPACT DAB kit (Vector Labs). Sections were counter stained with 0.1% Fast Green in PBS and examined by light microscopy.
Western blot analysis
Western blots were performed as described previously (Suzuki et al. 2015). Briefly, total proteins from cells were extracted with RIPA buffer (Sigma-Aldrich) containing protease inhibitor cocktail (Thermo Scientific, Rockford, IL). Mitochondrial fractions and cytosolic fractions were isolated using a mitochondria isolation kit for cultured cells (Thermo Scientific). Equal amounts of protein per lane (5–20 µg) were loaded onto Mini-Protean® TGX™ gels (Biorad, Hercules, CA), transferred to Trans-Blot Turbo Transfer nitrocellulose membranes (Biorad) and probed with primary antibodies. Primary and secondary antibodies are described in supplementary data. Enhanced chemiluminescence was performed with SuperSignal West Pico (Thermo Scientific), and signal was detected by myECL imager (Thermo Scientific). Bands were quantified by densitometry using Quantity One software (Biorad). And values obtained were expressed as a ratio to each lane’s loading control value. At least three biological replicates for each experiment were assayed and representative images are shown.
Real-time quantitative PCR (qPCR) analysis
Total RNA was extracted from cells using Direct-zol RNA Mini Prep (Zymo Research Corp., Irvine, CA). Total RNA was reverse-transcribed into cDNA using a Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics, Minneapolis, MN). The cDNA was subjected to qPCR amplification on QuantStudio 3 (Thermo Scientific). Primer sequences are described in supplementary data. The relative expression of target genes was determined by the 2−ΔΔCT method (Pfaffl 2001). The internal reference control genes were Gapdh for LS8Sirt1/KO cells and B2m for WT and LS8Sirt1/over cells because of their consistent expression with experimental treatments. To compare WT, LS8Sirt1/KO and LS8Sirt1/over cells, geometric mean of Gapdh and B2m was used for normalization (Fig. 6). At least three biological replicates were analyzed for each experiment.
Cell proliferation assay
To assess cell proliferation, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays were performed. Cells were cultured overnight in 96-well plates and then the indicated concentrations of NaF were added. After 24 h, MTT assays (Sigma-Aldrich) were performed as described previously (Kubota et al. 2005). Four wells were assayed for each experimental treatment and six separate experiments were performed.
Statistical analysis
Differences between two groups were analyzed by Student’s t test. Multiple group comparison was performed by one-way ANOVA with Bonferroni/Dunn post hoc test. For Cdkn1a/p21 expression, regression analysis was performed (Fig. 4b). Significance was assessed at P < 0.05.
Results
CRISPR/Cas9-mediated Sirt1 gene editing caused efficient removal of SIRT1 expression in LS8 cells
Cas9 was expressed by LS8Sirt1/KO clones (#321 and #325) and by LacZ negative controls, but not in parent WT cells (Fig. 1a). This demonstrated that Cas9 was present in the appropriate cell lines. The CRISPR/Cas9 system can produce insertions and/or deletions (indels) in the genomic target site via the non-homologous end-joining DNA repair pathway. To determine the nature of the genetic lesions caused by the Sirt1-targeted CRISPR/Cas9 construct, clones lacking SIRT1 protein expression were subjected to indel identification followed by Sanger sequencing. LS8Sirt1/KO clones (#321 and #325) have unique indels with bi-allelic mutations (Fig. 1b). Indels consisting of an insertion of one nucleotide and a deletion of three to four nucleotides were detected in both #321 and #325 clones. LS8Sirt1/KO clones show a significant decrease in SIRT1 transcripts (Fig. 1c) and no detectable protein (Fig. 1d). In contrast, SIRT1 was expressed at the transcript and protein levels in the LacZ negative control and WT cells.

CRISPR/Cas9-targeted disruption of SIRT1 in LS8 cells. a Whole cell lysates were subjected to Western blot analysis for Cas9 (160 kDa) expression in WT, negative control (LacZ), and SIRT1 Knockout (KO) clones (321 and 325). β-actin (44 kDa) was used as a loading control. Numbers show the Cas9/β-actin loading control ratio after scanning densitometry. b Unique bi-allelic indels were detected by sequence analysis in each knockout clone. The sgRNA targeting site was PCR-amplified from genomic DNA, cloned, multiple clones sequenced, and compared to the reference sequence for mouse WT Sirt1 (RefSeq Accession NC_000076.6). Nucleotide insertions are marked in blue, mutations are marked in green and deletions are marked by dashes. c Sirt1 mRNA in WT, LacZ and selected KO clones (321 and 325) was quantified by qPCR. Gapdh was the internal reference control gene. Data were presented as the mean ± SD (NS; not significant, *P < 0.05, **P < 0.01 vs LacZ control). d Whole cell lysates were subjected to Western blot analysis for SIRT1 (120 kDa) expression in WT, LacZ and KO clones (321 and 325). β-actin (44 kDa) was the loading control. Numbers show the SIRT1/β-actin loading control ratio
dCas9/SAM-mediated overexpression of SIRT1 at transcript and protein levels in LS8 cells
We performed real-time PCR to quantify Sirt1 mRNA and analyzed SIRT1 protein expression by Western blot. The clone (LS8Sirt1/over) that most efficiently overexpressed SIRT1 was selected. dCas9 protein was expressed in the selected LS8Sirt1/over clone and LacZ-negative control cells, but not in parent WT cells (Fig. 2a). The schema shows dCas9-SAM target site (Fig. 2b). The target sequence that increased Sirt1 expression is in the promoter region from − 125 to − 106 bp from the Sirt1 transcription start site (TSS). Relative to WT, LS8Sirt1/over showed a 45-fold increase in Sirt1 mRNA levels (Fig. 2c) and at least a 10-fold increase in SIRT1 protein levels (Fig. 2d), whereas neither mRNA nor protein levels increased over WT in the LacZ control cells.

CRISPR/dCas9/SAM-mediated overexpression of SIRT1 in LS8 cells. a Whole cell lysates were subjected to Western blot analysis for dCas9 (160 kDa) expression in WT, LS8Sirt1/over and negative control (LacZ) cells. β-actin (44 kDa) was the loading control. Numbers show the dCas9/β-actin loading control ratio. b The schema shows dCas9-SAM target site (green) in promoter region at -125 bp to -106 bp from the transcription start site (TSS). c Sirt1 mRNA in WT, LS8Sirt1/over and LacZ control cells were quantified by qPCR. B2m was the internal reference control gene. Data were presented as the mean ± SD (*P < 0.05, **P < 0.01). d Whole cell lysates were subjected to Western blot analysis for SIRT1(120 kDa) expression in WT, LS8Sirt1/over and LacZ control cells. α-tubulin (52 kDa) was the loading control. Numbers show the SIRT1/α-tubulin loading control ratio
Fluoride induced Acetyl-p53 (Ac-p53) and Cdkn1a/p21 expression in WT cells in vitro and in rat ameloblasts
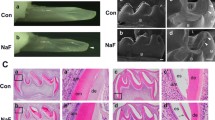
Following DNA damage p53 is acetylated at Lys379 and Ac-p53 regulates cell cycle arrest. We asked if Ac-p53 was increased by fluoride treatment in WT cells. WT cells were treated with 5 mM NaF for indicated times (Fig. 3a). Ac-p53 expression was increased at 2–18 h after fluoride treatment in WT cells. Fluoride also increased Ac-p53 expression after 6 h in a dose-dependent manner (Fig. 3b). Next, we performed immunohistochemistry on rat maxillary incisors. H&E staining clearly identify ameloblasts and Ac-p53 staining was performed on subsequent sections. Compared to control (0 ppm fluoride), Ac-p53 was more highly expressed in rat ameloblasts treated with 100 ppm fluoride for 6 weeks (Fig. 3c). Accumulation of Ac-p53 in nuclei was observed after fluoride treatment. Since Ac-p53 regulates Cdkn1a/p21 expression, we assessed if Cdkn1a/p21 mRNA was increased by fluoride treatment in WT cells. Fluoride treatment did up-regulate Cdkn1a/p21 at 24 h (Fig. 4a) and at 24 h, Cdkn1a/p21 expression increased in a dose-dependent manner (Fig. 4b). These results suggest that following DNA damage, Ac-p53 may contribute to subsequent Cdkn1a/p21-mediated cell cycle arrest as a result of fluoride toxicity.

Fluoride induced acetylation of p53 in WT cells and in rat enamel organ. Whole cell lysates were subjected to Western blot analysis. Acetylated p53 (Ac-p53) and total p53 were detected. β-actin (44 kDa) was the loading control. Numbers show the Ac-p53 or total p53/β-actin loading control ratio. a WT cells were treated with 5 mM fluoride for indicated times. Ac-p53 formation increased from 2 to 18 h after fluoride treatment. b WT cells were treated with fluoride at indicated concentrations for 6 h. Fluoride increased Ac-p53 in a dose-dependent manner. c Rats were treated with 0 or 100 ppm fluoride in their drinking water for 6 weeks. Hematoxylin and eosin (H&E) staining (upper panel) and immunohistochemistry (lower panel) were performed on maturation stage incisor sections. More Ac-p53 (Lys370) was formed in rat ameloblasts treated with 100 ppm fluoride compared to control ameloblasts (0 ppm). Shown are representative images from three rats. Scale bar represents 20 μm. Brackets denote ameloblasts (Am)

Fluoride increased Cdkn1a/p21 expression in WT cells. a Cdkn1a/p21 expression was quantified by qPCR. B2m was the internal reference control gene. WT cells were treated with 5 mM fluoride for indicated times. Fluoride treatment significantly increased Cdkn1a/p21 expression at 24 h. b WT cells were treated with fluoride at indicated concentration for 24 h. Cdkn1a/p21 expression increased in a dose-dependent manner. Data are presented as the mean ± SD (*P < 0.05). A regression analysis was performed and the results demonstrated that fluoride dose dependently induced Cdkn1a/p21 expression (*P < 0.0001)
Acetyl-p53 in WT, LS8Sirt1/KO and LS8Sirt1/over
SIRT1 deacetylates p53 to inhibit p53-dependent apoptosis (Vaziri et al. 2001). To assess if SIRT1 ablation or overexpression alters fluoride-induced Ac-p53 formation, we evaluated by Western blot Ac-p53 formation in WT, LS8Sirt1/KO and LS8Sirt1/over cells. The SIRT1 activator resveratrol (RES) suppressed fluoride-induced Ac-p53 in WT cells (Fig. 5a). Figure 5b shows p53 acetylation status in cells cultured without fluoride treatment. Compared to WT cells, Ac-p53 is constitutively augmented in LS8Sirt1/KO clones (321 and 325), whereas it was diminished in LS8Sirt1/over cells, suggesting that SIRT1 is important for constitutive deacetylation of p53 in LS8 cells. Fluoride treatment for 6 h increased Ac-p53 in LacZ control (LS8LacZ/over) cells, but this increase was suppressed in LS8Sirt1/over cells (Fig. 5c). Compared to control LS8LacZ/over cells, total p53 was decreased in LS8Sirt1/over cells (Fig. 5c), suggesting that deacetylation by SIRT1 decreases p53 protein stability leading to p53 degradation in LS8Sirt1/over cells. Figure 5D shows that fluoride elevated Ac-p53 in control LS8LacZ/KO cells, however Ac-p53 was not significantly altered by fluoride treatment in LS8Sirt1/KO cells. Perhaps hyperacetylation of p53 in LS8Sirt1/KO cells (Fig. 5b) already reached a plateau without fluoride.

Acetylation of p53 in LS8Sirt1/over and LS8Sirt1/KO cells. Whole cell lysates were subjected to Western blot. a WT cells were treated with 5 mM fluoride with or without 50 µM resveratrol (RES) for 6 h. RES suppressed fluoride-induced Ac-p53 formation. b Cultured cell lysates from WT, LS8Sirt1/KO clones (321 and 325) and LS8Sirt1/Over cells were subjected to Western blotting. Compared to WT cells, Ac-p53 formation was increased in LS8Sirt1/KO clones (321 and 325), but was diminished in LS8Sirt1/Over cells. c WT, LS8Sirt1/Over and control LS8LacZ/Over cells were treated with 5 mM fluoride for 6 h. Fluoride-induced Ac-p53 formation was decreased in LS8Sirt1/Over cells compared to control LS8LacZ/Over cells. d WT, LS8Sirt1/KO and control LS8LacZ/KO cells were treated with 5 mM fluoride for 6 h. Ac-p53 formation was elevated in control LS8LacZ/KO cells, but Ac-p53 was not significantly altered by fluoride treatment. α-tubulin (52 kDa) or β-actin (44 kDa) were the loading controls. The numbers show the Ac-p53 or Total p53/loading control ratio
Cdkn1a/p21 expression and cell growth inhibition in LS8Sirt1/KO and LS8Sirt1/over
Next we asked if Cdkn1a/p21 expression is altered in SIRT1 knockout or overexpressing LS8 cells. Cells were treated with 5 mM fluoride for 18 h and Cdkn1a/p21 mRNA was quantified by qPCR. Fluoride significantly increased Cdkn1a/p21 expression in LS8Sirt1/KO (P < 0.05), but did not in WT, LS8Sirt1/over and LacZ control cells (Fig. 6a). Figure 6b shows the net ΔΔCt increase between no treatment and fluoride treatment. Cdkn1a/p21 expression was significantly elevated in LS8Sirt1/KO cells compared to WT or control LS8LacZ/KO cells, whereas there was no significant difference among WT, LS8Sirt1/over and control LS8LacZ/Over cells (Fig. 6b). These results suggest that SIRT1 suppresses Ac-p53-mediated Cdkn1a/p21 expression. But, SIRT1 overexpression did not suppress Cdkn1a/p21 expression more than controls.

Cdkn1a/p21 expression and cell growth inhibition in WT, LS8Sirt1/Over and LS8Sirt1/KO. a WT, LS8Sirt1/Over, LS8Sirt1/KO, LACZ control LS8LacZ/Over and LS8LacZ/KO cells were treated with 5 mM fluoride for 18 h. Cdkn1a/p21 expression was quantified by qPCR. Fluoride significantly increased Cdkn1a/p21 expression in LS8Sirt1/KO, but not in WT, LS8Sirt1/Over and LacZ control cells. Geometric means of B2m and Gapdh were used for normalization. b Net ΔΔCt increase was calculated [(ΔΔCt fluoride treated) minus (ΔΔCt non-fluoride treated)]. Cdkn1a/p21 expression was significantly elevated in LS8Sirt1/KO cells compared to WT or control cells, but there was no significant difference among WT, LS8Sirt1/Over and control cells. Data were presented as the mean ± SD. Multiple group comparison was performed by one-way ANOVA with Bonferroni/Dunn post hoc test. *P < 0.05. NS not significant. c WT (open square) vs LS8Sirt1/KO (filled square) cells and d and WT (open square) vs LS8Sirt1/over (filled square) cells were seeded into 96-well plates and treated with NaF at the indicated concentrations for 24 h. Cell proliferation percentage was measured by MTT assays. Fluoride-induced cell growth inhibition was enhanced in LS8Sirt1/KO cells, but was mitigated in LS8Sirt1/over cells. Four wells were assayed for each experimental treatment and six separate experiments were performed. Data are expressed as mean ± SE. Differences between WT vs LS8Sirt1/KO, or WT vs LS8Sirt1/over were analyzed by Student’s t test. *P < 0.05, **P < 0.01
Next we investigated how fluoride effected cell growth in WT, LS8Sirt1/KO and LS8Sirt1/over cells. Cells were treated with fluoride at the indicated concentrations for 24 h and cell growth ratios were evaluated by MTT assays. Compared to WT, LS8Sirt1/KO cells were more susceptible to 1 mM (P < 0.01) and 3 mM (P < 0.05) fluoride-induced cell growth inhibition (Fig. 6c). Conversely, LS8Sirt1/over cells were resistant to 2 mM and 2.5 mM fluoride-induced cell growth inhibition (P < 0.05) when compared to WT (Fig. 6d). These results suggest that SIRT1 protects LS8 cells from fluoride-mediated cell growth inhibition.
SIRT1 overexpression attenuated fluoride-induced mitochondrial damage, apoptosis and DNA damage
Next we assessed if SIRT1 overexpression mitigates fluoride-induced cytochrome-c release and caspase-3 activation. WT and LacZ negative control cells treated with 5 mM fluoride for 18 h increased cytochrome-c release into the cytosol fraction and reduced cytochrome-c levels in the mitochondrial fraction. In contrast, cytochrome-c release from mitochondria into the cytosol fraction was suppressed in fluoride treated LS8Sirt1/over cells (Fig. 7a). Fluoride treatment for 18 h induced caspase-3 cleavage (activation) in WT and LacZ negative control cells and this was suppressed in LS8Sirt1/over cells (Fig. 7b). These results suggest that augmentation of SIRT1 expression protects LS8 cells from fluoride-induced mitochondrial damage and apoptosis.

Fluoride-induced mitochondrial damage, apoptosis and DNA damage were ameliorated in LS8Sirt1/over cells. WT, LS8Sirt1/over, LS8Sirt1/KO or control LacZ cells were treated with NaF (0 or 5 mM) for 18 h. Whole cell lysates were subjected to Western blot analysis. a Cytochrome-c (Cyto-c, 12 kDa) from the cytosol fraction or from the mitochondrial fraction were detected by Western blots. Fluoride-induced Cyto-c release to cytosol was suppressed in LS8Sirt1/over cells. β-actin (44 kDa) was the cytosol loading control and VDAC/porin (31 kDa) was the mitochondrial loading control protein. The numbers show the Cyto-c/loading control ratio. b Cleaved caspase-3 (19 kDa) was assessed. Fluoride-induced caspase-3 (Casp3) cleavage was suppressed in LS8Sirt1/over cells compared to WT and LacZ control cells. The numbers show the cleaved Casp3/α-tubulin loading control ratio. c DNA double strand break marker γH2AX (17 kDa) was assessed. Fluoride significantly elevated γH2AX formation in WT, but γH2AX induction was attenuated in LS8Sirt1/over cells and was substantially augmented in LS8Sirt1/KO cells. α-tubulin (52 kDa) was the loading control. The numbers show the γH2AX/α-tubulin loading control ratio
γH2AX is a surrogate marker of DNA double strand breaks (DSB) and the presence of prolonged high levels of γH2AX indicate that DNA repair mechanisms are impaired (Bourton et al. 2011). To explore the effects of SIRT1 function on fluoride-induced DNA damage, γH2AX induction was assessed in WT, LS8Sirt1/over and LS8Sirt1/KO cells. Fluoride treatment for 18 h significantly elevated γH2AX in WT, whereas the γH2AX induction was attenuated in LS8Sirt1/over cells and was substantially augmented in LS8Sirt1/KO cells (Fig. 7c). These results suggest that SIRT1 plays a protective role against fluoride-induced DNA damage in LS8 cells.
Discussion
We used pharmacological activators or inhibitors to characterize SIRT1 down-stream pathways. However, studies have reported that resveratrol (RES) directly potentiates SIRT1 activity (Kaeberlein et al. 2005), whereas other studies suggest that RES is not a direct activator of SIRT1 (Pacholec et al. 2010). Therefore, in addition to pharmacological studies, genetic strategies are also important to evaluate if modulation of SIRT1 pathways can account for resistance to toxicity. In this study, we established LS8Sirt1/KO cells and LS8Sirt1/over cells by CRISPR/Cas9 and CRISPR/dCas9/SAM, respectively, to determine if SIRT1 is directly responsible for mitigating fluoride toxicity.
Previously, we demonstrated fluoride-induced DNA damage accompanied ROS generation (Suzuki et al. 2015). Following DNA damage, p53 is acetylated at Lys379 (Sakaguchi et al. 1998) and Ac-p53 regulates cell cycle arrest via CDKN1A/p21 and apoptosis (Bao et al. 2016). However, p53 function and the molecular mechanisms in fluoride-induced DNA damage responses are not clearly delineated. Here, we identified for the first time that fluoride treatment increased acetylation of p53 in LS8 cells in vitro and in ameloblasts in vivo (Fig. 3), and fluoride increased the expression of Ac-p53 target Cdkn1a/p21 in LS8 cells (Fig. 4). These results suggest that fluoride-mediated DNA damage elicits acetylation of p53 to increase Cdkn1a/p21 expression resulting in cell growth inhibition. Six acetyltransferases including p300/CBP/PCAF (Wang et al. 2003) and Tip60/MOF/MOZ (Rokudai et al. 2013) have been identified that acetylate p53. SIRT1 deacetylates p53 to inhibit p53-dependent apoptosis (Vaziri et al. 2001). Without fluoride treatment, greater than normal Ac-p53 formation was observed in LS8Sirt1/KO cells, whereas Ac-p53 was diminished in LS8Sirt1/over cells (Fig. 5b). This indicates that SIRT1 plays a pivotal role in homeostatic p53 deacetylation. Fluoride-induced Cdkn1a/p21 expression was augmented in LS8Sirt1/KO cells (Fig. 6a, b) and fluoride-induced cell growth inhibition was enhanced in LS8Sirt1/KO cells, but was mitigated in LS8Sirt1/over cells (Fig. 6c, d). These data suggest that SIRT1-mediated p53 deacetylation is critical to alleviate p53-dependent cell growth inhibition during fluoride toxicity.
Fluoride induced cytochrome-c release in WT and in LacZ negative control cells, but this was suppressed in LS8Sirt1/over cells (Fig. 7a), suggesting that SIRT1 augmentation protects LS8 cells from fluoride-induced mitochondrial damage. Recent studies demonstrated that SIRT1 promotes mitochondrial biogenesis by deacetylation of target proteins such as PGC-1α (Wenz 2013) and HIF-1α (Gomes et al. 2013). These findings suggest that PGC-1α and HIF-1α may be potential SIRT1 targets to protect ameloblasts from fluoride-induced mitochondrial damage.
Previous studies have highlighted a unique feature of SIRT1 in regulating DNA damage repair and genomic stability (Rajendran et al. 2011). Upon genotoxic stress, SIRT1 moves from silent promoters to sites of DNA damage to deacetylate histones H1 (Lys26) and H4 (Lys16) and contribute to the recruitment of DNA damage factors. After DNA double strand break formation, γH2AX contributes to DNA damage repair. On the other hand, prolonged γH2AX expression impairs DNA repair (Bourton et al. 2011) and represents an apoptotic signal (Ding et al. 2016). Fluoride-induced γH2AX levels were decreased relative to controls in LS8Sirt1/over cells (Fig. 7c). This result is in concordance with a previous study showing that pharmacological SIRT1 activation reduced γH2AX levels and protected neurons against in vivo DNA damage (Dobbin et al. 2013). For LS8Sirt1/KO cells, fluoride-induced γH2AX expression was increased compared to WT, suggesting that SIRT1 deletion impairs DNA repair mechanisms (Fig. 7c). Alternatively, it was reported that SIRT1 ablation in mouse embryonic fibroblasts led to defective γH2AX formation that caused DNA repair failure (Wang et al. 2008). In Sirt1-deleted neurons, despite elevated amounts of DNA damage, etoposide-induced γH2AX was decreased (Dobbin et al. 2013). This result is somewhat different from our result. However, this discrepancy could be because fluoride causes DNA damage indirectly through ROS generation and/or caspase activation or it could be because SIRT1’s contribution to DNA damage repair depends on the kind of stress and/or cell/tissue types.
Conclusion
This is the first report implicating acetylation of p53 followed by Cdkn1a/p21 expression in fluoride toxicity. Our results support the hypothesis that SIRT1 plays a role in the adaptive response protecting against fluoride toxicity in ameloblast-derived cells. The SIRT1/p53 pathway may be a potential therapeutic target for dental fluorosis.
References
Adedara IA, Abolaji AO, Idris UF et al (2016) Neuroprotective influence of taurine on fluoride-induced biochemical and behavioral deficits in rats. Chem Biol Interact 261:1–10. https://doi.org/10.1016/j.cbi.2016.11.011
Aoba T, Fejerskov O (2002) Dental fluorosis: chemistry and biology. Crit Rev Oral Biol Med 13(2):155–170
Bao L, Diao H, Dong N et al (2016) Histone deacetylase inhibitor induces cell apoptosis and cycle arrest in lung cancer cells via mitochondrial injury and p53 up-acetylation. Cell Biol Toxicol. https://doi.org/10.1007/s10565-016-9347-8
Beltran-Aguilar ED, Barker L, Dye BA (2010) Prevalence and severity of dental fluorosis in the United States, 1999–2004. NCHS Data Brief (53):1–8
Bernier M, Paul RK, Martin-Montalvo A et al (2011) Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein. J Biol Chem 286(22):19270–19279. https://doi.org/10.1074/jbc.M110.200311
Boivin G, Chavassieux P, Chapuy MC, Baud CA, Meunier PJ (1989) Skeletal fluorosis: histomorphometric analysis of bone changes and bone fluoride content in 29 patients. Bone 10(2):89–99
Bourton EC, Plowman PN, Smith D, Arlett CF, Parris CN (2011) Prolonged expression of the gamma-H2AX DNA repair biomarker correlates with excess acute and chronic toxicity from radiotherapy treatment. Int J Cancer J Int du cancer 129(12):2928–2934. doi:https://doi.org/10.1002/ijc.25953
Braunstein M, Sobel RE, Allis CD, Turner BM, Broach JR (1996) Efficient transcriptional silencing in Saccharomyces cerevisiae requires a heterochromatin histone acetylation pattern. Mol Cell Biol 16(8):4349–4356
Chen LS, Couwenhoven RI, Hsu D, Luo W, Snead ML (1992) Maintenance of amelogenin gene expression by transformed epithelial cells of mouse enamel organ. Arch Oral Biol 37(10):771–778
Cong L, Ran FA, Cox D et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823. doi:https://doi.org/10.1126/science.1231143
Conrad E, Polonio-Vallon T, Meister M et al (2016) HIPK2 restricts SIRT1 activity upon severe DNA damage by a phosphorylation-controlled mechanism. Cell Death Differ 23(1):110–122. doi:https://doi.org/10.1038/cdd.2015.75
DenBesten PK (1999) Biological mechanisms of dental fluorosis relevant to the use of fluoride supplements. Community Dent Oral Epidemiol 27(1):41–47
Ding D, Zhang Y, Wang J et al (2016) Induction and inhibition of the pan-nuclear gamma-H2AX response in resting human peripheral blood lymphocytes after X-ray irradiation. Cell Death Discov 2:16011. doi:https://doi.org/10.1038/cddiscovery.2016.11
Dobbin MM, Madabhushi R, Pan L et al (2013) SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat Neurosci 16(8):1008–1015. https://doi.org/10.1038/nn.3460
Dutta M, Rajak P, Khatun S, Roy S (2017) Toxicity assessment of sodium fluoride in Drosophila melanogaster after chronic sub-lethal exposure. Chemosphere 166:255–266. https://doi.org/10.1016/j.chemosphere.2016.09.112
Gomes AP, Price NL, Ling AJ et al (2013) Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155(7):1624–1638. https://doi.org/10.1016/j.cell.2013.11.037
He M, Yuan H, Tan B et al. (2016) SIRT1-mediated downregulation of p27Kip1 is essential for overcoming contact inhibition of Kaposi. Oncotarget https://doi.org/10.18632/oncotarget.12359
Health, USDo, Human Services Federal Panel on Community Water F (2015) US Public Health Service Recommendation for Fluoride Concentration in Drinking Water for the Prevention of Dental Caries. Public Health Rep 130(4):318–331 doi:https://doi.org/10.1177/003335491513000408
Imai S, Johnson FB, Marciniak RA, McVey M, Park PU, Guarente L (2000) Sir2: an NAD-dependent histone deacetylase that connects chromatin silencing, metabolism, and aging. Cold Spring Harb Symp Quant Biol 65:297–302
Kaeberlein M, McDonagh T, Heltweg B et al (2005) Substrate-specific activation of sirtuins by resveratrol. J Biol Chem 280(17):17038–17045. https://doi.org/10.1074/jbc.M500655200
Konermann S, Brigham MD, Trevino AE et al (2015) Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517(7536):583–588. https://doi.org/10.1038/nature14136
Kubota K, Lee DH, Tsuchiya M et al (2005) Fluoride induces endoplasmic reticulum stress in ameloblasts responsible for dental enamel formation. J Biol Chem 280(24):23194–23202. https://doi.org/10.1074/jbc.M503288200
Pacholec M, Bleasdale JE, Chrunyk B et al (2010) SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem 285(11):8340–8351. https://doi.org/10.1074/jbc.M109.088682
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9):e45
Poulose N, Raju R (2015) Sirtuin regulation in aging and injury. Biochim Biophys Acta 1852(11):2442–2455. https://doi.org/10.1016/j.bbadis.2015.08.017
Rajendran P, Ho E, Williams DE, Dashwood RH (2011) Dietary phytochemicals, HDAC inhibition, and DNA damage/repair defects in cancer cells. Clin Epigenetics 3(1):4. https://doi.org/10.1186/1868-7083-3-4
Reed SM, Quelle DE (2014) p53 Acetylation: Regulation and Consequences. Cancers (Basel) 7(1):30–69. https://doi.org/10.3390/cancers7010030
Rokudai S, Laptenko O, Arnal SM, Taya Y, Kitabayashi I, Prives C (2013) MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc Natl Acad Sci USA 110(10):3895–900. https://doi.org/10.1073/pnas.1300490110
Sakaguchi K, Herrera JE, Saito S et al (1998) DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev 12(18):2831–2841
Salminen A, Kaarniranta K, Kauppinen A (2013) Crosstalk between oxidative stress and SIRT1: impact on the aging process. Int J Mol Sci 14(2):3834–3859. https://doi.org/10.3390/ijms14023834
Shalem O, Sanjana NE, Hartenian E et al (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343(6166):84–87. https://doi.org/10.1126/science.1247005
Sharma R, Tsuchiya M, Bartlett JD (2008) Fluoride induces endoplasmic reticulum stress and inhibits protein synthesis and secretion. Environ Health Perspect 116(9):1142–1146. https://doi.org/10.1289/ehp.11375
Sm S, Mahaboob Basha P (2016) Fluoride exposure aggravates the testicular damage and sperm quality in diabetic mice: protective role of Ginseng and Banaba. Biol Trace Elem Res. https://doi.org/10.1007/s12011-016-0893-y
Sun Z, Niu R, Wang B et al (2011) Fluoride-induced apoptosis and gene expression profiling in mice sperm in vivo. Arch Toxicol 85(11):1441–1452. https://doi.org/10.1007/s00204-011-0672-7
Suzuki M, Shin M, Simmer JP, Bartlett JD (2014a) Fluoride affects enamel protein content via TGF-beta1-mediated KLK4 inhibition. J Dent Res 93(10):1022–1027. https://doi.org/10.1177/0022034514545629
Suzuki M, Sierant ML, Antone JV, Everett ET, Whitford GM, Bartlett JD (2014b) Uncoupling protein-2 is an antioxidant that is up-regulated in the enamel organ of fluoride-treated rats. Connect Tissue Res 55(1):25–28. https://doi.org/10.3109/03008207.2014.923854
Suzuki M, Bandoski C, Bartlett JD (2015) Fluoride induces oxidative damage and SIRT1/autophagy through ROS-mediated JNK signaling. Free Radic Biol Med 89:369–378. https://doi.org/10.1016/j.freeradbiomed.2015.08.015
Suzuki M, Everett ET, Whitford GM, Bartlett JD (2017) 4-phenylbutyrate Mitigates Fluoride-Induced Cytotoxicity in ALC Cells. Front Physiol 8:302. https://doi.org/10.3389/fphys.2017.00302
Vaziri H, Dessain SK, Ng Eaton E et al (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107(2):149–159
Wang YH, Tsay YG, Tan BC, Lo WY, Lee SC (2003) Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J Biol Chem 278(28):25568–25576. https://doi.org/10.1074/jbc.M212574200
Wang RH, Sengupta K, Li C et al (2008) Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 14(4):312–23. https://doi.org/10.1016/j.ccr.2008.09.001
Wenz T (2013) Regulation of mitochondrial biogenesis and PGC-1alpha under cellular stress. Mitochondrion 13(2):134–142. https://doi.org/10.1016/j.mito.2013.01.006
Acknowledgements
We thank Dr. Malcolm L. Snead for generously providing us with LS8 cells, Dr. Atsuko Ogino for her expert advice on CRISPR/Cas9 techniques and Addgene for providing us with vectors. Research reported in this publication was supported by the National Institute of Dental and Craniofacial Research of the National Institutes of Health under award number R01DE018106 (JDB), and was supported by a Seed Grant from The Ohio State University, College of Dentistry under award number 21-100300 (MS).
Author information
Authors and Affiliations
Contributions
MS, contributed to conception, design, data acquisition, analysis, interpretation and draft of the manuscript; AI, contributed to data acquisition, analysis and interpretation; JDB, contributed to design, data interpretation and editing of the manuscript text. All authors critically revised the manuscript, gave final approval and agree to be accountable for all aspects of this work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Suzuki, M., Ikeda, A. & Bartlett, J.D. Sirt1 overexpression suppresses fluoride-induced p53 acetylation to alleviate fluoride toxicity in ameloblasts responsible for enamel formation. Arch Toxicol 92, 1283–1293 (2018). https://doi.org/10.1007/s00204-017-2135-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-017-2135-2