
Abstract
BAL30072 is a new monocyclic β-lactam antibiotic under development which provides a therapeutic option for the treatment of severe infections caused by multi-drug-resistant Gram-negative bacteria. Despite the absence of liver toxicity in preclinical studies in rats and marmosets and in single dose clinical studies in humans, increased transaminase activities were observed in healthy subjects in multiple-dose clinical studies. We, therefore, initiated a comprehensive program to find out the mechanisms leading to hepatocellular injury using HepG2 cells (human hepatocellular carcinoma cell line), HepaRG cells (inducible hepatocytes derived from a human hepatic progenitor cell line), and human liver microtissue preparations. Our investigations demonstrated a concentration- and time-dependent reduction of the ATP content of BAL30072-treated HepG2 cells and liver microtissues. BAL30072 impaired oxygen consumption by HepG2 cells at clinically relevant concentrations, inhibited complexes II and III of the mitochondrial electron transport chain, increased the production of reactive oxygen species (ROS), and reduced the mitochondrial membrane potential. Furthermore, BAL 30072 impaired mitochondrial fatty acid metabolism, inhibited glycolysis, and was associated with hepatocyte apoptosis. Co-administration of N-acetyl-l-cysteine partially protected hepatocytes from BAL30072-mediated toxicity, underscoring the role of oxidative damage in the observed hepatocellular toxicity. In conclusion, BAL30072 is toxic for liver mitochondria and inhibits glycolysis at clinically relevant concentrations. Impaired hepatic mitochondrial function and inhibition of glycolysis can explain liver injury observed in human subjects receiving long-term treatment with this compound.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Drug-induced liver injury is a major problem in all phases of drug development, representing a significant challenge for patients, clinicians, the pharmaceutical industry, and regulatory agencies (Kaplowitz 2005). It is the most common reason for post-marketing warnings or withdrawal of an approved drug (Kaplowitz 2005; Temple and Himmel 2002). Since preclinical studies in animals cannot always reliably predict DILI in humans, close monitoring during clinical studies and post-marketing surveillance are critical for avoiding damage to patients, with ALT or alkaline phosphatase levels >5 upper limits of normal (ULN) or >2 ULN, respectively, proposed as a signal for concern (Aithal et al. 2011).
BAL30072 is a new chemical entity, a sulfactam belonging to the monocyclic β-lactam group of antibiotics with a molecular weight of 518 Da (see Fig. 1). The compound is currently under development for the treatment of severe infections caused by multi-drug-resistant (MDR) Gram-negative pathogens, especially P. aeruginosa and Acinetobacter spp., in hospitalized patients (Page et al. 2010).

Chemical structure of BAL30072 and BAL104936
So far unpublished phase I studies by Basilea revealed that BAL30072 has a dose-proportional pharmacokinetics with a half-life in the range of 1.5–2 h, a volume distribution of approximately 13 L, and a serum protein binding of 49%. Approximately 45–50% of a single dose was recovered in urine within 24 h. During an infusion of a daily dose of 3 g of BAL30072 over 22 h, stable plasma concentrations were reached which averaged 30.5 mg/L (total) and 22.2 mg/L (free drug concentration). The metabolism occurs mainly by opening of the β-lactam ring, leading to BAL104936 (see Fig. 1). BAL104936 contributed 20–30% to total systemic exposure independent of the BAL30072 dose administered and approximately 12% of the BAL30072 dose could be recovered as BAL104936 in the urine.
Single doses up to 8 g of BAL30072 in healthy subjects were well tolerated, with no serious adverse events reported. Multiple doses of 1 g BAL30072 administered as 1 h i.v. infusions every 8 h (total daily dose 3 g) were also well tolerated. Unexpectedly, in a multiple-dose study with 2 g given as 1 h i.v. infusions every 8 h (total daily dose 6 g), ALT and AST values exceeding the upper limit of normal were observed in 6/6 healthy male subjects. In a subsequent study with multiple doses of 4 g BAL30072 administered as 22 h infusions, relevant transaminase elevations were observed in 5/6 subjects. Since no abnormalities were observed in serum levels of bilirubin, the Hy’s law criteria for severe drug-induced liver injury (DILI) were not met in any subject (Reuben 2004).
Pharmacokinetics and metabolism of BAL30072 were similar in animals and humans. No signs of liver toxicity had been observed in preclinical safety studies in rats and marmosets at intravenous doses up to 1 and 0.8 g/kg/day, respectively, for 4 weeks. Other signs of toxicity in these studies were generally mild and reversible, and limited to the highest dose administered. They included soft feces, anemia, deposition of pigments in proximal renal tubules and Kupffer cells, and minor signs of local irritation at the injection site.
In consideration of the diverse findings regarding hepatotoxicity between the preclinical and clinical studies, we decided to initiate a comprehensive program to investigate the mechanism of the observed transaminase elevations to improve the hepatic safety of this drug in patients. Taking into account the main mechanisms of hepatotoxicity postulated by Lee (2003), the program included toxicity associated with the production of active metabolites, assessment of the interaction of BAL30072 with hepatocyte drug transporting proteins, toxicity associated with activation of Kupffer cells, mitochondrial toxicity, and studies about the mechanism of cell death. The studies were carried out using HepG2 cells (human hepatocellular carcinoma cell line), HepaRG cells (terminally differentiated hepatic cells derived from a human hepatic progenitor cell line), and 3-dimensional (3D) human liver microtissue preparations.
The investigations revealed that BAL30072 inhibited glycolysis and both BAL30072 and BAL104936 were mitochondrial toxicants starting at concentrations that can possibly be reached in patients.
Materials and methods
Chemicals
BAL30072 (see Fig. 1a) and BAL104936 (the ring-opened product of BAL30072, see Fig. 1b) were prepared in the research laboratories of Basilea Pharmaceutica International Ltd (Basel, Switzerland). Because of the limited stability of BAL30072 in solution, the drug was re-dosed every 12 h in all experiments, unless otherwise indicated. All other chemicals were supplied by Sigma-Aldrich (Buchs, Switzerland), unless otherwise indicated.
Clinical study
In the context of a large phase I study program, a cohort of six healthy male subjects were treated in a multiple-dose study with 4 g per day of BAL30072 (n = 5 subjects) or placebo (n = 1 subject) infused intravenously over 22 h for 6 days. Blood samples were obtained daily before the start of the infusion for the determination of transaminases, alkaline phosphatase, and bilirubin, using routine methods of clinical chemistry. For the determination of the serum bile acids, blood plasma samples were obtained from patients in the fasted state every 2 days. Total bile acids were determined enzymatically as described previously (Taegtmeyer et al. 2014).
Mechanistic biomarkers of hepatotoxicity were quantified every second day. Levels of total HMGB1 [cell death not specific for hepatocytes; (Yang et al. 2013)], liver-specific miRNA122 [hepatocyte breakdown; (Bala et al. 2012)], as well as total caspase and cleaved cytokeratin 18 [cell death not specific for hepatocytes and apoptosis, respectively; (Yilmaz 2009)] were also assessed. Quantification of HMGB1 was carried out as described by the manufacturer (IBL International; Ontario, Canada), but with slight modifications. Two additional low standards (1.25 and 0.625 ng/mL) were added to the “normal range” curve and measurements were made on a SpectraMax plate reader (Sunnyvale, CA). miRNA122 was measured in RNA extracted from serum using the miRNeasy serum/plasma kit (Qiagen, Valencia, CA) following the manufacturer’s protocol. Spike-in caenorhabditis elegans miRNA39 (Qiagen) was added during isolation to control the extraction efficiency. Total RNA was reverse transcribed utilizing a Taqman miRNA Reverse Transcription Kit and miRNA-specific primer sets (Life Technologies, Foster City, CA) according to the manufacturer’s specifications, in parallel with standard curves created with synthetic miRNA122 and miRNA39 (Qiagen). Absolute quantitative real-time PCR (qRT-PCR) was performed utilizing TaqMan Universal PCR Master Mix II (no Uracil N-Glycosylase) and miRNA-specific Taqman assays (Life Technologies) on an Applied Biosystems 7900HT RT-PCR system (Life Technologies). Normalization for extraction efficiency was conducted as previously described (Kroh et al. 2010).
Cytokeratin-18 measurements were performed with ELISA kits (M30 CytoDeath™ and M65 EpiDeath® ELISA; Peviva), according to the manufacturer’s instructions.
Cell culture
The human hepatocellular carcinoma cell line HepG2 was provided by American type culture collection (ATCC, Manassas, VA, USA). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, containing 1 g/L glucose, 4 mM l-glutamine, and 1 mM pyruvate) from Invitrogen (Basel, Switzerland) supplemented with 10% (v/v) heat-inactivated fetal bovine serum, 2 mM GlutaMax, 10 mM HEPES buffer, 10 mM non-essential amino acids, 100 units/mL penicillin, and 100 μg/mL streptomycin.
The HepaRG cell line—a terminally differentiated human hepatic progenitor cell line—was provided by Biopredic International (Saint-Gregoire, France). Cells were cultured and differentiated as described (Aninat et al. 2006). Cells were first seeded in 96-well plates (10,000 cells/well) and grown for 2 weeks in culture medium [William’s E Medium, Invitrogen (Basel, Switzerland) supplemented with 10% (v/v) fetal bovine serum (not inactivated by heat), 2 mM l-glutamine, 5 μg/mL bovine insulin, 50 μM hemisuccinate, 100 units/mL penicillin, and 100 μg/mL streptomycin]. For differentiation, cells were cultured for 2 weeks in culture medium supplemented with 2% DMSO. Induction of different CYPs was achieved by pre-incubation of differentiated HepaRG cells with 20 μM rifampicin for 72 h (Berger et al. 2016).
All cells were kept at 37 °C in a humidified 5% CO2 cell culture incubator and passaged using trypsin.
3D human liver microtissue preparation
Cryopreserved primary human hepatocytes (lot IZT) and non-parenchymal cells (NPCs) were obtained from BioreclamationIVT. A combination of hepatocytes and NPCs were seeded in a 96-well hanging drop culture platform (GravityPLUS™, InSphero AG, Schlieren, Switzerland) in liver microtissue re-aggregation medium (InSphero AG). After microtissue formation, the resulting 3D human liver microtissues were transferred to a spheroid-specific 96-well microtissue receiver plate (GravityTRAP™, InSphero AG). 3D human liver microtissues were further cultured in the GravityTRAP™ plates with a tissue-specific serum-free maintenance medium (#CS-07-001-01, InSphero AG) in a culture volume of 50 µL per well. The 3D human liver microtissues obtained contain approximately 1000 hepatocytes per microtissue and incorporated functional Kupffer cells (Messner et al. 2013).
Intracellular ATP content
The intracellular ATP content, a marker for mitochondrial function and cell viability, was determined using the CellTiter-Glo® kit from Promega (Madison, USA) following the manufacturer’s instructions. Cells were grown in 96-well plates or GravityTRAP™ plates and exposed to a range of BAL30072 or paracetamol concentrations. Triton X (0.5%) was used as a positive control. The same amount of assay buffer was added to each well containing culture medium. After incubation in the dark for 15 min, luminescence was measured using a Tecan M200 Pro Infinity plate reader (Männedorf, Switzerland).
3D human liver microtissue albumin production
The concentration of human albumin in supernatants collected from microtissue cultures (after medium replenishment every 12 h) was determined using the human Albumin ELISA Quantification Set (E80-129/E101, Bethyl Laboratories Inc., Montgomery, USA), according to the manufacturer’s instructions.
Glutathione content
Glutathione (GSH) content was measured by the GSH/GSSG-Glo™ Glutathione assay (Promega, Madison, USA) according to the manufacturer’s instructions. Human liver microtissues were pre-treated for 40 h with or without 100 μM buthionine-sulphoximine (BSO) and further cultured with or without 100 μM BSO in the presence of BAL30072.
After exposure to BAL30072, the cell culture medium was removed and 50 μL of Total Glutathione Lysis Reagent was added to each 96-well plate and transferred into a white half-area assay plate. The plate was shaken at room temperature for 15 min before addition of 50 µl Luciferin Generation Reagent and then further incubated on a shaker for 30 min before addition of 100 µL Luciferin Detection Reagent. After shaking for another 15 min, luminescence was recorded on a Tecan M200 Pro Infinity plate reader (Männedorf, Switzerland).
Toxicity associated with stimulation of Kupffer cells
Kupffer cell stimulation was performed with lipopolysaccharide (LPS) (#L6529, Sigma-Aldrich, St. Louis, USA). 3D human liver microtissues were incubated in culture medium with or without 10 µg/mL LPS for 120 h with ascending BAL30072 concentrations. 50% of the medium was replenished every 12 h. Cellular ATP content was assessed with the CellTiter-Glo® ATP-assay and normalized to controls.
In vitro efflux- and uptake transporter study
The transport experiments were conducted by Xenotech according to published protocols (Xenotech.com/services/drug-transporters). Individual data points were determined in triplicate.
The ability of BAL30072 (up to 3700 µM) to inhibit human efflux or uptake transporters was determined by assessing the effect of BAL30072 on the transport of probe substrates by the respective transporting proteins expressed in suitable cell lines. Results are expressed as the IC50 values.
To determine whether BAL30072 (up to 100 µM) is a substrate of human efflux transporters, the bidirectional permeability of BAL30072 across MDCKII cells expressing the specific transporter was determined. A ratio >2 of the transport activities (higher activity/lower activity of the bidirectional transport experiments) was regarded as an indication for active transport.
Uptake experiments were performed by measuring the transport of BAL30072 into cell lines expressing a specific transporter and corresponding mock-transfected cells. A ratio of the transport activity >2 (transfected cells/mock-transfected cells) was regarded as an indication for active transport.
Oxygen consumption and extracellular acidification rate by HepG2 cells
Cellular respiration in intact cells was measured with a Seahorse XF24 analyzer (Seahorse Biosciences, North Billerica, MA, USA) as described previously (Felser et al. 2013). HepG2 cells were seeded in Seahorse XF 24-well culture plates at 100,000 cells/well in DMEM growth medium and then incubated for 1 or 72 h with BAL30072 or BAL104936. Before assessing the cellular respiration, the medium was replaced with 750 μL unbuffered DMEM medium (4 mM l-glutamate, 1 mM pyruvate, 1 g/L glucose, 63.3 mM sodium chloride, pH 7.4) and equilibrated at 37 °C in a CO2-free incubator for at least 30 min. Plates were transferred to the XF24 analyzer. Basal oxygen consumption rate (OCR) was determined in the presence of glutamate/pyruvate (4 and 1 mM, respectively). The oxidative leak was determined after inhibition of the mitochondrial phosphorylation by adding 1 μM oligomycin. The mitochondrial electron transport chain was stimulated maximally by the addition of 2 μM FCCP, and the extra-mitochondrial respiration was determined after the addition of complex I inhibitor rotenone (1 μM). To determine the basal respiration, oxidative leak, and maximum respiration, the extra-mitochondrial respiration was subtracted.
The extracellular acidification rate (ECAR), which approximates glycolysis, was determined in the same incubations as used for oxygen consumption rate as described by Zhang et al. (2012).
Activity of specific enzyme complexes of the mitochondrial electron transport chain in HepG2 cells
The activity of specific enzyme complexes of the respiratory chain was analyzed using an Oxygraph-2 k-high-resolution respirometer equipped with DataLab software (Oroboros instruments, Innsbruck, Austria) (Felser et al. 2013). HepG2 cells were treated for 72 h with BAL30072, suspended in MiR05 (mitochondrial respiration medium containing 0.5 mM EGTA, 3 mM magnesium chloride, 20 mM taurine, 10 mM potassium dihydrogen phosphate, 20 mM HEPES, 110 mM sucrose, 1 g/L fatty acid free bovine serum albumin, and 60 mM lactobionic acid, pH 7.1) and transferred to the pre-calibrated Oxygraph chamber (Pesta and Gnaiger 2012).
Respiratory capacities through complexes I, II, III, and IV were assessed in HepG2 cells permeabilized with digitonin (10 μg/million cells). Complexes I and III were analyzed using l-glutamate/malate (10 and 2 mM, respectively) as substrates followed by the addition of adenosine diphosphate (ADP; 2.5 mM) and rotenone (0.5 μM) as inhibitor of complex I. Duroquinol (500 μM) was then added to investigate complex III.
Complexes II and IV were analyzed using succinate/rotenone (10 mM and 0.5 μM, respectively) as substrates, followed by the addition of ADP (2.5 mM) and the complex III inhibitor antimycin A (2.5 μM). N,N,N′,N′-tetramethyl-1,4-phenylendiamine (TMPD)/ascorbate (0.5 and 2 mM, respectively) was added to investigate complex IV.
The integrity of the outer mitochondrial membrane was confirmed by the absence of a stimulatory effect of exogenous cytochrome c (10 μM) on respiration, expressed as oxygen consumption per mg protein.
Accumulation of mitochondrial reactive oxygen species in HepG2 cells
Generation of mitochondrial reactive oxygen species (ROS) was assessed using MitoSOX Red (Invitrogen, Basel, Switzerland). 50 μM amiodarone was used as a positive control. After the treatment of HepG2 cells (duration up to 120 h), cell culture medium was removed, and 2.5 μM MitoSOX dissolved in DPBS was added. After incubation for 10 min at 37 °C in the dark, fluorescence was measured (excitation 510 nm, emission 580 nm) using a Tecan M200 Pro Infinity plate reader (Männedorf, Switzerland).
Lactate production
Lactate production was measured with the Lactate Assay Kit (MAK064, Sigma-Aldrich, St. Louis, USA) using an enzymatic reaction. Supernatants from cell cultures or from human liver microtissues from 12 h exposures were harvested, with 5 µL subjected to the enzymatic reaction. The resulting fluorescent signal was calculated according to the standard curve of the Lactate Assay Kit.
3D human liver microtissue histology
Human liver microtissues were harvested at defined endpoints, washed with PBS-buffer, and fixed with 4% paraformaldehyde for 2 h at room temperature. Specimens were further paraffinized in a histo-kineter. Formalin-fixed paraffin-embedded sections were stained with hematoxylin and eosin (H and E).
Lipid accumulation in HepG2 cells
Using the method developed by Donato and colleagues (2009), HepG2 cells were exposed for 24 h to exogenous lipids (DMEM medium containing 41.3 μM oleate and 20.7 μM palmitate). HepG2 cells were then treated with BAL30072 in lipid-free medium for 72 h. The intracellular lipid accumulation was measured using 3.75 ng/mL BODIPY 493/503, a non-polar derivative of the BODIPY fluorophore (Donato et al. 2009). After treatment, cells were detached and washed with lipid-free medium. The cell suspension was stained for 30 min at 37 °C in the dark with PBS containing BODIPY and 2.5 μg/mL propidium iodide (PI). PI was used for the determination of cell viability, with analysis restricted to live-cell populations. Stained cells were examined by flow cytometry using a FACSCalibur (BD Bioscience, Allschwil, Switzerland). Data were analyzed using CellQuest Pro 6.0 software (BD Bioscience, Allschwil, Switzerland).
Fatty acid metabolism by HepG2 cells
Metabolism of [1-14C] palmitic acid (60 mCi/mmol, Perkin Elmer, Schwerzenbach, Switzerland) was assessed by the formation of 14C-acid-soluble β-oxidation products (Felser et al. 2013). HepG2 cells were seeded in 6-well plates (500,000 cells/well) and treated with BAL30072 for 72 h. Amiodarone (50 μM) was used as a positive control. After treatment, HepG2 cells were permeabilized with digitonin (10 μg/million cells) in 225 μL assay buffer (70 mM sucrose, 43 mM potassium chloride, 3.6 mM magnesium chloride, 7.2 mM potassium dihydrogen phosphate, 36 mM TRIS, 0.2 mM ATP, 50 μM l-carnitine, 15 μM coenzyme A, 5 mM acetoacetate, pH 7.4) and incubated for 10 min at 37 °C. Afterwards, 25 μL [1-14C] palmitic acid (200 μM final concentration) was added to each sample and incubated at 37 °C. The reaction was stopped after 15 min by adding 250 μL of perchloric acid (6%). The samples were precipitated for 5 min on ice before centrifugation. Radioactivity was measured in the supernatant using a Packard 1900 TR liquid scintillation analyzer.
Caspase 3/7 activation in liver microtissues
Caspase 3/7 activity was determined using the luminescent Caspase-Glo 3/7 assay (Promega, Madison, USA) according to the manufacturer’s manual. 3D human liver microtissues were exposed to a range of BAL30072 concentrations in GravityTRAP™ plates, and 50 μL assay buffer was added to each well containing 50 μL culture medium. The lysates were transferred to an opaque half-area assay plate and shaken for 30 min in the dark at room temperature. Luminescence was measured using a Tecan M200 Pro Infinity plate reader (Männedorf, Switzerland).
Statistical analysis
Data are given as the mean ± SEM of at least three independent experiments. Statistical analyses used GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA). One-way analysis of variance (ANOVA) was used for the comparison of more than two groups, followed by the comparison between treatments containing test compounds and the control group using Dunnett’s post-test procedure. Differences between experiments with multiple conditions were compared using two-way ANOVA followed by Bonferroni’s post hoc test. P values <0.05 (*), <0.01 (**), or <0.001 (***) were considered significant.
Results
Hepatocellular injury in BAL30072-treated patients
Transaminase elevations were observed in 5 of 6 study subjects treated with 4 g of BAL30072 administered as continuous 22-h infusions for 6 consecutive days (Fig. 2a, b). The unaffected subjects had been treated with placebo, whereas all subjects treated with BAL30072 had elevated transaminases at day 6. In three subjects, ALT activity in serum was >200 U/L (upper limit of normal 50 U/L) and in the remaining two between 50 and 100 U/L. The transaminase activities gradually returned to normal after the treatment was stopped, and the latest normalization was observed at 51 days. The subjects with liver injury were asymptomatic, except for subject 2 (with the highest ALT elevations), who developed moderate abdominal pain, mild nausea, mild diarrhea, and palpitations. The serum bilirubin and total bile acid concentration as well as the activity of alkaline phosphatase were not increased by treatment with BAL30072 (data not shown).

Time course of established (transaminases) and exploratory biomarkers in five healthy male BAL30072-treated subjects and one placebo-treated subject in a phase I study. a ALT, b AST, c total HMGB1, d miRNA122, e total, and f caspase-cleaved cytokeratin-18 were analyzed repeatedly in patient serum samples. BAL30072 (4 g per day) was administered as continuous 22-h intravenous infusions from day 1 to 6
In addition to the transaminases, four exploratory liver biomarkers, the high-mobility group protein B1 [HMGB1, reflecting non-specific cellular breakdown (Yang et al. 2013)], miRNA122 [reflecting hepatocyte breakdown (Bala et al. 2012)], full-length cytokeratin 18, and caspase-cleaved cytokeratin 18 [reflecting non-hepatocyte-specific cellular breakdown and apoptosis, respectively (Yilmaz 2009)], were also determined. The subject with the highest ALT elevation (subject 2) was correctly identified by all exploratory biomarkers, whereas the identification of the other subjects with transaminase elevation was less evident due to a high background activity (Fig. 2c–f). Full-length cytokeratin 18 and caspase-cleaved cytokeratin 18 reflected the ALT activity better than HMGB1 and miRNA122. For full-length cytokeratin 18 and caspase-cleaved cytokeratin 18, the median time until the maximal activity in serum (T max) was 5 days compared to 6 days for the other biomarkers including ALT and AST.
The serum concentrations of BAL30072 obtained in this study were in the expected range (total concentration 50–80 µM) with little inter-individual variation (data not shown). Therefore, they do not explain inter-individual differences in the increase of the biomarkers determined.
The data indicated that BAL30072 were associated with hepatocellular injury starting at 2–3 days and reaching the maximum at 5–6 days after initiation of treatment. Hepatocellular injury was slowly reversible after stopping therapy and could at least partially be explained by hepatocyte apoptosis.
Effect of BAL30072 on hepatocyte viability
Hepatocyte viability in the presence of BAL30072 was first investigated using HepG2 cells and human 3D liver microtissues exposed for 120 h by assessing the cellular ATP content. The cellular ATP content started to drop at 500 and 1000 µM in HepG2 cells and human liver microtissues and the corresponding IC50 values were 2420 and 6520 µM, respectively (Fig. 3a, b).

Influence of CYP30072 on viability of HepG2 cells and on viability and albumin synthesis of 3D human liver microtissues. a ATP content of HepG2 cells after BAL30072 exposure for 120 h. b ATP content of 3D human liver microtissues after BAL30072 exposure for 120 h. c ATP content of 3D human liver microtissues after BAL30072 exposure for 7, 14, 21, and 28 days. d Albumin synthesis by 3D human liver microtissues at different concentrations of BAL30072 at different time points. All data are expressed as a percentage of control incubations. Data show the mean ± SEM of three independent experiments
Long-term treatment of human 3D liver microtissues with BAL30072 for up to 28 days was associated with a dose- and time-dependent decrease in the hepatocyte ATP content, but did not induce complete liver microtissue cell death (Fig. 3c). The onset of cytotoxicity at the lowest concentration (289 µM) was observed from day 7 onwards.
As shown in Fig. 3d, BAL30072 impaired albumin synthesis in human 3D liver microtissues. Similar to the effect on the cellular ATP pool, the effect of BAL 30072 albumin synthesis was clearly time- and dose-dependent.
These studies showed that BAL30072 impaired important hepatic functions and exhibited hepatocellular toxicity which was dependent on the concentration and time of exposure. Taking into account the publication by Lee (2003), we subsequently investigated hepatocellular toxicity associated with the production of reactive metabolites, inhibition of hepatocyte drug transporting proteins, increased toxicity in combination with activation of Kupffer cells, and mitochondrial toxicity as possible mechanisms of hepatocellular toxicity.
Hepatocellular toxicity associated with the production of reactive metabolites
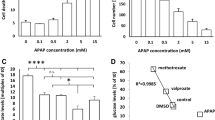
Since electrophilic metabolites can react with glutathione and decrease the cellular glutathione pool (Anderson 1997), we assessed the effect of BAL30072 on the glutathione pool of 3D human liver microtissues. As shown in Fig. 4a, incubation of 3D human liver microtissues with BAL30072 for 5 days resulted in decreased intracellular GSH levels at 2 mM, compatible with the formation of electrophilic metabolites reacting with GSH. The pretreatment with buthionine-sulfoximine (BSO), an inhibitor of γ-glutamyl-cysteine synthetase required for glutathione synthesis (Yang et al. 2015), depleted the cellular GSH content almost completely (Fig. 4a), but did not relevantly increase ATP depletion in the presence of BAL30072 (Fig. 4b).

Possible role of reactive metabolites for the toxicity of BAL30072. a Glutathione (GSH) content in 3D human liver microtissue preparations exposed to BAL30072 for 120 h with and without buthionine sulphoximine (BSO). Where indicated, the tissues were pre-treated with BSO for 40 h with 100 µM BSO (and further incubated with 100 µM BSO in the presence of BAL30072). b ATP content in human liver microtissue preparations exposed to BAL30072 for 120 h with and without BSO. c HepaRG cells were pretreated for 72 h with 20 µM rifampicin and then exposed to BAL30072 for 72 h. All data are expressed as a percentage of control incubations; data show the mean ± SEM of three independent experiments
Since these experiments suggested the possibility of the formation of reactive metabolites, we tested the formation of such metabolites directly using human liver microsomes. In these experiments, we used paracetamol as a positive control. Two high-resolution mass spectrometry-based approaches were applied using short-term microsomal incubations of BAL30072 with (1) GSH trapping and (2) total protein digestion with pronase. The experiments revealed the formation of GSH adducts with BAL30072 to a similar extent as with other monobactams (e.g., aztreonam) (data not shown). However, no covalent binding on BAL30072 metabolites could be demonstrated in the pronase assay (data not shown). This was in contrast to the positive control paracetamol, for which not only massive formation of GSH products but also covalent conjugates with proteins could be detected (data not shown).
To assess a possible effect of BAL30072 metabolites on hepatocyte viability, we performed experiments with HepaRG cells after cytochrome P450 (CYP) induction. We have shown previously that treatment of differentiated HepaRG cells with 20 µM rifampicin for 72 h increases the activity of CYP2B6, 2C19, and 3A4 by a factor of 4–8 (Berger et al. 2016). As shown in Fig. 4c, BAL30072 was toxic for HepaRG cells, but CYP induction by rifampicin did not increase this toxicity.
These results indicate that production of reactive metabolites and subsequent covalent binding to intracellular proteins is an unlikely cause for the liver toxicity caused by BAL30072.
Interference of BAL30072 with hepatocellular uptake and efflux transporters
Inhibition of canalicular export pumps by drugs can be associated with cholestasis which can impair the function of hepatocytes (Lee 2003). We, therefore, studied the interaction of BAL30072 with human hepatocyte transporters. These studies demonstrated that BAL30072 is a substrate of the hepatic uptake transporters OAT1 and OAT3, and possibly also of OATP1B1 and OATP1B3 (supplementary Tables S1 and S2). BAL30072 was a weak inhibitor of the canalicular transporter MRP2, but did not inhibit the bile salt efflux pump BSEP, the breast cancer related protein (BCRP), and P-glycoprotein (P-gp). In agreement with these findings, the serum bile acid concentration was not increased in human subjects treated with BAL30072 (data not shown).
These studies excluded interference with canalicular export pumps as a main mechanism of BAL30072-associated hepatotoxicity.
Effect on LPS-mediated toxicity
Lipopolysaccharides (LPS), also known as endotoxins, are the major components of the outer membrane of Gram-negative bacteria and can elicit strong inflammatory responses. Lipopolysaccharides can activate Kupffer cells, leading to cytokine production and secretion, and to an inflammatory state which can aggravate the toxic effects of drugs on hepatocytes (Tukov et al. 2007).
To investigate the possibility of aggravation of hepatotoxicity of BAL30072 by LPS in clinical studies with patients infected with Gram-negative bacteria, 3D human liver microtissues were co-incubated with BAL30072 and LPS. After 120 h of incubation, intracellular ATP levels in Kupffer cells containing 3D human liver microtissues showed that BAL30072-mediated hepatotoxicity was not increased by the addition of LPS (supplementary Fig. S1).
Based on these studies, we excluded Kupffer cell activation by endotoxin as a contributing factor for BAL30072-associated hepatocellular toxicity.
Effect on oxidative metabolism
Since mitochondria are important sources of ATP (glycolysis is another important source), we investigated the effect of BAL30072 on mitochondrial functions.
To investigate how BAL30072 affects oxidative metabolism, we used a Seahorse XF24 analyzer to determine changes in oxygen consumption (OCR) in HepG2 cells treated with BAL30072 and BAL104936 (the ring-opened product of BAL30072). Cellular consumption of oxygen mainly reflects mitochondrial metabolism (Felser et al. 2013). Acute exposure for 1 h up to 5000 µM of BAL30072 and BAL104936 did not reduce the OCR (Fig. 5a, b). After 72 h of exposure, the basal and maximal oxygen consumption rate started to decrease at 100 and 200 µM in the presence of BAL30072 and BAL104936, respectively (Fig. 5c–f). BAL30072 and BAL104936 did not increase the leak respiration after the addition of oligomycin, excluding an uncoupling effect (Fig. 5e, f).

Effect of BAL30072 and BAL104936 on oxygen consumption by HepG2 cells. Oxygen consumption rate (OCR) by HepG2 cells determined using a Seahorse XF24 analyzer after exposure to a BAL30072 and b BAL104936 for 1 h. OCR by HepG2 cells after exposure to c BAL30072 and d BAL104936 for 72 h. The values obtained for BAL30072 and for BAL104936 after exposure for 72 h are quantified in (e) and (f), respectively. The different concentrations used for the positive control amiodarone are due to the duration of exposure. Data show the mean ± SEM of three to four independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs. respective medium control
Effect on enzyme complexes of the electron transport chain
To analyze the mechanism of impaired oxidative metabolism, the respiratory capacities of HepG2 cells through the complexes of the electron transport chain were analyzed using a high-resolution respirometry system. After 72 h of exposure to BAL30072, we observed inhibition of the enzyme complexes II and III starting for both complexes at 500 µM (Fig. 6a).

Function of the respiratory chain, ROS production, acidification rate, and mitochondrial membrane potential in HepG2 cells, and lactate production in 3D human liver microtissues. a Effect on respiratory capacities through complexes I, II, III, and IV measured on the Oxygraph-2 k-high-resolution respirometer after BAL30072 incubation for 72 h. b ROS production after BAL30072 incubation for 72 h, with ROS data expressed as a percentage of control incubations. c, d Lactate quantification in the supernatants of 3D human liver microtissues after exposure to BAL30072 or meropenem for 24, 48, 72, 96, and 120 h. e Acidification rates determined using a Seahorse XF24 analyzer by HepG2 cells exposed to BAL30072 for 72 h. f Mitochondrial membrane potential in HepG2 cells exposed to BAL30072 for 72 h. Data show the mean ± SEM of three independent experiments. *p < 0.05, ***p < 0.001 vs. respective medium control
Consequences of the inhibition of the electron transport chain
Inhibition of the electron transport chain (especially inhibition of complexes I and III) can be associated with increased mitochondrial ROS production (Drose and Brandt 2012; Felser et al. 2013) and with increased production of lactate due to stimulation of glycolysis (Felser et al. 2013).
As expected from the inhibition of complex III, mitochondrial ROS accumulation started at 1000 µM BAL30072 for 72 h (Fig. 6b), further supporting the conclusion that BAL30072 affects mitochondria. Interestingly, lactate production by human hepatic microtissues (starting 12 h after incubation, Fig. 6c) and by HepG2 cells (starting 36 h after incubation) was decreased by BAL30072 (supplementary Fig. S2). In contrast, meropenem, which we used as a control substance, increased lactate production by human hepatic microtissues (Fig. 6d).
Impaired production of lactate suggested inhibition of glycolysis (Felser et al. 2013). We, therefore, determined the extracellular acidification rate (ECAR) using the Seahorse after 1 and 72 h of exposure to BAL30072. After 1 h of exposure, the ECAR was not impaired by BAL30072 (data not shown). However, after 72 h of exposure (Fig. 6e), BAL30072 reduced ECAR significantly starting at 200 µM, confirming impaired glycolysis (Zhang et al. 2012).
Impaired function of the mitochondrial electron transport chain can be associated with a decrease in the mitochondrial membrane potential (Kaufmann et al. 2005). As shown in Fig. 6f, after 72 h of exposure, BAL30072 was associated with a concentration-dependent decrease in mitochondrial membrane potential.
Effect of BAL30072 on mitochondrial β-oxidation and cellular fat accumulation
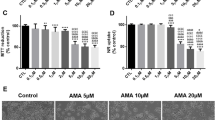
Long-term treatment with BAL30072 for up to 28 days induced a concentration- and time-dependent alteration of the 3D liver microtissue morphology, beginning after 14 days of exposure at 2893 µM (Fig. 7a). After 28 days, morphology changes could also be seen at 289 µM BAL30072, the lowest concentration tested (Fig. 7a). Changes in morphology included vacuolization of the cytoplasm of hepatocytes, with the nuclei in many instances displaced to the cell periphery. This kind of morphology is compatible with accumulation of fat in hepatocytes.

Effect on fat accumulation in HepG2 cells and 3D human liver microtissues, and on fatty acid metabolism by HepG2 cells. a H and E staining of 3D human liver microtissues treated with BAL30072 for the indicated time points; alterations in morphology are depicted by an arrow. Magnification: ×20. b Intracellular triglyceride accumulation in HepG2 cells after BAL30072 exposure for 72 h. c Metabolism of [1-14C] palmitic acid in HepG2 cells after BAL30072 exposure for 72 h (the activity of the control was 1.6 nmol/min/mg protein). Data show the mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs. respective medium control
To investigate this possibility, we studied the accumulation of triglycerides by HepG2 cells exposed to palmitate using the lipid-specific fluorescent dye BODIPY 493/503 (Felser et al. 2013). Using this technique, we could demonstrate intracellular accumulation of triglycerides in HepG2 cells incubated with BAL30072 for 72 h starting at 1000 µM (Fig. 7b).
To study the mechanism of lipid accumulation by HepG2 cells exposed to BAL30072, mitochondrial β-oxidation was quantified by monitoring the formation of 14C-acid-soluble β-oxidation products from 14C-palmitate. After an exposure of 72 h, a significant, concentration-dependent inhibition of β-oxidation by BAL30072 was observed starting at 2000 µM (Fig. 7c).
These findings showed that BAL30072 inhibited fatty acid metabolism in HepG2 cells, leading to triglyceride accumulation. These findings supported the idea that the vacuoles observed after long-term exposure of 3D liver microtissues to BAL30072 consisted of triglycerides.
Effect of BAL30072 on markers of apoptosis
A decrease in the mitochondrial membrane potential, mitochondrial production of ROS, and cellular accumulation of triglycerides are triggers for mitochondrial membrane permeabilization (Charlot et al. 2004; Zamzami et al. 1996a). Mitochondrial membrane permeabilization not only decreases the mitochondrial membrane potential, but is also associated with apoptosis and/or necrosis (Marchetti et al. 1996; Zamzami et al. 1995, 1996b). Accordingly, BAL30072 activated caspase-3/7 in 3D human liver microtissues in a concentration- and time-dependent manner, suggesting that apoptosis contributes to hepatotoxicity associated with BAL30072 (Fig. 8a, c). As shown previously, the cellular ATP concentration showed the expected drop in the presence of BAL30072, which was also time- and concentration-dependent (Fig. 8b, d).

Effect of BAL30072 on markers of apoptosis in 3D human liver microtissues and effect of NAC on the cellular ATP content. Human liver microtissue preparations were exposed to different concentrations of BAL30072 over different periods of time and the activity of caspase 3/7 was determined in the preparations. a Caspase 3/7 activity after exposure for 24 h. b Cellular ATP content after exposure for 24 h. c Caspase 3/7 activity after exposure for 72 h. d Cellular ATP content after exposure for 72 h. e Mitochondrial ROS production of HepG2 cells after 120 h exposure to BAL30072 in the absence or presence of 1, 2, or 5 mM N-acetylcysteine (NAC). f ATP content of 3D human liver microtissues after 120 h of exposure to BAL30072 in the absence or presence of 1, 2, or 5 mM NAC. All data are expressed as percentage of control incubations; data show the mean ± SEM of three independent experiments
Mitigation of BAL30072-mediated cell death
Since oxidative stress appears to be important for BAL30072-associated hepatotoxicity, we investigated whether the hepatocellular toxicity of BAL30072 could be mitigated by co-treatment with N-acetylcysteine (NAC), a well-established antioxidant (Bouitbir et al. 2012).
In HepG2 cells, a concentration-dependent protective effect of NAC for BAL30072-mediated mitochondrial ROS production was detected (Fig. 8e). Co-treatment with 5000 µM NAC reduced the mitochondrial ROS concentration by 61% at 10,000 µM BAL30072. A concentration-dependent protective effect of NAC for BAL30072-mediated hepatocellular toxicity was also observed in 3D human liver microtissues (Fig. 8f).
Discussion
In the current study, we tested the main mechanisms of hepatotoxicity proposed by Lee (2003) using different human hepatocyte models after having detected hepatocellular injury in healthy subjects treated with BAL30072 in a phase I clinical study. The investigations included the formation of reactive metabolites with covalent binding to intracellular proteins, interference with hepatocyte drug transport systems, toxicity associated with Kupffer cell activation, and mitochondrial toxicity. We did not test for immunological toxicity, because we considered this mechanism to have a low probability. Clinical signs for an immunological reaction were lacking and the time until the increase in transaminases was detectable (approximately 3 days) was considered to be too short for sensitization in naïve persons (Tailor et al. 2015).
The studies revealed that BAL30072 depleted cellular ATP stores in HepG2 cells and human liver microtissues, inhibited glycolysis, and interfered with the mitochondrial electron transport chain starting at concentrations in the range of 100–200 µM. The other types of toxicities investigated appeared either at concentrations >1 mM or could not be observed in the concentration range of BAL30072 investigated.
In the early stages of mitochondrial liver injury, an adaptive metabolic shift from predominantly oxidative phosphorylation to glycolysis can maintain the cellular ATP levels. When glycolysis is also impaired, hepatocytes may be unable to sustain sufficient cellular ATP levels and may undergo apoptosis and/or necrosis (Nishikawa et al. 2014). Treatment with BAL30072 was associated with impaired lactate generation by HepG2 cells and human liver microtissues, suggesting impaired glycolysis. In addition, BAL30072 also impaired the acidification rate of HepG2 cells at a concentration of 200 µM, supporting the notion that BAL30072 impaired glycolysis. Since mitochondria and glycolysis are the main sources of ATP, the observed decrease in the cellular ATP stores can be explained by these mechanisms.
After 72 h of incubation, both BAL30072 and BAL104936 significantly reduced oxygen uptake by HepG2 cells at concentrations starting between 100 and 200 µM, compatible with mitochondrial dysfunction. Based on preclinical and clinical data (data on file at Basilea), the hepatic BAL30072 concentration can be estimated to reach approximately 100 µM in patients treated with 4 g of BAL30072 infused over 22 h, which is close to the concentration where we started to observe inhibition of mitochondrial respiration and glycolysis. Since the plasma concentration of BAL104936 reached only 20–30% of the parent compound BAL30072, and BAL104396 was a less potent mitochondrial toxicant compared to BAL30072, it can be assumed that hepatic toxicity is mainly associated with the parent compound BAL30072. Importantly, short-term exposure (<24 h) of HepG2 cells with BAL30072 was not toxic (or only at concentrations >1 mM); toxicity at concentration <1 mM was observed only after exposure for 72 h or longer. This observation is in agreement with the clinical study, where toxicity started not before 3 days of exposure. Based on the available pharmacokinetic data, this delayed toxicity cannot be explained by hepatocellular accumulation of the parent compound or of a metabolite. Since mitochondria contain a distinct antioxidative defense system (Lim et al. 2014), the delay until toxicity occurs may reflect the time needed to overcome mitochondrial antioxidative defense.
Further analysis of the mitochondrial function, using the Oxygraph, demonstrated that BAL30072 inhibited complex II and III of the electron transport chain, which was associated with increased ROS production and with a drop in the mitochondrial membrane potential and cellular ATP content. The BAL30072 concentrations needed to impair mitochondrial function using the Oxygraph were higher compared with the Seahorse, which can possibly be explained by the different experimental settings. With the Seahorse, cells are investigated as an intact layer, whereas with the Oxygraph, cells are investigated in suspension after treatment with digitonin to open the plasma membrane.
A reduction of the activity of complex III is a well-known cause for an increase in mitochondrial ROS production and for a decrease in ATP production (Drose and Brandt 2012). An increase in mitochondrial ROS production was observed in HepG2 cells and a drop in the ATP content in both HepG2 cells and human liver microtissues exposed to BAL30072. Mitochondrial ROS production exceeding a certain extent is associated with mitochondrial membrane permeabilization, which leads to a drop in the mitochondrial membrane potential and induction of apoptosis and/or necrosis (Kaufmann et al. 2005; Zamzami et al. 1996b). We could demonstrate mitochondrial membrane depolarization on HepG2 cells and an increase in markers of apoptosis both in human liver microtissues and in human subjects. The important role of cellular ROS accumulation in hepatocellular toxicity of BAL30072 was further demonstrated by co-incubations with N-acetylcysteine. N-acetylcysteine could at least partially prevent the ROS accumulation in HepG2 cells and cytotoxicity in human liver microtissues exposed to BAL30072. Mitochondrial dysfunction (in combination with impaired glycolysis) appears, therefore, to be an important mechanism in BAL30072-associated liver injury.
Treatment with BAL30072 was also associated with accumulation of intracellular triglycerides in hepatocytes of human liver microtissues, which were stored as large droplets, shifting the nucleus to the cell periphery. Triglyceride accumulation, which was also shown in HepG2 cells, can be regarded as a consequence of impaired fatty acid metabolism and impaired function of the electron transport chain (Krahenbuhl et al. 1994). Since hepatocellular steatosis has been reported to be associated with increased ROS production and induction of mitochondrial membrane permeabilization (Rial et al. 2010), cellular triglyceride accumulation may have contributed to hepatocellular damage.
Formation of reactive metabolites with covalent binding to cellular proteins could be excluded as a major mechanism of BAL30072 hepatocellular toxicity for two reasons. First, cytochrome P450 (CYP) induction by rifampicin in HepaRG cells did not increase BAL30072-associated toxicity, and, second, covalent protein binding of BAL30072 and/or metabolites was not detected in human microsomes exposed to BAL30072. The observed decrease in the hepatocellular glutathione content in 3D human microtissues incubated with BAL30072 was, therefore, more likely the result of ROS detoxification and not of a reaction with metabolites of BAL30072. GSH is an important endogenous antioxidant, participating directly in the neutralization of free radicals (Anderson 1997).
The in vitro investigation of the interaction of BAL30072 with hepatic drug transporters revealed that BAL30072 was a substrate of the hepatic uptake transporters OATP1B1 and OATP1B3. Importantly, BAL30072 did not interact with the bile salt export pump (BSEP) or other canalicular export pumps. In agreement with these findings, serum bile acids were not increased in patients treated with BAL30072. Accordingly, inhibition of canalicular bile acid excretion with intracellular accumulation of bile acids could be excluded as a mechanism of hepatotoxicity by BAL30072. Inhibition of the BSEP with hepatocellular accumulation of (toxic) bile acids has been reported to represent the main mechanism of the hepatic toxicity of bosentan (Fattinger et al. 2001).
We also investigated the possibility that activation of Kupffer cells could increase the toxicity of BAL30072. Since 3D human liver microtissues contain Kupffer cells, we could stimulate them by the administration of LPS. LPS is a major component of the outer membrane of Gram-negative bacteria and can elicit strong inflammatory reactions in humans (Cui et al. 2014), thereby aggravating liver injury associated with certain drugs (Tukov et al. 2007).Since pretreatment with LPS did not increase hepatocellular toxicity of BAL30072, we could exclude this possibility.
Since mitochondria are considered to have evolved from endosymbiotic α-proteobacteria which are potentially vulnerable to antibiotics (Chandel and Budinger 2013), it is not unexpected to detect mitochondrial toxicity during the pharmaceutical development of antibiotics. It is well-established that antibiotics, including quinolones, aminoglycosides, and β-lactams, can cause both DILI (Leitner et al. 2010) and mitochondrial dysfunction (Kalghatgi et al. 2013). Antibiotics have been described to affect mitochondrial DNA and protein synthesis, as well as activities of the enzyme complexes of the respiratory chain (Duewelhenke et al. 2007) as observed in the current investigation.
In conclusion, long-term exposure to BAL30072 inhibited the mitochondrial electron transport chain and β-oxidation in HepG2 cells and human liver microtissues and caused mitochondrial ROS accumulation and a drop in the mitochondrial membrane potential. Since BAL30072 also inhibited glycolysis, the cellular ATP level dropped and hepatocytes eventually underwent apoptosis and/or necrosis. Mitochondrial dysfunction and impaired glycolysis are likely mechanisms contributing to hepatocellular injury in subjects treated with BAL30072.
Abbreviations
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- BSO:
-
Buthionine sulphoximine
- CYP:
-
Cytochrome P450
- DILI:
-
Drug-induced liver injury
- DMSO:
-
Dimethylsulfoxide
- DPBS:
-
Dulbecco’s phosphate buffered saline
- ECAR:
-
Extracellular acidification rate
- FCCP:
-
Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone
- FDA:
-
Food and drug administration (US)
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GSH:
-
Reduced glutathione
- H and E:
-
Hematoxylin and Eosin
- HEPES:
-
4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid
- HMGB1:
-
High-mobility group protein B1
- LDH:
-
Lactate dehydrogenase
- LPS:
-
Lipopolysaccharide
- NAC:
-
N-acetyl-l-cysteine
- OCR:
-
Oxygen consumption rate
- PBS:
-
Phosphate-buffered saline
- ROS:
-
Reactive oxygen species
- ULN:
-
Upper limit of normal
References
Aithal GP, Watkins PB, Andrade RJ et al (2011) Case definition and phenotype standardization in drug-induced liver injury. Clin Pharmacol Ther 89(6):806–815. doi:10.1038/clpt.2011.58
Anderson ME (1997) Glutathione and glutathione delivery compounds. Adv Pharmacol 38:65–78
Aninat C, Piton A, Glaise D et al (2006) Expression of cytochromes P450, conjugating enzymes and nuclear receptors in human hepatoma HepaRG cells. Drug Metab Dispos 34(1):75–83. doi:10.1124/dmd.105.006759
Bala S, Petrasek J, Mundkur S et al (2012) Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology 56(5):1946–1957. doi:10.1002/hep.25873
Berger B, Donzelli M, Maseneni S et al (2016) Comparison of liver cell models using the basel phenotyping cocktail. Front Pharmacol 7:443. doi:10.3389/fphar.2016.00443
Bouitbir J, Charles AL, Echaniz-Laguna A et al (2012) Opposite effects of statins on mitochondria of cardiac and skeletal muscles: a ‘mitohormesis’ mechanism involving reactive oxygen species and PGC-1. Eur Heart J 33(11):1397–1407. doi:10.1093/eurheartj/ehr224
Chandel NS, Budinger GR (2013) The good and the bad of antibiotics. Sci Transl Med 5(192):192fs25. doi:10.1126/scitranslmed.3006567
Charlot JF, Pretet JL, Haughey C, Mougin C (2004) Mitochondrial translocation of p53 and mitochondrial membrane potential (Delta Psi m) dissipation are early events in staurosporine-induced apoptosis of wild type and mutated p53 epithelial cells. Apoptosis 9(3):333–343
Cui J, Chen Y, Wang HY, Wang RF (2014) Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum Vaccin Immunother 10(11):3270–3285. doi:10.4161/21645515.2014.979640
Donato MT, Martinez-Romero A, Jimenez N et al (2009) Cytometric analysis for drug-induced steatosis in HepG2 cells. Chem Biol Interact 181(3):417–423. doi:10.1016/j.cbi.2009.07.019
Drose S, Brandt U (2012) Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol 748:145–169. doi:10.1007/978-1-4614-3573-0_6
Duewelhenke N, Krut O, Eysel P (2007) Influence on mitochondria and cytotoxicity of different antibiotics administered in high concentrations on primary human osteoblasts and cell lines. Antimicrob Agents Chemother 51(1):54–63. doi:10.1128/AAC.00729-05
Fattinger K, Funk C, Pantze M et al (2001) The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther 69(4):223–231. doi:10.1067/mcp.2001.114667
Felser A, Blum K, Lindinger PW, Bouitbir J, Krahenbuhl S (2013) Mechanisms of hepatocellular toxicity associated with dronedarone—a comparison to amiodarone. Toxicological sciences: an official journal of the Society of Toxicology 131(2):480–490. doi:10.1093/toxsci/kfs298
Kalghatgi S, Spina CS, Costello JC et al (2013) Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci Transl Med 5(192):192ra85. doi:10.1126/scitranslmed.3006055
Kaplowitz N (2005) Idiosyncratic drug hepatotoxicity. Nat Rev Drug Discov 4(6):489–499. doi:10.1038/nrd1750
Kaufmann P, Torok M, Hanni A, Roberts P, Gasser R, Krahenbuhl S (2005) Mechanisms of benzarone and benzbromarone-induced hepatic toxicity. Hepatology 41(4):925–935. doi:10.1002/hep.20634
Krahenbuhl S, Talos C, Reichen J (1994) Mechanisms of impaired hepatic fatty acid metabolism in rats with long-term bile duct ligation. Hepatology 19(5):1272–1281
Kroh EM, Parkin RK, Mitchell PS, Tewari M (2010) Analysis of circulating microRNA biomarkers in plasma and serum using quantitative reverse transcription-PCR (qRT-PCR). Methods 50(4):298–301. doi:10.1016/j.ymeth.2010.01.032
Lee WM (2003) Drug-induced hepatotoxicity. N Engl J Med 349(5):474–485. doi:10.1056/NEJMra021844
Leitner JM, Graninger W, Thalhammer F (2010) Hepatotoxicity of antibacterials: pathomechanisms and clinical. Infection 38(1):3–11. doi:10.1007/s15010-009-9179-z
Lim JL, Wilhelmus MM, de Vries HE, Drukarch B, Hoozemans JJ, van Horssen J (2014) Antioxidative defense mechanisms controlled by Nrf2: state-of-the-art and clinical perspectives in neurodegenerative diseases. Arch Toxicol 88(10):1773–1786. doi:10.1007/s00204-014-1338-z
Marchetti P, Castedo M, Susin SA et al (1996) Mitochondrial permeability transition is a central coordinating event of apoptosis. J Exp Med 184(3):1155–1160
Messner S, Agarkova I, Moritz W, Kelm JM (2013) Multi-cell type human liver microtissues for hepatotoxicity testing. Arch Toxicol 87(1):209–213. doi:10.1007/s00204-012-0968-2
Nishikawa T, Bellance N, Damm A et al (2014) A switch in the source of ATP production and a loss in capacity to perform glycolysis are hallmarks of hepatocyte failure in advance liver disease. J Hepatol 60(6):1203–1211. doi:10.1016/j.jhep.2014.02.014
Page MG, Dantier C, Desarbre E (2010) In vitro properties of BAL30072, a novel siderophore sulfactam with activity against multiresistant Gram-negative bacilli. Antimicrob Agents Chemother 54(6):2291–2302. doi:10.1128/AAC.01525-09
Pesta D, Gnaiger E (2012) High-resolution respirometry: oXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol Biol 810:25–58. doi:10.1007/978-1-61779-382-0_3
Reuben A (2004) Hy’s law. Hepatology 39(2):574–578. doi:10.1002/hep.20081
Rial E, Rodriguez-Sanchez L, Gallardo-Vara E, Zaragoza P, Moyano E, Gonzalez-Barroso MM (2010) Lipotoxicity, fatty acid uncoupling and mitochondrial carrier function. Biochim Biophys Acta 1797(6–7):800–806. doi:10.1016/j.bbabio.2010.04.001
Taegtmeyer AB, Haschke M, Tchambaz L et al (2014) A study of the relationship between serum bile acids and propranolol pharmacokinetics and pharmacodynamics in patients with liver cirrhosis and in healthy controls. PLoS One 9(6):e97885. doi:10.1371/journal.pone.0097885
Tailor A, Faulkner L, Naisbitt DJ, Park BK (2015) The chemical, genetic and immunological basis of idiosyncratic drug-induced liver injury. Hum Exp Toxicol 34(12):1310–1317. doi:10.1177/0960327115606529
Temple RJ, Himmel MH (2002) Safety of newly approved drugs: implications for prescribing. JAMA 287(17):2273–2275
Tukov FF, Luyendyk JP, Ganey PE, Roth RA (2007) The role of tumor necrosis factor alpha in lipopolysaccharide/ranitidine-induced inflammatory liver injury. Toxicol Sci 100(1):267–280. doi:10.1093/toxsci/kfm209
Yang H, Antoine DJ, Andersson U, Tracey KJ (2013) The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol 93(6):865–873. doi:10.1189/jlb.1212662
Yang X, Yao H, Chen Y et al (2015) Inhibition of Glutathione production induces macrophage CD36 expression and enhances cellular-oxidized low density lipoprotein (oxLDL) uptake. J Biol Chem 290(36):21788–21799. doi:10.1074/jbc.M115.654582
Yilmaz Y (2009) Systematic review: caspase-cleaved fragments of cytokeratin 18—the promises and challenges of a biomarker for chronic liver disease. Aliment Pharmacol Ther 30(11–12):1103–1109. doi:10.1111/j.1365-2036.2009.04148.x
Zamzami N, Marchetti P, Castedo M et al (1995) Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J Exp Med 181(5):1661–1672
Zamzami N, Marchetti P, Castedo M et al (1996a) Inhibitors of permeability transition interfere with the disruption of the mitochondrial transmembrane potential during apoptosis. FEBS Lett 384(1):53–57
Zamzami N, Susin SA, Marchetti P et al (1996b) Mitochondrial control of nuclear apoptosis. J Exp Med 183(4):1533–1544
Zhang J, Nuebel E, Wisidagama DR et al (2012) Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat Protoc 7(6):1068–1085. doi:10.1038/nprot.2012.048
Acknowledgements
Scott Eaddy (UNC Institute for Drug Safety Sciences) provided laboratory assistance for quantification of miRNA122 and Total HMGB1 in clinical samples. The authors acknowledge the provision of medical writing services by David Main, Basilea Pharmaceutica International Ltd.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SK was a member of the Data and Safety Monitoring Board (DSMB) for the phase 1 studies of BAL30072. FP has acted as a consultant for Basilea Pharmaceutica International Ltd.
Financial support
SK was supported by a grant of the Swiss National Science Foundation (31003A_156270).
The studies investigating the potential of BAL30072 to form reactive metabolites that conjugate covalently to proteins were funded with Federal funds from the Department of Health and Human Services; Office of the Assistant Secretary for Preparedness and Response; Biomedical Advanced Research and Development Authority, under Contract No. HHSO100201300010C.
Ethical conduct of clinical studies
The clinical studies of BAL30072 referred to in this manuscript were conducted in accordance with the International Conference on Harmonisation Guideline for Good Clinical Practice E6, with ethical principles consistent with those laid down in the Declaration of Helsinki, and with applicable local laws and regulations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Paech, F., Messner, S., Spickermann, J. et al. Mechanisms of hepatotoxicity associated with the monocyclic β-lactam antibiotic BAL30072. Arch Toxicol 91, 3647–3662 (2017). https://doi.org/10.1007/s00204-017-1994-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-017-1994-x