
Abstract
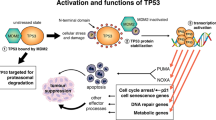
The tumour suppressor p53 is a central player in cellular DNA damage responses. P53 is upregulated and activated by genotoxic stress and induces a transcriptional programme with effectors promoting apoptosis, cell cycle arrest, senescence and DNA repair. For the best part of the last three decades, these DNA damage-related programmes triggered by p53 were unequivocally regarded as the major if not sole mechanism by which p53 exerts its tumour suppressor function. However, this interpretation has been challenged by a number of recent in vivo studies, demonstrating that mice which are defective in inducing p53-dependent apoptosis, cell cycle arrest and senescence suppress thymic lymphoma as well as wild-type p53 expressing animals. Consequently, the importance of DNA damage responses for p53-mediated tumour suppression has been questioned. In this review, I summarize current knowledge on p53-controlled DNA damage responses and argue that these activities, while their role has certainly changed, remain an important feature of p53 biology with relevance for cancer therapy and tumour suppression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
DNA damage affects the integrity of the genetic information in a cell and thus can be mutagenic. To counteract this, mammalian cells have developed a sophisticated network of complex signalling programmes that respond to DNA damage with the aim of restoring genomic integrity (Fig. 1). This can be either achieved by DNA repair or elimination of the damaged cell from the organism/proliferating cell pool through induced cell death or senescence. If both types of programmes fail, mutations can manifest in the genome and, if a damaged cell proliferates, be duplicated during DNA replication and cell division. With repeated failures and the accumulation of too many mutations, there is a high risk that genes which regulate cell growth and death are disrupted in their proper function. This can lead to cancer.

Cellular responses to DNA damage. Different sources induce different types of DNA damage. Depending on the type and extent of damage cells respond in two fundamentally different ways. Transient activation of cell cycle checkpoints coupled with DNA repair can restore genomic integrity. Such cells survive and resume their normal life cycle. In cases where repair is not completely accurate and/or the damage too severe, cells can be eliminated from the organism by cell death programmes. Alternatively, their growth is irreversibly stopped by the senescence programme (terminal growth arrest). If DNA repair is not accurate and elimination programmes fail, mutations and chromosomal aberrations manifest in the genome. This can result in malignant transformation and cancer
Indeed, the prevalence of mutations and genomic instability—that is a propensity to easily acquire more mutations—are hallmarks of cancer cells and common features across the vast majority of cancers (Hanahan and Weinberg 2011; Roberts and Gordenin 2014). Therefore, cellular DNA damage responses have been firmly established as a crucial component of the mechanisms that suppress cancer from developing. Consistently, genes that control DNA damage response programmes are particularly frequently mutated and disrupted in cancer.
The Tp53 gene, encoding for the tumour suppressor protein p53, has, for a long time, been regarded a prime example illustrating the crucial relationship between DNA responses and tumour suppression (Lane 1992; Vogelstein et al. 2000). Tp53 is the most frequently mutated gene in cancer and a central player in cellular DNA damage responses (Kandoth et al. 2013; Vousden and Lane 2007). Numerous studies have demonstrated that p53 can induce transient cell cycle arrest, senescence and apoptosis in response to genotoxic stress, while cells deficient for p53 show significant defects in their response to DNA damage (Vousden 2006; Vousden and Prives 2009). There is also no doubt on the potent tumour-suppressive function of p53 which is revealed impressively by, for example, the fact that p53-null mice die quickly and with nearly 100 % penetrance of cancer (mostly thymic lymphomas) at around 6 months of age (Donehower et al. 1992; Jacks et al. 1994; Kemp et al. 1994; Purdie et al. 1994). Moreover, patients suffering from Li-Fraumeni syndrome, a condition often associated with germline p53 mutations, have a much higher incidence of developing cancer than the normal population (Malkin et al. 1990).
For the best part of the last thirty-five years since the discovery of p53, DNA damage-response-related functions of p53 have been regarded the primary if not sole relevant mechanism to exert its potent tumour-suppressive effect. However, as it turns out, this view was too simplistic. Indeed, a number of recent studies described efficient p53-mediated tumour suppression in vivo despite severe defects in the mechanisms or genes involved in p53-mediated DNA damage responses. In this article, I will review data supporting the different views on the importance of p53-mediated DNA damage responses with a focus on cancer therapy and tumour suppression and discuss the implications for our understanding of p53 and its role in cancer biology.
Activation of p53 by DNA damage
The idea that p53 is involved in cellular DNA damage responses relates back to observations made in the 1980s and 1990s, showing that p53 is massively upregulated by ultraviolet (UV) light (Maltzman and Czyzyk 1984), ionizing radiation (Kastan et al. 1991; Lowe et al. 1993b) and chemotherapeutic agents (Fritsche et al. 1993), all causing different types of DNA damage. Upregulation of p53 occurs rapidly (within 30–60 min) and mainly at the level of the protein, which is stabilized (Fritsche et al. 1993; Kastan et al. 1991; Maltzman and Czyzyk 1984). Many years later, we know that more or less all DNA damaging agents (and beyond that, many other stimuli that cause cellular stress) lead to p53 upregulation.
In addition to being stabilized and upregulated, p53 is also heavily modified upon DNA damage, including phosphorylation of serine 15 by the ATM and ATR kinases [reviewed in (Meek and Anderson 2009)]. These two events (stabilization and phosphorylation at serine 15) are well-established measures of DNA damage-induced p53 activation and can be assayed by Western blot analysis (Fig. 2). Moreover, phosphorylation of p53 contributes to its upregulation, because DNA damage-induced stabilization of p53 is primarily caused by disruption of its complex with Mdm2—a ubiquitin E3 ligase that targets p53 for proteasomal degradation (Haupt et al. 1997; Kubbutat et al. 1997). In normal unstressed cells, p53 protein levels are kept very low, providing an efficient way to keep p53 in check. Posttranslational modifications of both p53 and Mdm2, including phosphorylation of p53 at serine 20 and phosphorylation of Mdm2 at serine 395, are rapidly triggered upon DNA damage, disrupt the p53-Mdm2 complex and inhibit Mdm2 by other means (Chehab et al. 1999; Cheng et al. 2009; Hu et al. 2012; Maya et al. 2001; Momand et al. 1992; Oliner et al. 1993; Shieh et al. 1997). Because p53 is then no longer targeted for proteasomal degradation, its levels rise quickly. However, it should be noted that p53 has also some activity in unstressed cells, as reflected by its presence at the p21 promoter and the different expression profiles of unstressed isogenic wild-type and p53-null cell pairs (Allen et al. 2014; Espinosa et al. 2003; Tang et al. 1998). Notably, too, some cancerous cell lines display readily detectable levels of p53 protein without showing obvious signs of its anti-proliferative activity, cell cycle arrest or apoptosis in particular. Together, this suggests that the pure abundance of the p53 protein is not the sole determinant of its activity. Nevertheless, upregulation of wild-type p53 in response to DNA damage is normally a reliable indicator of its activation.

DNA damage-induced activation of p53. Upregulation of p53, primarily caused by stabilization of the protein, and phosphorylation at serine 15 (serine 18 in mouse) are hallmarks of DNA damage-induced activation of p53 and can be assayed by Western blot analysis. Shown here are Western blot analyses of NIH3T3 cells treated with a low and high dose of UV and ionizing radiation, respectively. Note that the extent and type of DNA damage have an effect on the extent and kinetics of p53 stabilization and phosphorylation. Figure modified from Speidel et al. (2006)
Usually, p53 activity relates to changes in expression of p53-responsive target genes. A protein that can bind DNA in a sequence-specific manner (Bargonetti et al. 1991; El-Deiry et al. 1992; Kern et al. 1991), p53 transactivates or represses an ever growing list of genes and micro-RNAs that operate in numerous pathways, including apoptosis, cell cycle arrest, DNA repair, metabolism, autophagy, differentiation and others (Vousden and Prives 2009). The direct role of p53 in inducing any of these significant cell fate-determining processes has been confirmed in many experimental systems, most simply by overexpression and knock-down studies. These studies also highlighted the importance of the context, as overexpression of p53 would, for example, induce apoptosis in some cell systems but cell cycle arrest in others (Agarwal et al. 1995; Chen et al. 1996; Liu et al. 1995; Polyak et al. 1996; Shaw et al. 1992; Stewart et al. 1995; Yonish-Rouach et al. 1991).
Transcriptional functions are clearly the main mode of action for the p53 protein against which the majority of cancer cells select. Indeed, most p53 mutations found in human cancers affect codons in the central DNA binding domain of p53 and thus impair its transcriptional activity (Olivier et al. 2010). However, it is worth noting that p53 exerts activity also through non-transcriptional mechanisms, in particular protein–protein interactions. These activities have been particularly well characterized in the context of apoptosis induction (Speidel 2010). As discussed further below, p53 activities triggered by DNA damage have high relevance from a clinical perspective. This applies not only to their role in tumour suppression but also in the context of cancer therapy with cytotoxic agents, most of which trigger activation of p53.
P53-Mediated apoptosis
P53 can be a strong inducer of apoptosis in response to DNA damage. This function is dependent on the cell type as well as type and extent of the DNA damaging agent. Thus, the apoptotic function of p53 becomes most obvious in cell systems that are completely dependent on functional wild-type p53 for apoptosis in response to certain stressors. Mouse thymocytes provide a prominent example. These cells respond to ionizing radiation with massive apoptosis but only when harbouring wild-type p53 (Clarke et al. 1993; Lowe et al. 1993b). Similarly, human colon carcinoma cells (HCT116 cell line) undergo apoptotic cell death when treated with 5-fluorouracil, but an isogenic p53-deficient cell line is protected (Bunz et al. 1999). Clearly, however, apoptosis can occur also in the absence of p53, and in many cases, the loss of or inactivating mutations in p53 will even increase susceptibility of cells to apoptosis induced by cytotoxic agents. This can be seen in fibroblasts receiving high-dose gamma irradiation or the above-mentioned HCT116 cells after treatment with adriamycin (Bunz et al. 1999; Speidel et al. 2006). In these systems, wild-type p53 promotes a pro-survival response, cell cycle arrest/senescence in particular (discussed further below) whereas p53-null cells die (Bunz et al. 1999; Speidel et al. 2006). There will also be cases where a certain stressor will induce apoptosis in a particular type of cell regardless of the presence of wild-type p53. However, the presence of functional wild-type p53 may modulate the signalling pathways leading to cell death and sometimes change the efficiency of apoptotic cell destruction. For example, high-dose UV-irradiation induces apoptosis in mouse fibroblasts with wild-type p53, p53-null or mutant p53 genotype, but only wild-type and certain mutant p53 lines will show activation of Bax as measured by its translocation and aggregation in the outer mitochondrial membrane (Speidel et al. 2006). Furthermore, the extent of apoptotic cell death in a panel of human lymphoma cell lines that were treated with chemotherapeutic drugs depended on the cells’ p53 status with wild-type p53-expressing lines being generally more sensitive than mutant p53 or p53-deficient cell lines (Fan et al. 1994). Together, the exact circumstances, i.e. type of cell, stress and other signalling pathways, will determine whether or not p53 will induce apoptosis in response to DNA damage.
Mechanistically, p53 can promote apoptosis through three distinct ways: transcriptional activation, transcriptional repression and transcription-independent mechanisms.
Among p53-responsive target genes that are upregulated by p53 and known to promote apoptosis are Puma (Nakano and Vousden 2001; Yu et al. 2001), Noxa (Oda et al. 2000a) and Bax (Miyashita and Reed 1995 (all belonging to the Bcl-2 family—the master controller of the intrinsic pathway of apoptosis), CD95/Fas (Muller et al. 1998; Owen-Schaub et al. 1995) and DR5 (Wu et al. 1997 (part of the extrinsic receptor-mediated pathway of apoptosis) and other factors, including Apaf1, Perp and Pidd (Attardi et al. 2000; Lin et al. 2000; Moroni et al. 2001; Robles et al. 2001). Puma and Noxa appear as the major target genes mediating DNA damage-induced and p53-dependent apoptosis as demonstrated by a number of knockout studies in which deficiency in these genes caused protection against apoptotic cell death (Jeffers et al. 2003; Nakano and Vousden 2001; Oda et al. 2000a; Shao et al. 2010; Villunger et al. 2003; Yu et al. 2003, 2010). Significantly, fibroblasts and thymocytes derived from Puma/Noxa double knockout mice showed similar resistance to apoptosis as cells from p53-null mice when treated with agents that induced p53-mediated cell death (Michalak et al. 2008).
In addition to the effects related to upregulation of proapoptotic target genes, p53 promotes apoptosis by direct protein–protein interactions with members of the Bcl-2 family at the cytosol and mitochondrial membrane (Chipuk et al. 2004; Leu et al. 2004; Mihara et al. 2003). These non-transcriptional activities lead to activation of either Bax or Bak and thereby promote mitochondrial membrane permeabilization and apoptosis (Speidel 2010). Because p53 accumulates primarily in the nucleus after genotoxic stress (Fritsche et al. 1993), it seems that a high extent of p53 stabilization leading to high cellular levels of p53 protein is required for the cytosolic and mitochondrial function (Speidel et al. 2006). This mode of apoptosis induction may be specifically used under certain physiological conditions, such as in response to high-dose UV-irradiation (Speidel et al. 2006) when it would be fatal if a severely damaged cell required normal operation of transcription and translation to carry out the apoptotic programme. In addition, it may also be employed to complement and augment the transcriptional response of p53 (Chipuk et al. 2005; Erster et al. 2004).
The significance of p53-mediated transcriptional repression in apoptosis induction is not completely clear. However, p53 response elements are present at the promoters of at least two bona fide anti-apoptotic factors, Bcl-2 and survivin, and both genes have been shown to be negatively regulated by p53 (Haldar et al. 1994; Hoffman et al. 2002; Mirza et al. 2002; Miyashita et al. 1994). Presumably more significant, p53 regulates microRNAs and thereby represses the expression of genes in an indirect manner [reviewed in (He et al. 2007b; Hunten et al. 2013; Otsuka and Ochiya 2014)]. Members of the mir34 family (mir34a, in particular) have clear relevance for p53-mediated apoptosis induction, as they are upregulated upon DNA damage in a p53-dependent manner and silence several bona fide anti-apoptotic genes, including Bcl-2 (Bommer et al. 2007; Chang et al. 2007; He et al. 2007a; Raver-Shapira et al. 2007; Tarasov et al. 2007). Consistently, downregulation of miR34-a attenuated p53-dependent apoptosis, whereas overexpression could promote cell death in some cell systems (Chang et al. 2007; Raver-Shapira et al. 2007; Tarasov et al. 2007). However, cells derived from miR34-deficient mice showed a normal p53 response suggesting that gene regulation through the miR-34 family provides a redundant form of regulating DNA damage responses (Concepcion et al. 2012).
P53-Mediated cell cycle arrest, senescence and DNA repair
While some cells respond to high p53 levels with apoptosis, numerous others will stop their growth, either transiently or permanently. Like with apoptosis, this can be observed in systems with enforced overexpression of p53 but also in response to DNA damage (Agarwal et al. 1995; Di Leonardo et al. 1994; Heinlein et al. 2010; Speidel et al. 2006; Stewart et al. 1995; Sugrue et al. 1997; Taylor et al. 1999). Which parameters decide whether activated p53 induces transient cell cycle arrest, permanent growth arrest (senescence) or cell death is still not fully understood—despite much effort [reviewed in (Carvajal and Manfredi 2013)]. However, several factors, including p53-levels, modification, type and extent of stress, co-factors, microenvironment and/or cell type specifics appear to play a role (Carvajal and Manfredi 2013; Chen et al. 1996; Das et al. 2007; Kracikova et al. 2013; Oda et al. 2000b; Speidel et al. 2006). There is also some evidence that in many cell types, cell cycle arrest is the primary p53 response with the induction of (p53-dependent) apoptosis being more ‘difficult’ and requiring additional signals, such as higher p53 concentrations and cooperative binding of several p53 tetramers (Chen et al. 1996; Kracikova et al. 2013; Schlereth et al. 2010; Speidel et al. 2006).
Traditionally, cell cycle arrest has been seen as an immediate response to DNA damage that gives cells time to conduct DNA repair and, if this is not successful, targets cells to either apoptosis or halts the cell cycle permanently by the senescence programme (Massague 2004; Medema and Macurek 2012; Weinert and Hartwell 1988). DNA repair activities have therefore been associated with cell cycle arrest, and several proteins, including the p53 targets p21 and Gadd45a, that induce cell cycle arrest have also distinct functions in DNA repair (McDonald et al. 1996; Smith et al. 1994, 2000; Stivala et al. 2001).
P53 can impair progression through the cycle by several mechanisms and induce arrest at the G1/S border (G1 arrest) and the G2/M border (G2 arrest) as detailed below. In addition, p53 can have an impact on S-phase and attenuate replication (Dutta et al. 1993; Ogryzko et al. 1997; Waga et al. 1994; Zhou and Prives 2003). While p53-dependent transient cell cycle arrest is already triggered by relatively low or moderate damage, more severe damage can either result in apoptosis or a terminal growth arrest, also known as cellular senescence. The latter can be observed in a variety of cells derived from solid tumours after treatment with DNA damaging drugs and is driven to a major part by p53 and its target gene p21 (Chang et al. 1999a, b). Cellular senescence is also induced by high-dose gamma irradiation in human and mouse fibroblasts where it seems completely dependent on functional wild-type p53 and p21 (Speidel et al. 2006; Suzuki et al. 2001). Moreover, there is clear evidence for p53-dependent senescence occurring as a DNA damage response in vivo as demonstrated, for example, in Eµ-myc-driven lymphomas treated with the chemotherapeutic agent cyclophosphamide (Schmitt et al. 2002).
A transient arrest in response to DNA damage stops cells at the G1/S and/or G2/M borders, thereby preventing the amplification of damaged material by replication and cell division, respectively. P53 has been shown to promote cell cycle arrest in G1 and G2 through upregulation of several micro-RNAs, including miR-34a-c and miR-192/215. In turn, these micro-RNAs repress a number of important factors necessary for cell cycle progression resulting in transient cell cycle arrest or senescence (Braun et al. 2008; Georges et al. 2008; He et al. 2007a; Tarasov et al. 2007).
P53 has a particularly crucial role in DNA damage-induced G1 arrest. This is reflected by the fact that fibroblasts that lack functional wild-type p53 are also deficient in displaying G1 arrest in response to DNA damage (Kastan et al. 1992; Lowe et al. 1993a). The same phenotype is observed in cells (MEFs and HCT116) lacking the p53 target gene p21 (Brugarolas et al. 1995; Deng et al. 1995; Waldman et al. 1995). Hence, p53-mediated transcriptional upregulation of p21 is essentially required for DNA damage-induced G1 arrest, at least in cell culture settings (Dulic et al. 1994; El-Deiry et al. 1993, 1994). P21 is an inhibitor of cyclin-dependent kinases (CDKs) which are the drivers of cell cycle progression (Xiong et al. 1993). In particular, p21 strongly inhibits the Cyclin 2-CDK2 kinase which promotes transition from G1 into S-phase (Harper et al. 1993). Upregulation of p21 that is induced by p53 in response to DNA damage therefore leads to an efficient G1 arrest (Dulic et al. 1994; El-Deiry et al. 1993, 1994). In addition, another target gene, Ptprv, encoding for a transmembrane tyrosine phosphatase contributes to p53-induced G1 arrest in response to DNA damage and its deficiency resulted in a compromised ability of MEFs to induce G1 arrest in responses to gamma radiation (Doumont et al. 2005). However, the mechanism by which Ptprv exerts its cell cycle regulatory function remains to be elucidated.
P53 plays also a role in inducing G2 arrest, and this is mediated by upregulation of various target genes, including p21, Gadd45alpha, 14-3-3σ, Btg2, PCBP4/MCG10, GTSE-1/B99 and reprimo (Bunz et al. 1998; Hermeking et al. 1997; Kastan et al. 1992; Niculescu et al. 1998; Ohki et al. 2000; Rouault et al. 1996; Utrera et al. 1998; Zhu and Chen 2000). The mechanisms by which upregulation of these target genes effect G2 arrest are very diverse and include interactions with CDK1 (Gadd45alpha and Btg2; Ryu et al. 2004; Wang et al. 1999; Zhan et al. 1999), sequestration of Cyclin B1 and CDK1 (14-3-3σ; Chan et al. 1999), regulation of p21 mRNA stability (PCBP4/MCG10; Scoumanne et al. 2011) as well as other yet to be clarified means. In addition, there is evidence that p53 inhibits G2-M progression through direct repression of genes such as CDK1, Cyclin B1, cdc25 and others (Azzam et al. 1997; de Toledo et al. 1998; Passalaris et al. 1999; Spurgers et al. 2006; St Clair et al. 2004; Taylor et al. 1999). Moreover, p53 represses cell cycle progression also through upregulation of several microRNAs as mentioned before. Clearly, however, induction of G2 arrest in response to DNA damage does not require p53 and can be observed in p53-deficient cells (Bunz et al. 1998; Hirose et al. 2001; Kastan et al. 1991; Passalaris et al. 1999). Nevertheless, p53 and its target genes p21 and 14-3-3σ appear to be required to sustain an arrest in G2 (Bunz et al. 1998; Chan et al. 1999; Hirose et al. 2001).
The importance of p53 for DNA repair is reflected by the defects of p53-deficient cells. This was first noted in UV-irradiated fibroblasts where damage is repaired via the nucleotide excision repair (NER) pathway and UV-induced photo-products persist longer when p53 is not functional (Ford and Hanawalt 1995, 1997; Smith et al. 1995; Therrien et al. 1999; Wang et al. 1995). Similarly, compromised p53 function affected removal of DNA-adducts induced by tobacco carcinogens (Lloyd and Hanawalt 2000, 2002; Wani et al. 2000). However, p53 is also involved in the base excision repair (BER) and mismatch repair pathways (Chen and Sadowski 2005; de Souza-Pinto et al. 2004; Offer et al. 1999, 2001a, b; Scherer et al. 2000; Seo et al. 2002; Zhou et al. 2001; Zurer et al. 2004). Similarly, p53 regulates the repair of DNA double-strand breaks by homologous recombination (HR) and non-homologous end-joining (NHEJ) [reviewed in (Gatz and Wiesmuller 2006; Sengupta and Harris 2005)]. Of note, p53 can either suppress or promote DNA repair activities, revealing a somehow irritating complexity. This becomes particularly obvious with regard to the NHEJ processes that repair most DNA double-strand breaks in mammalian cells (Lieber et al. 2003). Here, p53 was shown to either stimulate (Lin et al. 2003; Tang et al. 1999; Yang et al. 1997) or inhibit (Akyuz et al. 2002; Bill et al. 1997; Bristow et al. 1998; Dahm-Daphi et al. 2005; Okorokov et al. 2002) DNA end-joining. Similarly, p53 would either enhance or reduce the activity of 3-methyladenine (3-MeAde) DNA glycosylase, an important enzyme in the BER pathway, and this was dependent on the type of DNA damage (Zurer et al. 2004). In addition, the phosphatase Wip1, also known as PPM1D, is induced by p53 in response to genotoxic stress and inhibits BER (Lu et al. 2004), although there is compelling evidence from several laboratories that p53 stimulates BER (Offer et al. 1999; Seo et al. 2002; Zhou and Prives 2003). Clearly, there are plausible reasons as to why a tumour suppressor would not only facilitate DNA repair but suppress it under certain conditions. The latter makes sense to efficiently kill cells after severe DNA damage, limit error-prone repair and/or switch off increased repair activity at completion of the DNA damage response.
Besides facilitating DNA repair by inducing and maintaining cell cycle arrest, p53 can promote DNA repair directly by upregulating the expression of DNA repair genes. These include Gadd45a (Smith et al. 1994), DBB2 (Hwang et al. 1999), XPC (Adimoolam and Ford 2002), p53R2 (Nakano et al. 2000; Tanaka et al. 2000), KARP-1 (Myung et al. 1998), thymine DNA glycosylase (da Costa et al. 2012) and MGMT (Grombacher et al. 1998; Rafferty et al. 1996). Notably, MGMT, a gene that is essential for converting the mutagenic DNA lesion O 6-methylguanine back to guanine, can also be downregulated by p53 as a means to efficiently kill cells in response to DNA damaging agents, alkylating drugs in particular (Grombacher et al. 1998; Harris et al. 1996). Similarly, p53 can suppress transcription of 3-methyladenine (3-MeAde) DNA glycosylase, an important enzyme in the BER pathway, in response to nitric oxide (Zurer et al. 2004). In addition to the mentioned genes, p53-responsive elements have also been identified in other genes related to DNA repair, including FANCC (Liebetrau et al. 1997), MSH2 (Scherer et al. 1996, 2000), MLH1 (Chen and Sadowski 2005) and PMS2 (Chen and Sadowski 2005; Gatz and Wiesmuller 2006).
Equally important for its role in DNA repair are transcription-independent activities of p53 (Sengupta and Harris 2005). P53 facilitates DNA repair in particular through direct protein–protein interactions with repair proteins such as the helicases XPB and XPD (Wang et al. 1995), the ribonucleotide reductase p53R2 (Xue et al. 2003), DNA polymerase beta (Zhou et al. 2001) and the homologous recombination factor RAD51 (Linke et al. 2003; Sturzbecher et al. 1996). In addition, p53 was shown to bind to damaged DNA in vitro in a non-sequence-specific manner via its C-terminus (Jayaraman and Prives 1995; Lee et al. 1995; Reed et al. 1995). Furthermore, p53 binds Holliday junctions and heteroduplex joints, which are both intermediate DNA structures of homologous recombination (Dudenhoffer et al. 1998; Janz and Wiesmuller 2002; Lee et al. 1997). Such binding of p53 to ‘unusual’ DNA structures has been associated with DNA repair; however, the underlying mechanisms remain incompletely understood. Much of this is caused by limitations in the methodology to assess the repair of chromatin-packed DNA in vivo.
The role of p53 responses in cancer therapy
The vast majority of cancer patients receive treatment with DNA damaging agents such as radiation and chemotherapeutic drugs. The preferred result of this treatment is the induction of cancer cell death as this outcome irreversibly eliminates these cells from the body. Most of the clinically used cytotoxic agents will activate p53 and induce p53-mediated DNA damage responses in cancer cells expressing wild-type p53. Because the vast majority of p53-mutations found in cancer patients impair the proper function of the p53 protein (Olivier et al. 2010), it is to expect that the p53 status has an influence on the outcome of cancer therapy. While early xenograft studies in mice have shown a clear correlation between p53 status and apoptotic cell death after treatment with adriamycin or gamma radiation (Lowe et al. 1994), the question of whether human wild-type p53 cancers respond generally better to therapy than cancers with mutated or deleted p53 does not have an easy general answer. This has mainly two reasons: as discussed before, p53 can either promote or suppress cell death with the outcome depending on the cell type and therapeutic agent. Secondly, the term ‘mutant p53’ describes a quite heterogeneous group of proteins that have biological activity themselves (Brosh and Rotter 2009). Although the vast majority of mutations found in cancer patients cluster in the DNA binding domain of p53 and hence impair its transcriptional activity, the exact type of mutation can make significant differences with regard to cellular DNA damage responses. Some mutations leave the p53 proteins with residual wild-type p53 activity. This could, for instance, mean that apoptotic functions are compromised but the ability to induce cell cycle arrest is retained (Ludwig et al. 1996; Rowan et al. 1996). Moreover, some mutations result in a ‘gain of function’, being it unique transactivation activities (e.g. induction of the multidrug resistance 1 gene by mutant p53 (Chin et al. 1992)) or interactions with other cellular proteins, such as p63, p73 or AMP-activated protein kinase (Brosh and Rotter 2009; Lozano 2007; Zhou et al. 2014). Together, this complexity makes it difficult to dissect which effects with regard to cancer therapy are truly related to a loss of wild-type p53 function rather than a consequence of biological activity exerted by the various mutated p53 proteins. Consequently, the real impact of p53 for the success of cancer therapy is not finally understood yet.
Many clinical studies have been conducted to determine whether and for which cancers the p53 status has prognostic and/or predictive relevance and this has been reviewed in detail by Brosh and Rotter (2009) and Tchelebi et al. (2014). A full, annotated and regularly updated list of studies that have assessed p53 mutations and their association with prognosis is available at the IARC Tp53 database (http://p53.iarc.fr/) (Petitjean et al. 2007). Of note, many older studies must be interpreted with caution. These studies would have determined the p53 status simply by means of the staining intensity in immunohistochemical analysis of tumour samples. An established practise in times when DNA sequencing was not as easy and cost-effective as it is today, this approach followed the simplified and therefore sometimes false concept that mutant p53 is more stable and thus more abundant than wild-type p53. More recent studies that used sequencing to determine the p53 status provide a more reliable basis for analysis of the prognostic value of p53.
As it turns out, there is a clear trend for certain cancer types, suggesting that mutations in p53 indeed segregate with poor prognosis. This is true for haematologic malignancies as well as cancers of the breast and colorectum (IARC database, version R17 from November 2013; Petitjean et al. 2007). Not surprisingly, this association is weaker for other tumour types, such as cancers of the bladder, brain or lungs. Inconsistencies and conflicting results may be caused by looking at different groups of tumours (e.g. ‘non-small cell lung cancer’ versus ‘lung adenocarcinoma’), different treatment regiments or differences in other yet to be clarified parameters. Clearly, some studies do not support an influence of p53 status on overall patient survival and therapy response at all. Notably too, for many cancers, the prognostic impact of p53 mutations has not been thoroughly studied at all. Table 1 shows the number of studies on the prognostic value of p53 mutations and their results as downloaded from the most current version (R17 from November 2013) of the IARC database (Petitjean et al. 2007).
Whereas a considerable number of studies have looked at the relationship between p53-status and survival, less is known about the impact that p53 mutations have on the direct and immediate response to chemotherapy. The IARC database (version R17) lists only 38 peer-reviewed studies that have explicitly looked at the response to chemotherapy in vivo. Eleven studies concern breast cancer and seven colorectal/rectum cancer, whereas other malignancies have been analysed by less than five studies each. A closer look at the breast cancer studies reveals strong heterogeneity in the patient cohorts, treatment regimens and measures of chemotherapy response or resistance (Table 2). These differences can explain some apparent discrepancies in the results. More importantly, this comparison highlights the need for more studies that assess therapy resistance in a standardized manner. This will be an essential requirement to decide on optimal therapy for individual patients. Notwithstanding these limitations, it seems clear that the disruption of p53 has a measurable and mostly negative impact on the success of cancer therapy in many cases.
While conventional chemo and radiation therapies are still the most important treatment for the majority of cancer patients, considerable efforts are made to substitute or complement these therapies with non-genotoxic agents. In this context, p53 has been regarded as an extremely promising target (Brown et al. 2009). Ideally, pharmacologic induction of p53-mediated DNA damage responses in a non-genotoxic way could induce apoptosis but reduce the side effects of treatment with conventional radiation and chemotherapy. Currently, more than 20 different p53-activating drugs are under pre-clinical and clinical evaluation and pursued by different companies [reviewed in (Khoo et al. 2014)]. These agents aim to either re-activate mutant p53 and thereby restore wild-type p53 functions in mutant p53-expressing cells or activate p53 in cancer cells expressing wild-type p53. The majority of drugs currently in clinical trials (all phase 1) are so-called Mdm2 antagonists (Khoo et al. 2014). These drugs disrupt the interaction of p53 and Mdm2 and thereby trigger stabilization and activation of wild-type p53. Although very promising, it is important to note that use of some of these drugs (e.g. nutlin-3) but not all of them (e.g. RITA) may also induce de novo p53 mutations and drug resistance, as demonstrated in tissue culture experiments (Aziz et al. 2011; Cinatl et al. 2014; Michaelis et al. 2011, 2012).
The role of p53-mediated DNA damage responses in tumour suppression
Any cellular programme that restricts the growth of cells with potentially mutagenic lesions and restores genomic integrity qualifies in principle as tumour suppressive. Because p53 triggers many of these programmes (transient cell cycle arrest, DNA repair, apoptosis and senescence), it seemed rather safe to assume that these activities of p53 (all of them being classic DNA damage responses) were responsible for its tumour suppressor function. Indeed, this concept remained unchallenged until recently. Many animal models were created to prove which target gene and/or programme regulated by p53 is responsible for its powerful tumour-suppressive effect. In these models, either target genes of p53 were deleted or the p53 gene itself mutated to impair some or all of its transcriptional activities. The details on most relevant animal models addressing p53-mediated tumour suppression have been summarized and expertly reviewed in two excellent articles (Bieging and Attardi 2012; Bieging et al. 2014) and hence do not need to be replicated here. In the paragraphs below, I rather focus on the aspects relevant for the role of p53-controlled DNA damage response programmes.
When assessing the tumour-suppressive effect of individual p53 targets and/or programmes in vivo, three approaches/read-outs were used. Firstly, spontaneous tumour formation was assessed. Here, p53-null mice provide the benchmark. These mice succumb to cancer with almost 100 % penetrance around 6 months of age with most of them developing thymic T cell lymphoma and occasionally B cell lymphoma or sarcoma (Donehower et al. 1992; Jacks et al. 1994; Kemp et al. 1994; Purdie et al. 1994). Consequently, mouse models with a deficiency in a target gene/cellular programme regulated by p53 were studied with regard to the onset of early thymic lymphoma development. The second approach relies on mouse models where the formation of cancer is driven by oncogenes including Eμ-myc (B cell lymphoma), KrasG12D (non-small cell lung cancer), SV40 T-antigen/T121 (brain cancer) or other gene defects that confer predisposition to cancer and where p53 is known to counteract tumorigenesis. Here, enhanced tumorigenesis or quicker progression of cancer (relative to wild-type control animals) would reveal the contribution of a specific p53-regulated target gene/mechanism to tumour suppression. The third approach is of immediate significance when discussing the role of DNA damage responses because it directly uses DNA damage, mostly gamma irradiation applied to very young mice (2 days–7 weeks old) but also UV-irradiation and chemicals, as a cancer-initiating stimulus. Significantly, irradiation of mice heterozygous (p53+/−)or deficient for both alleles of p53 (p53−/−) with low doses of ionizing radiation (1–4 Gy) showed a marked acceleration of tumorigenesis when compared to unirradiated controls (Kemp et al. 1994). Hence, this early study provides one of the clearest and most direct demonstrations for the importance of p53-mediated DNA damage responses for tumour suppression in vivo. However, in this context, it must be noted that the immediate response to acute DNA damage triggered by p53 might actually not contribute much to tumour suppression as suggested by studies with mouse models, in which p53 could be selectively activated and/or inactivated at different time points before or after irradiation (Christophorou et al. 2006; Hinkal et al. 2009). Both studies reported that the absence or presence of p53 during radiation treatment had no effect on radiation-induced lymphoma latency. Rather, p53-activity is required at much later time points to counteract the proliferation of abnormal cells with activated oncogenes. Therefore, persistent low levels of DNA damage and/or oncogene activation—both of which can of course be a consequence of a more severe genotoxic insult—appear crucial to trigger p53-mediated suppression of radiation-induced tumours in mice (Christophorou et al. 2006; Hinkal et al. 2009).
Conclusions that appear as conflicting at the first view have been drawn regarding the significance of the classic DNA damage response programmes apoptosis, cell cycle arrest and senescence for p53-mediated tumour suppression. At least in parts, this is due to the different experimental approaches used to dissect the importance of individual p53-regulated target genes and mechanisms for tumour suppression. However, two things seem clear: (1) the transcriptional activity of p53 is crucially important for its tumour suppressor function. This was demonstrated most compellingly in mice expressing a transcriptionally dead mutant p53 (p5325,26,53,54). These mice, in which both transactional activation domains (TADs) of the trp53 gene were mutated, resembled p53-null animals and did not show any protection against cancer formation (Brady et al. 2011; Jiang et al. 2011). (2) None of the existing knockout mice with deficiency in one or more p53-responsive target genes fully phenocopies p53-null mice (Bieging and Attardi 2012). Therefore, the most important target(s) with regard to tumour suppression have not been identified yet, or p53-mediated tumour suppression is achieved by the combination of the many processes and genes controlled by p53.
Clearly, there is strong evidence supporting an important contribution of the classic DNA damage response programmes to p53-mediated tumour suppression. For example, such evidence comes from mice, in which the p53 gene was mutated to impair its apoptotic activity but not its ability to induce cell cycle arrest, senescence and other effector programmes in response to DNA damage. This was done in independent laboratories using different strategies. The team around Gigi Lozano engineered a mutp53 R172P mouse that is the homologue of a missense point mutation found in cancer patients (R175P; Liu et al. 2004). Apoptosis deficiency was achieved by another modification in the Braithwaite laboratory. Here, mice expressing a deletion mutant of p53 (mΔpro p53) that lacks the proline-rich domain were studied (Slatter et al. 2010). Similar to the mutp53 R172P model, cells from these mice could induce p53-mediated cell cycle arrest but not apoptosis in response to DNA damage (Liu et al. 2004; Slatter et al. 2010). A third and again different approach was taken recently by a team led by Thorsten Stiewe. They generated mice expressing a cooperativity mutant of p53 (p53E177R) (Timofeev et al. 2013). This mutant p53 protein is impaired for interactions between adjacent p53 DNA binding domains in the tetrameric p53-DNA complex and thereby defective in transactivating proapoptotic target genes (Schlereth et al. 2010). When compared to p53-null mice, all of these mutant p53 mice (R172P, mΔpro p53 and p53E177R) showed a longer latency and reduced incidence of thymic lymphoma but eventually succumbed to other malignancies (Liu et al. 2004; Slatter et al. 2010; Timofeev et al. 2013). Together, these studies support the conclusion that apoptotic activities are important for p53-mediated tumour suppression in vivo because the deficiency in p53 apoptosis results in some form of cancer. However, the complete or partial protection against spontaneously developing thymic lymphoma in these mouse models (R172P, mΔpro p53 and p53E177R) highlights the fact that other activities of p53 (not apoptosis) are responsible for suppressing this type of cancer in mice.
In addition to apoptosis, also cell cycle arrest and senescence were shown to contribute to p53-mediated tumour suppression. For example, loss of p21—the crucial target gene for DNA damage-induced and p53-dependent G1 arrest and senescence—accelerated tumour development in mutp53 (R172P) and MMTV/v-Ha-ras transgenic mice (Adnane et al. 2000; Barboza et al. 2006), provoked renal carcinomas in Apc knockout mice (Cole et al. 2010) and increased sensitivity to carcinogen-induced cancer formation (Jackson et al. 2002). However, it should be noted that p21-null mice are generally not tumour-prone and only subtly sensitized to irradiation-induced cancer (Deng et al. 1995; Jackson et al. 2003; Martin-Caballero et al. 2001). Conversely, mice deficient in Gadd45a—another p53 target gene involved in cell cycle regulation and DNA repair—were markedly sensitized to gamma and UV-radiation-induced cancers (Hildesheim et al. 2002; Hollander et al. 1999). Interestingly, loss of gadd45a accelerated tumorigenesis in ras-driven mammary tumours but delayed cancer development in myc-driven breast cancer (Tront et al. 2006, 2010). Hence, different mechanisms are required for tumour suppression, and parameters such as cancer type, tissue and cancer-driving signal determine what is necessary to counteract cancer development and progression. Accordingly, it makes sense that specific target genes or DNA damage response programme controlled by p53 are crucial for tumour suppression under some conditions but not required in other settings. Results from the many other studies dissecting p53-mediated tumour suppression in vivo [reviewed in (Bieging and Attardi 2012)] fit into this concept as well.
Whereas the studies highlighted above are consistent with the idea that p53-mediated apoptosis, senescence and cell cycle arrest are important for tumour suppression, a few other reports challenged the view that these programmes are the sole effectors in preventing cancer (Brady et al. 2011; Li et al. 2012; Valente et al. 2013). Spectacular insights came from a model recently established by Wei Gu’s group (Li et al. 2012). The so-called p533KR mice have three mutations in the p53 gene preventing p53 from being acetylated at three lysines (K117, K161, K162) that are crucial for transactivating a substantial subset of p53-target genes. Cells from these mice behaved like p53-null cells with regard to the induction of cell cycle arrest, senescence and apoptosis upon treatment with DNA damaging agents. Surprisingly, p533KR mice did not develop thymic lymphoma or any other spontaneous tumours when monitored up to 16 months (Li et al. 2012). This unexpected result suggested that the classic DNA damage responses triggered by p53 were completely dispensable for suppression of spontaneous tumours. A similar conclusion was made on the basis of another mouse model, in which not p53 was modified but three crucial target genes, Puma, Noxa and p21, deleted. Cells from these mice were defective for p53-dependent and DNA damage-induced apoptosis and senescence, but again the mice did not develop spontaneous tumours (Valente et al. 2013). Similarly, an earlier mouse model expressing a mutant p53 protein that is unable to elicit responses to acute DNA damage (p53 L25Q; W26S, also referred to as p5325,26) showed a remarkable ability to suppress tumour formation (Brady et al. 2011).
Clearly, these studies demonstrate compellingly that for suppression of spontaneous tumours in mice, the classic DNA damage response programmes regulated by p53 are dispensable. Hence, other cellular programmes and/or target genes that have been underappreciated or not identified yet appear to play a powerful role in tumour suppression. Their identification is currently a major focus of several laboratories with regulation of metabolism and/or the coordination of DNA repair emerging as particularly interesting effector processes (Berkers et al. 2013; Li et al. 2012; Timofeev et al. 2013; Valente et al. 2013). What the aforementioned studies do not support (and also do not claim) is the conclusion that p53-mediated apoptosis, senescence and cell cycle arrest are generally dispensable for tumour suppression, although the titles ‘Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence’ (Li et al. 2012) and ‘p53 Efficiently Suppresses Tumor Development in the Complete Absence of Its Cell-Cycle Inhibitory and Proapoptotic Effectors p21, Puma, and Noxa’ (Valente et al. 2013) may suggest so. Indeed, it can be expected that both of the mentioned mouse models (p533KR and puma−/−, noxa−/−, p21−/−) will show impaired tumour-suppressive activity in another context such as oncogene activation and/or exposure to DNA damage. Notably, the puma single-gene knockout mouse showed enhanced tumorigenesis in myc-driven cancers (Michalak et al. 2009). Together, the numerous existing mouse models underscore the somewhat trivial, yet often underappreciated concept that, depending on the context, effective tumour suppression requires different genes and cellular programmes. The classic p53-mediated DNA damage response programmes play certainly an important part, but other effector processes, p53-dependent and independent ones, are necessary complements to suppress cancer.
Concluding remarks
P53 is a very complex and multi-faceted protein with many functions to which DNA damage-related responses provide only one albeit crucial component. Despite the many studies carried out in the past, our knowledge still has substantial gaps. This is illustrated for example by the lack of compelling data on the predictive power of p53 mutations with regard to therapy response and survival in most cancers. A central aspect in all areas of p53 research is the crucial importance of the cellular context. We can only understand p53 if parameters like cell type and stress signal are taken into account. With these being considered, many apparently conflicting results are no longer inconsistent (as discussed for the tumour suppressor function) but simply reflect the extreme versatility and multi-functionality of p53.
References
Adimoolam S, Ford JM (2002) p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc Natl Acad Sci USA 99(20):12985–12990. doi:10.1073/pnas.202485699
Adnane J, Jackson RJ, Nicosia SV, Cantor AB, Pledger WJ, Sebti SM (2000) Loss of p21WAF1/CIP1 accelerates Ras oncogenesis in a transgenic/knockout mammary cancer model. Oncogene 19(47):5338–5347. doi:10.1038/sj.onc.1203956
Agarwal ML, Agarwal A, Taylor WR, Stark GR (1995) p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc Natl Acad Sci USA 92(18):8493–8497
Akyuz N, Boehden GS, Susse S et al (2002) DNA substrate dependence of p53-mediated regulation of double-strand break repair. Mol Cell Biol 22(17):6306–6317
Allen MA, Andrysik Z, Dengler VL et al (2014) Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. eLife 3:e02200. doi:10.7554/eLife.02200
Attardi LD, Reczek EE, Cosmas C et al (2000) PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev 14(6):704–718
Aziz MH, Shen H, Maki CG (2011) Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene 30(46):4678–4686. doi:10.1038/onc.2011.185
Azzam EI, de Toledo SM, Pykett MJ, Nagasawa H, Little JB (1997) CDC2 is down-regulated by ionizing radiation in a p53-dependent manner. Cell Growth Differ Mol Biol J Am Assoc Cancer Res 8(11):1161–1169
Barboza JA, Liu G, Ju Z, El-Naggar AK, Lozano G (2006) p21 delays tumor onset by preservation of chromosomal stability. Proc Natl Acad Sci USA 103(52):19842–19847. doi:10.1073/pnas.0606343104
Bargonetti J, Friedman PN, Kern SE, Vogelstein B, Prives C (1991) Wild-type but not mutant p53 immunopurified proteins bind to sequences adjacent to the SV40 origin of replication. Cell 65(6):1083–1091
Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH (2013) Metabolic regulation by p53 family members. Cell Metab 18(5):617–633. doi:10.1016/j.cmet.2013.06.019
Berns EM, Foekens JA, Vossen R et al (2000) Complete sequencing of TP53 predicts poor response to systemic therapy of advanced breast cancer. Cancer Res 60(8):2155–2162
Bertheau P, Turpin E, Rickman DS et al (2007) Exquisite sensitivity of TP53 mutant and basal breast cancers to a dose-dense epirubicin-cyclophosphamide regimen. PLoS Med 4(3):e90. doi:10.1371/journal.pmed.0040090
Bieging KT, Attardi LD (2012) Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol 22(2):97–106. doi:10.1016/j.tcb.2011.10.006
Bieging KT, Mello SS, Attardi LD (2014) Unravelling mechanisms of p53-mediated tumour suppression. Natl Rev Cancer 14(5):359–370. doi:10.1038/nrc3711
Bill CA, Yu Y, Miselis NR, Little JB, Nickoloff JA (1997) A role for p53 in DNA end rejoining by human cell extracts. Mutat Res 385(1):21–29
Bommer GT, Gerin I, Feng Y et al (2007) p53-Mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol CB 17(15):1298–1307. doi:10.1016/j.cub.2007.06.068
Brady CA, Jiang D, Mello SS et al (2011) Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 145(4):571–583. doi:10.1016/j.cell.2011.03.035
Braun CJ, Zhang X, Savelyeva I et al (2008) p53-Responsive micrornas 192 and 215 are capable of inducing cell cycle arrest. Cancer Res 68(24):10094–10104. doi:10.1158/0008-5472.can-08-1569
Bristow RG, Hu Q, Jang A et al (1998) Radioresistant MTp53-expressing rat embryo cell transformants exhibit increased DNA-dsb rejoining during exposure to ionizing radiation. Oncogene 16(14):1789–1802. doi:10.1038/sj.onc.1201935
Brosh R, Rotter V (2009) When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 9(10):701–713
Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP (2009) Awakening guardian angels: drugging the p53 pathway. Natl Rev Cancer 9(12):862–873. doi:10.1038/nrc2763
Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ (1995) Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 377(6549):552–557
Bunz F, Dutriaux A, Lengauer C et al (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282(5393):1497–1501
Bunz F, Hwang PM, Torrance C et al (1999) Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Investig 104(3):263–269. doi:10.1172/jci6863
Carvajal LA, Manfredi JJ (2013) Another fork in the road—life or death decisions by the tumour suppressor p53. EMBO Rep 14(5):414–421. doi:10.1038/embor.2013.25
Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B (1999) 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature 401(6753):616–620. doi:10.1038/44188
Chang BD, Broude EV, Dokmanovic M et al (1999a) A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res 59(15):3761–3767
Chang BD, Xuan Y, Broude EV et al (1999b) Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene 18(34):4808–4818. doi:10.1038/sj.onc.1203078
Chang TC, Wentzel EA, Kent OA et al (2007) Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 26(5):745–752. doi:10.1016/j.molcel.2007.05.010
Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD (1999) Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Natl Acad Sci USA 96(24):13777–13782
Chen J, Sadowski I (2005) Identification of the mismatch repair genes PMS2 and MLH1 as p53 target genes by using serial analysis of binding elements. Proc Natl Acad Sci USA 102(13):4813–4818. doi:10.1073/pnas.0407069102
Chen X, Ko LJ, Jayaraman L, Prives C (1996) p53 Levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev 10(19):2438–2451
Cheng Q, Chen L, Li Z, Lane WS, Chen J (2009) ATM activates p53 by regulating MDM2 oligomerization and E3 processivity. EMBO J 28(24):3857–3867. doi:10.1038/emboj.2009.294
Chin KV, Ueda K, Pastan I, Gottesman MM (1992) Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science 255(5043):459–462
Chipuk JE, Kuwana T, Bouchier-Hayes L et al (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303(5660):1010–1014
Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR (2005) PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 309(5741):1732–1735
Chrisanthar R, Knappskog S, Lokkevik E et al (2008) CHEK2 mutations affecting kinase activity together with mutations in TP53 indicate a functional pathway associated with resistance to epirubicin in primary breast cancer. PLoS ONE 3(8):e3062. doi:10.1371/journal.pone.0003062
Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI (2006) The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 443(7108):214–217. doi:10.1038/nature05077
Cinatl J, Speidel D, Hardcastle I, Michaelis M (2014) Resistance acquisition to MDM2 inhibitors. Biochem Soc Trans 42(4):752–757. doi:10.1042/bst20140035
Clarke AR, Purdie CA, Harrison DJ et al (1993) Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 362(6423):849–852. doi:10.1038/362849a0
Cole AM, Ridgway RA, Derkits SE et al (2010) p21 loss blocks senescence following Apc loss and provokes tumourigenesis in the renal but not the intestinal epithelium. EMBO Mol Med 2(11):472–486. doi:10.1002/emmm.201000101
Concepcion CP, Han YC, Mu P et al (2012) Intact p53-dependent responses in miR-34-deficient mice. PLoS Genet 8(7):e1002797. doi:10.1371/journal.pgen.1002797
da Costa NM, Hautefeuille A, Cros MP et al (2012) Transcriptional regulation of thymine DNA glycosylase (TDG) by the tumor suppressor protein p53. Cell Cycle 11(24):4570–4578. doi:10.4161/cc.22843
Dahm-Daphi J, Hubbe P, Horvath F et al (2005) Nonhomologous end-joining of site-specific but not of radiation-induced DNA double-strand breaks is reduced in the presence of wild-type p53. Oncogene 24(10):1663–1672. doi:10.1038/sj.onc.1208396
Das S, Raj L, Zhao B et al (2007) Hzf Determines cell survival upon genotoxic stress by modulating p53 transactivation. Cell 130(4):624–637. doi:10.1016/j.cell.2007.06.013
de Souza-Pinto NC, Harris CC, Bohr VA (2004) p53 Functions in the incorporation step in DNA base excision repair in mouse liver mitochondria. Oncogene 23(39):6559–6568. doi:10.1038/sj.onc.1207874
de Toledo SM, Azzam EI, Keng P, Laffrenier S, Little JB (1998) Regulation by ionizing radiation of CDC2, cyclin A, cyclin B, thymidine kinase, topoisomerase IIalpha, and RAD51 expression in normal human diploid fibroblasts is dependent on p53/p21Waf1. Cell growth Differ Mol Biol J Am Assoc Cancer Res 9(11):887–896
Deng C, Zhang P, Harper JW, Elledge SJ, Leder P (1995) Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82(4):675–684
Di Leonardo A, Linke SP, Clarkin K, Wahl GM (1994) DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev 8(21):2540–2551
Donehower LA, Harvey M, Slagle BL et al (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356(6366):215–221. doi:10.1038/356215a0
Doumont G, Martoriati A, Beekman C et al (2005) G1 checkpoint failure and increased tumor susceptibility in mice lacking the novel p53 target Ptprv. EMBO J 24(17):3093–3103. doi:10.1038/sj.emboj.7600769
Dudenhoffer C, Rohaly G, Will K, Deppert W, Wiesmuller L (1998) Specific mismatch recognition in heteroduplex intermediates by p53 suggests a role in fidelity control of homologous recombination. Mol Cell Biol 18(9):5332–5342
Dulic V, Kaufmann WK, Wilson SJ et al (1994) p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 76(6):1013–1023
Dutta A, Ruppert JM, Aster JC, Winchester E (1993) Inhibition of DNA replication factor RPA by p53. Nature 365(6441):79–82. doi:10.1038/365079a0
El-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B (1992) Definition of a consensus binding site for p53. Nat Genet 1(1):45–49. doi:10.1038/ng0492-45
El-Deiry WS, Tokino T, Velculescu VE et al (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75(4):817–825
El-Deiry WS, Harper JW, O’Connor PM et al (1994) WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res 54(5):1169–1174
Erster S, Mihara M, Kim RH, Petrenko O, Moll UM (2004) In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol Cell Biol 24(15):6728–6741
Espinosa JM, Verdun RE, Emerson BM (2003) p53 Functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol Cell 12(4):1015–1027
Fan S, El-Deiry WS, Bae I et al (1994) p53 Gene mutations are associated with decreased sensitivity of human lymphoma cells to DNA damaging agents. Cancer Res 54(22):5824–5830
Ford JM, Hanawalt PC (1995) Li-Fraumeni syndrome fibroblasts homozygous for p53 mutations are deficient in global DNA repair but exhibit normal transcription-coupled repair and enhanced UV resistance. Proc Natl Acad Sci USA 92(19):8876–8880
Ford JM, Hanawalt PC (1997) Expression of wild-type p53 is required for efficient global genomic nucleotide excision repair in UV-irradiated human fibroblasts. J Biol Chem 272(44):28073–28080
Fritsche M, Haessler C, Brandner G (1993) Induction of nuclear accumulation of the tumor-suppressor protein p53 by DNA-damaging agents. Oncogene 8(2):307–318
Gatz SA, Wiesmuller L (2006) p53 in recombination and repair. Cell Death Differ 13(6):1003–1016. doi:10.1038/sj.cdd.4401903
Geisler S, Lonning PE, Aas T et al (2001) Influence of TP53 gene alterations and c-erbB-2 expression on the response to treatment with doxorubicin in locally advanced breast cancer. Cancer Res 61(6):2505–2512
Geisler S, Borresen-Dale AL, Johnsen H et al (2003) TP53 Gene mutations predict the response to neoadjuvant treatment with 5-fluorouracil and mitomycin in locally advanced breast cancer. Clin Cancer Res 9(15):5582–5588
Georges SA, Biery MC, Kim SY et al (2008) Coordinated regulation of cell cycle transcripts by p53-Inducible microRNAs, miR-192 and miR-215. Cancer Res 68(24):10105–10112. doi:10.1158/0008-5472.can-08-1846
Grombacher T, Eichhorn U, Kaina B (1998) p53 is involved in regulation of the DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) by DNA damaging agents. Oncogene 17(7):845–851. doi:10.1038/sj.onc.1202000
Haldar S, Negrini M, Monne M, Sabbioni S, Croce CM (1994) Down-regulation of bcl-2 by p53 in breast cancer cells. Cancer Res 54(8):2095–2097
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75(4):805–816
Harris LC, Remack JS, Houghton PJ, Brent TP (1996) Wild-type p53 suppresses transcription of the human O6-methylguanine-DNA methyltransferase gene. Cancer Res 56(9):2029–2032
Harris LN, Broadwater G, Lin NU et al (2006) Molecular subtypes of breast cancer in relation to paclitaxel response and outcomes in women with metastatic disease: results from CALGB 9342. Breast Cancer Res BCR 8(6):R66. doi:10.1186/bcr1622
Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387(6630):296–299. doi:10.1038/387296a0
He L, He X, Lim LP et al (2007a) A microRNA component of the p53 tumour suppressor network. Nature 447(7148):1130–1134. doi:10.1038/nature05939
He L, He X, Lowe SW, Hannon GJ (2007b) microRNAs join the p53 network—another piece in the tumour-suppression puzzle. Nat Rev Cancer 7(11):819–822. doi:10.1038/nrc2232
Heinlein C, Deppert W, Braithwaite AW, Speidel D (2010) A rapid and optimization-free procedure allows the in vivo detection of subtle cell cycle and ploidy alterations in tissues by flow cytometry. Cell Cycle 9(17):3584–3590
Hermeking H, Lengauer C, Polyak K et al (1997) 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell 1(1):3–11
Hildesheim J, Bulavin DV, Anver MR et al (2002) Gadd45a protects against UV irradiation-induced skin tumors, and promotes apoptosis and stress signaling via MAPK and p53. Cancer Res 62(24):7305–7315
Hinkal G, Parikh N, Donehower LA (2009) Timed somatic deletion of p53 in mice reveals age-associated differences in tumor progression. PLoS ONE 4(8):e6654. doi:10.1371/journal.pone.0006654
Hirose Y, Berger MS, Pieper RO (2001) p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res 61(5):1957–1963
Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M (2002) Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem 277(5):3247–3257. doi:10.1074/jbc.M106643200
Hollander MC, Sheikh MS, Bulavin DV et al (1999) Genomic instability in Gadd45a-deficient mice. Nat Genet 23(2):176–184. doi:10.1038/13802
Hu W, Feng Z, Levine AJ (2012) The regulation of multiple p53 stress responses is mediated through MDM2. Genes Cancer 3(3–4):199–208. doi:10.1177/1947601912454734
Hunten S, Siemens H, Kaller M, Hermeking H (2013) The p53/microRNA network in cancer: experimental and bioinformatics approaches. Adv Exp Med Biol 774:77–101. doi:10.1007/978-94-007-5590-1_5
Hwang BJ, Ford JM, Hanawalt PC, Chu G (1999) Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc Natl Acad Sci USA 96(2):424–428
Jacks T, Remington L, Williams BO et al (1994) Tumor spectrum analysis in p53-mutant mice. Curr Biol CB 4(1):1–7
Jackson RJ, Adnane J, Coppola D, Cantor A, Sebti SM, Pledger WJ (2002) Loss of the cell cycle inhibitors p21(Cip1) and p27(Kip1) enhances tumorigenesis in knockout mouse models. Oncogene 21(55):8486–8497. doi:10.1038/sj.onc.1205946
Jackson RJ, Engelman RW, Coppola D, Cantor AB, Wharton W, Pledger WJ (2003) p21Cip1 nullizygosity increases tumor metastasis in irradiated mice. Cancer Res 63(12):3021–3025
Janz C, Wiesmuller L (2002) Wild-type p53 inhibits replication-associated homologous recombination. Oncogene 21(38):5929–5933. doi:10.1038/sj.onc.1205757
Jayaraman J, Prives C (1995) Activation of p53 sequence-specific DNA binding by short single strands of DNA requires the p53 C-terminus. Cell 81(7):1021–1029
Jeffers JR, Parganas E, Lee Y et al (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4(4):321–328
Jiang D, Brady CA, Johnson TM et al (2011) Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proc Natl Acad Sci USA 108(41):17123–17128. doi:10.1073/pnas.1111245108
Kandioler-Eckersberger D, Ludwig C, Rudas M et al (2000) TP53 Mutation and p53 overexpression for prediction of response to neoadjuvant treatment in breast cancer patients. Clin Cancer Res 6(1):50–56
Kandoth C, McLellan MD, Vandin F et al (2013) Mutational landscape and significance across 12 major cancer types. Nature 502(7471):333–339. doi:10.1038/nature12634
Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW (1991) Participation of p53 protein in the cellular response to DNA damage. Cancer Res 51(23 Part 1):6304–6311
Kastan MB, Zhan Q, El-Deiry WS et al (1992) A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71(4):587–597
Kemp CJ, Wheldon T, Balmain A (1994) p53-Deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat Genet 8(1):66–69. doi:10.1038/ng0994-66
Kern SE, Kinzler KW, Bruskin A et al (1991) Identification of p53 as a sequence-specific DNA-binding protein. Science 252(5013):1708–1711
Khoo KH, Verma CS, Lane DP (2014) Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov 13(3):217–236. doi:10.1038/nrd4236
Kracikova M, Akiri G, George A, Sachidanandam R, Aaronson SA (2013) A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ 20(4):576–588. doi:10.1038/cdd.2012.155
Kubbutat MH, Jones SN, Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387(6630):299–303. doi:10.1038/387299a0
Lane DP (1992) Cancer. p53, guardian of the genome. Nature 358(6381):15–16
Lee S, Elenbaas B, Levine A, Griffith J (1995) p53 and its 14 kDa C-terminal domain recognize primary DNA damage in the form of insertion/deletion mismatches. Cell 81(7):1013–1020
Lee S, Cavallo L, Griffith J (1997) Human p53 binds holliday junctions strongly and facilitates their cleavage. J Biol Chem 272(11):7532–7539
Leu JI, Dumont P, Hafey M, Murphy ME, George DL (2004) Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 6(5):443–450
Li T, Kon N, Jiang L et al (2012) Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149(6):1269–1283. doi:10.1016/j.cell.2012.04.026
Lieber MR, Ma Y, Pannicke U, Schwarz K (2003) Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 4(9):712–720. doi:10.1038/nrm1202
Liebetrau W, Budde A, Savoia A, Grummt F, Hoehn H (1997) p53 Activates Fanconi anemia group C gene expression. Hum Mol Genet 6(2):277–283
Lin Y, Ma W, Benchimol S (2000) Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat Genet 26(1):122–127. doi:10.1038/79102
Lin Y, Waldman BC, Waldman AS (2003) Suppression of high-fidelity double-strand break repair in mammalian chromosomes by pifithrin-alpha, a chemical inhibitor of p53. DNA Repair 2(1):1–11
Linke SP, Sengupta S, Khabie N et al (2003) p53 Interacts with hRAD51 and hRAD54, and directly modulates homologous recombination. Cancer Res 63(10):2596–2605
Liu TJ, El-Naggar AK, McDonnell TJ et al (1995) Apoptosis induction mediated by wild-type p53 adenoviral gene transfer in squamous cell carcinoma of the head and neck. Cancer Res 55(14):3117–3122
Liu G, Parant JM, Lang G et al (2004) Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet 36(1):63–68. doi:10.1038/ng1282
Lloyd DR, Hanawalt PC (2000) p53-dependent global genomic repair of benzo[a]pyrene-7,8-diol-9,10-epoxide adducts in human cells. Cancer Res 60(3):517–521
Lloyd DR, Hanawalt PC (2002) p53 Controls global nucleotide excision repair of low levels of structurally diverse benzo(g)chrysene-DNA adducts in human fibroblasts. Cancer Res 62(18):5288–5294
Lowe SW, Ruley HE, Jacks T, Housman DE (1993a) p53-Dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 74(6):957–967
Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T (1993b) p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 362(6423):847–849. doi:10.1038/362847a0
Lowe SW, Bodis S, McClatchey A et al (1994) p53 Status and the efficacy of cancer therapy in vivo. Science 266(5186):807–810
Lozano G (2007) The oncogenic roles of p53 mutants in mouse models. Curr Opin Genet Dev 17(1):66–70
Lu X, Bocangel D, Nannenga B, Yamaguchi H, Appella E, Donehower LA (2004) The p53-induced oncogenic phosphatase PPM1D interacts with uracil DNA glycosylase and suppresses base excision repair. Mol Cell 15(4):621–634. doi:10.1016/j.molcel.2004.08.007
Ludwig RL, Bates S, Vousden KH (1996) Differential activation of target cellular promoters by p53 mutants with impaired apoptotic function. Mol Cell Biol 16(9):4952–4960
Malkin D, Li FP, Strong LC et al (1990) Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250(4985):1233–1238
Maltzman W, Czyzyk L (1984) UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol Cell Biol 4(9):1689–1694
Martin-Caballero J, Flores JM, Garcia-Palencia P, Serrano M (2001) Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res 61(16):6234–6238
Massague J (2004) G1 cell-cycle control and cancer. Nature 432(7015):298–306. doi:10.1038/nature03094
Maya R, Balass M, Kim ST et al (2001) ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev 15(9):1067–1077. doi:10.1101/gad.886901
McDonald ER 3rd, Wu GS, Waldman T, El-Deiry WS (1996) Repair defect in p21WAF1/CIP1-/-human cancer cells. Cancer Res 56(10):2250–2255
Medema RH, Macurek L (2012) Checkpoint control and cancer. Oncogene 31(21):2601–2613. doi:10.1038/onc.2011.451
Meek DW, Anderson CW (2009) Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol 1(6):a000950. doi:10.1101/cshperspect.a000950
Michaelis M, Rothweiler F, Barth S et al (2011) Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis 2:e243
Michaelis M, Rothweiler F, Agha B et al (2012) Human neuroblastoma cells with acquired resistance to the p53 activator RITA retain functional p53 and sensitivity to other p53 activating agents. Cell Death Dis 3:e294. doi:10.1038/cddis.2012.35
Michalak EM, Villunger A, Adams JM, Strasser A (2008) In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ 15(6):1019–1029. doi:10.1038/cdd.2008.16
Michalak EM, Jansen ES, Happo L et al (2009) Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ 16(5):684–696. doi:10.1038/cdd.2008.195
Mihara M, Erster S, Zaika A et al (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11(3):577–590
Mirza A, McGuirk M, Hockenberry TN et al (2002) Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene 21(17):2613–2622. doi:10.1038/sj.onc.1205353
Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80(2):293–299
Miyashita T, Harigai M, Hanada M, Reed JC (1994) Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer Res 54(12):3131–3135
Momand J, Zambetti GP, Olson DC, George D, Levine AJ (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69(7):1237–1245
Moroni MC, Hickman ES, Lazzerini Denchi E et al (2001) Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol 3(6):552–558. doi:10.1038/35078527
Muller M, Wilder S, Bannasch D et al (1998) p53 Activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med 188(11):2033–2045
Myung K, Braastad C, He DM, Hendrickson EA (1998) KARP-1 is induced by DNA damage in a p53- and ataxia telangiectasia mutated-dependent fashion. Proc Natl Acad Sci USA 95(13):7664–7669
Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7(3):683–694
Nakano K, Balint E, Ashcroft M, Vousden KH (2000) A ribonucleotide reductase gene is a transcriptional target of p53 and p73. Oncogene 19(37):4283–4289
Niculescu AB 3rd, Chen X, Smeets M, Hengst L, Prives C, Reed SI (1998) Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol 18(1):629–643
Oda E, Ohki R, Murasawa H et al (2000a) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288(5468):1053–1058
Oda K, Arakawa H, Tanaka T et al (2000b) p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 102(6):849–862
Offer H, Wolkowicz R, Matas D, Blumenstein S, Livneh Z, Rotter V (1999) Direct involvement of p53 in the base excision repair pathway of the DNA repair machinery. FEBS Lett 450(3):197–204
Offer H, Milyavsky M, Erez N et al (2001a) Structural and functional involvement of p53 in BER in vitro and in vivo. Oncogene 20(5):581–589. doi:10.1038/sj.onc.1204120
Offer H, Zurer I, Banfalvi G et al (2001b) p53 Modulates base excision repair activity in a cell cycle-specific manner after genotoxic stress. Cancer Res 61(1):88–96
Ogryzko VV, Wong P, Howard BH (1997) WAF1 retards S-phase progression primarily by inhibition of cyclin-dependent kinases. Mol Cell Biol 17(8):4877–4882
Ohki R, Nemoto J, Murasawa H et al (2000) Reprimo, a new candidate mediator of the p53-mediated cell cycle arrest at the G2 phase. J Biol Chem 275(30):22627–22630. doi:10.1074/jbc.C000235200
Okorokov AL, Warnock L, Milner J (2002) Effect of wild-type, S15D and R175H p53 proteins on DNA end joining in vitro: potential mechanism of DNA double-strand break repair modulation. Carcinogenesis 23(4):549–557
Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B (1993) Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362(6423):857–860. doi:10.1038/362857a0
Olivier M, Hollstein M, Hainaut P (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2(1):a001008. doi:10.1101/cshperspect.a001008
Otsuka K, Ochiya T (2014) Genetic networks lead and follow tumor development: microRNA regulation of cell cycle and apoptosis in the p53 pathways. BioMed Res Int 2014:749724. doi:10.1155/2014/749724
Owen-Schaub LB, Zhang W, Cusack JC et al (1995) Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol Cell Biol 15(6):3032–3040
Passalaris TM, Benanti JA, Gewin L, Kiyono T, Galloway DA (1999) The G(2) checkpoint is maintained by redundant pathways. Mol Cell Biol 19(9):5872–5881
Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M (2007) TP53 Mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene 26(15):2157–2165
Polyak K, Waldman T, He TC, Kinzler KW, Vogelstein B (1996) Genetic determinants of p53-induced apoptosis and growth arrest. Genes Dev 10(15):1945–1952
Purdie CA, Harrison DJ, Peter A et al (1994) Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene 9(2):603–609
Rafferty JA, Clarke AR, Sellappan D, Koref MS, Frayling IM, Margison GP (1996) Induction of murine O6-alkylguanine-DNA-alkyltransferase in response to ionising radiation is p53 gene dose dependent. Oncogene 12(3):693–697
Raver-Shapira N, Marciano E, Meiri E et al (2007) Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell 26(5):731–743. doi:10.1016/j.molcel.2007.05.017
Reed M, Woelker B, Wang P, Wang Y, Anderson ME, Tegtmeyer P (1995) The C-terminal domain of p53 recognizes DNA damaged by ionizing radiation. Proc Natl Acad Sci USA 92(21):9455–9459
Roberts SA, Gordenin DA (2014) Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer 14(12):786–800. doi:10.1038/nrc3816
Robles AI, Bemmels NA, Foraker AB, Harris CC (2001) APAF-1 is a transcriptional target of p53 in DNA damage-induced apoptosis. Cancer Res 61(18):6660–6664
Rouault JP, Falette N, Guehenneux F et al (1996) Identification of BTG2, an antiproliferative p53-dependent component of the DNA damage cellular response pathway. Nat Genet 14(4):482–486. doi:10.1038/ng1296-482
Rowan S, Ludwig RL, Haupt Y et al (1996) Specific loss of apoptotic but not cell-cycle arrest function in a human tumor derived p53 mutant. EMBO J 15(4):827–838
Ryu MS, Lee MS, Hong JW, Hahn TR, Moon E, Lim IK (2004) TIS21/BTG2/PC3 is expressed through PKC-delta pathway and inhibits binding of cyclin B1-Cdc2 and its activity, independent of p53 expression. Exp Cell Res 299(1):159–170. doi:10.1016/j.yexcr.2004.05.014
Scherer SJ, Welter C, Zang KD, Dooley S (1996) Specific in vitro binding of p53 to the promoter region of the human mismatch repair gene hMSH2. Biochem Biophys Res Commun 221(3):722–728. doi:10.1006/bbrc.1996.0663
Scherer SJ, Maier SM, Seifert M et al (2000) p53 and c-Jun functionally synergize in the regulation of the DNA repair gene hMSH2 in response to UV. J Biol Chem 275(48):37469–37473. doi:10.1074/jbc.M006990200
Schlereth K, Beinoraviciute-Kellner R, Zeitlinger MK et al (2010) DNA binding cooperativity of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 38(3):356–368. doi:10.1016/j.molcel.2010.02.037
Schmitt CA, Fridman JS, Yang M et al (2002) A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 109(3):335–346
Scoumanne A, Cho SJ, Zhang J, Chen X (2011) The cyclin-dependent kinase inhibitor p21 is regulated by RNA-binding protein PCBP4 via mRNA stability. Nucleic Acids Res 39(1):213–224. doi:10.1093/nar/gkq778
Sengupta S, Harris CC (2005) p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol 6(1):44–55. doi:10.1038/nrm1546
Seo YR, Fishel ML, Amundson S, Kelley MR, Smith ML (2002) Implication of p53 in base excision DNA repair: in vivo evidence. Oncogene 21(5):731–737. doi:10.1038/sj.onc.1205129
Shao L, Sun Y, Zhang Z et al (2010) Deletion of proapoptotic Puma selectively protects hematopoietic stem and progenitor cells against high-dose radiation. Blood 115(23):4707–4714. doi:10.1182/blood-2009-10-248872
Shaw P, Bovey R, Tardy S, Sahli R, Sordat B, Costa J (1992) Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc Natl Acad Sci USA 89(10):4495–4499
Shieh SY, Ikeda M, Taya Y, Prives C (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91(3):325–334
Slatter TL, Ganesan P, Holzhauer C et al (2010) p53-Mediated apoptosis prevents the accumulation of progenitor B cells and B-cell tumors. Cell Death Differ 17(3):540–550
Smith ML, Chen IT, Zhan Q et al (1994) Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science 266(5189):1376–1380
Smith ML, Chen IT, Zhan Q, O’Connor PM, Fornace AJ Jr (1995) Involvement of the p53 tumor suppressor in repair of u.v.-type DNA damage. Oncogene 10(6):1053–1059
Smith ML, Ford JM, Hollander MC et al (2000) p53-Mediated DNA repair responses to UV radiation: studies of mouse cells lacking p53, p21, and/or gadd45 genes. Mol Cell Biol 20(10):3705–3714
Speidel D (2010) Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol 20(1):14–24
Speidel D, Helmbold H, Deppert W (2006) Dissection of transcriptional and non-transcriptional p53 activities in the response to genotoxic stress. Oncogene 25(6):940–953
Spurgers KB, Gold DL, Coombes KR et al (2006) Identification of cell cycle regulatory genes as principal targets of p53-mediated transcriptional repression. J Biol Chem 281(35):25134–25142. doi:10.1074/jbc.M513901200
St Clair S, Giono L, Varmeh-Ziaie S et al (2004) DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: one involves direct binding to the cdc25C promoter. Mol Cell 16(5):725–736. doi:10.1016/j.molcel.2004.11.002
Stewart N, Hicks GG, Paraskevas F, Mowat M (1995) Evidence for a second cell cycle block at G2/M by p53. Oncogene 10(1):109–115
Stivala LA, Riva F, Cazzalini O, Savio M, Prosperi E (2001) p21(waf1/cip1)-null human fibroblasts are deficient in nucleotide excision repair downstream the recruitment of PCNA to DNA repair sites. Oncogene 20(5):563–570. doi:10.1038/sj.onc.1204132
Sturzbecher HW, Donzelmann B, Henning W, Knippschild U, Buchhop S (1996) p53 is linked directly to homologous recombination processes via RAD51/RecA protein interaction. EMBO J 15(8):1992–2002
Sugrue MM, Shin DY, Lee SW, Aaronson SA (1997) Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proc Natl Acad Sci USA 94(18):9648–9653
Suzuki K, Mori I, Nakayama Y, Miyakoda M, Kodama S, Watanabe M (2001) Radiation-induced senescence-like growth arrest requires TP53 function but not telomere shortening. Radiat Res 155(1 Pt 2):248–253
Tanaka H, Arakawa H, Yamaguchi T et al (2000) A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 404(6773):42–49. doi:10.1038/35003506
Tang HY, Zhao K, Pizzolato JF, Fonarev M, Langer JC, Manfredi JJ (1998) Constitutive expression of the cyclin-dependent kinase inhibitor p21 is transcriptionally regulated by the tumor suppressor protein p53. J Biol Chem 273(44):29156–29163
Tang W, Willers H, Powell SN (1999) p53 Directly enhances rejoining of DNA double-strand breaks with cohesive ends in gamma-irradiated mouse fibroblasts. Cancer Res 59(11):2562–2565
Tarasov V, Jung P, Verdoodt B et al (2007) Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 6(13):1586–1593
Taylor WR, DePrimo SE, Agarwal A et al (1999) Mechanisms of G2 arrest in response to overexpression of p53. Mol Biol Cell 10(11):3607–3622
Tchelebi L, Ashamalla H, Graves PR (2014) Mutant p53 and the response to chemotherapy and radiation. Sub-cell Biochem 85:133–159. doi:10.1007/978-94-017-9211-0_8
Therrien JP, Drouin R, Baril C, Drobetsky EA (1999) Human cells compromised for p53 function exhibit defective global and transcription-coupled nucleotide excision repair, whereas cells compromised for pRb function are defective only in global repair. Proc Natl Acad Sci USA 96(26):15038–15043
Timofeev O, Schlereth K, Wanzel M et al (2013) p53 DNA binding cooperativity is essential for apoptosis and tumor suppression in vivo. Cell Rep 3(5):1512–1525. doi:10.1016/j.celrep.2013.04.008
Tront JS, Hoffman B, Liebermann DA (2006) Gadd45a suppresses Ras-driven mammary tumorigenesis by activation of c-Jun NH2-terminal kinase and p38 stress signaling resulting in apoptosis and senescence. Cancer Res 66(17):8448–8454. doi:10.1158/0008-5472.can-06-2013
Tront JS, Huang Y, Fornace AJ Jr, Hoffman B, Liebermann DA (2010) Gadd45a functions as a promoter or suppressor of breast cancer dependent on the oncogenic stress. Cancer Res 70(23):9671–9681. doi:10.1158/0008-5472.can-10-2177
Utrera R, Collavin L, Lazarevic D, Delia D, Schneider C (1998) A novel p53-inducible gene coding for a microtubule-localized protein with G2-phase-specific expression. EMBO J 17(17):5015–5025. doi:10.1093/emboj/17.17.5015
Valente LJ, Gray DH, Michalak EM et al (2013) p53 Efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep 3(5):1339–1345. doi:10.1016/j.celrep.2013.04.012
Villunger A, Michalak EM, Coultas L et al (2003) p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302(5647):1036–1038
Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408(6810):307–310
Vousden KH (2006) Outcomes of p53 activation—spoilt for choice. J Cell Sci 119(Pt 24):5015–5020
Vousden KH, Lane DP (2007) p53 in health and disease. Nat Rev Mol Cell Biol 8(4):275–283
Vousden KH, Prives C (2009) Blinded by the light: the Growing Complexity of p53. Cell 137(3):413–431
Waga S, Hannon GJ, Beach D, Stillman B (1994) The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 369(6481):574–578. doi:10.1038/369574a0
Waldman T, Kinzler KW, Vogelstein B (1995) p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 55(22):5187–5190
Wang XW, Yeh H, Schaeffer L et al (1995) p53 Modulation of TFIIH-associated nucleotide excision repair activity. Nat Genet 10(2):188–195. doi:10.1038/ng0695-188
Wang XW, Zhan Q, Coursen JD et al (1999) GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci USA 96(7):3706–3711
Wani MA, Zhu Q, El-Mahdy M, Venkatachalam S, Wani AA (2000) Enhanced sensitivity to anti-benzo(a)pyrene-diol-epoxide DNA damage correlates with decreased global genomic repair attributable to abrogated p53 function in human cells. Cancer Res 60(8):2273–2280
Weinert TA, Hartwell LH (1988) The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 241(4863):317–322
Wu GS, Burns TF, McDonald ER 3rd et al (1997) KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet 17(2):141–143. doi:10.1038/ng1097-141
Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D (1993) p21 is a universal inhibitor of cyclin kinases. Nature 366(6456):701–704. doi:10.1038/366701a0
Xue L, Zhou B, Liu X, Qiu W, Jin Z, Yen Y (2003) Wild-type p53 regulates human ribonucleotide reductase by protein—protein interaction with p53R2 as well as hRRM2 subunits. Cancer Res 63(5):980–986
Yang T, Namba H, Hara T et al (1997) p53 Induced by ionizing radiation mediates DNA end-jointing activity, but not apoptosis of thyroid cells. Oncogene 14(13):1511–1519. doi:10.1038/sj.onc.1200979
Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M (1991) Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 352(6333):345–347. doi:10.1038/352345a0
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 7(3):673–682
Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L (2003) PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci USA 100(4):1931–1936. doi:10.1073/pnas.2627984100
Yu H, Shen H, Yuan Y et al (2010) Deletion of Puma protects hematopoietic stem cells and confers long-term survival in response to high-dose gamma-irradiation. Blood 115(17):3472–3480. doi:10.1182/blood-2009-10-248278
Zhan Q, Antinore MJ, Wang XW et al (1999) Association with Cdc2 and inhibition of Cdc2/Cyclin B1 kinase activity by the p53-regulated protein Gadd45. Oncogene 18(18):2892–2900. doi:10.1038/sj.onc.1202667
Zhou J, Prives C (2003) Replication of damaged DNA in vitro is blocked by p53. Nucleic Acids Res 31(14):3881–3892
Zhou J, Ahn J, Wilson SH, Prives C (2001) A role for p53 in base excision repair. EMBO J 20(4):914–923. doi:10.1093/emboj/20.4.914
Zhou G, Wang J, Zhao M et al (2014) Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol Cell 54(6):960–974. doi:10.1016/j.molcel.2014.04.024
Zhu J, Chen X (2000) MCG10, a novel p53 target gene that encodes a KH domain RNA-binding protein, is capable of inducing apoptosis and cell cycle arrest in G(2)-M. Mol Cell Biol 20(15):5602–5618
Zurer I, Hofseth LJ, Cohen Y et al (2004) The role of p53 in base excision repair following genotoxic stress. Carcinogenesis 25(1):11–19. doi:10.1093/carcin/bgg186
Acknowledgments
Work on DNA damage responses in my laboratory has been supported by Grants from Cure Cancer Australia Foundation, Cancer Institute New South Wales, Anthony Rothe Memorial Trust and Tour de Cure.
Conflict of interest
The author declares that he has no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Speidel, D. The role of DNA damage responses in p53 biology. Arch Toxicol 89, 501–517 (2015). https://doi.org/10.1007/s00204-015-1459-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-015-1459-z