
Abstract
Simvastatin is effective and well tolerated, with adverse reactions mainly affecting skeletal muscle. Important mechanisms for skeletal muscle toxicity include mitochondrial impairment and increased expression of atrogin-1. The aim was to study the mechanisms of toxicity of simvastatin on H9c2 cells (a rodent cardiomyocyte cell line) and on the heart of male C57BL/6 mice. After, exposure to 10 μmol/L simvastatin for 24 h, H9c2 cells showed impaired oxygen consumption, a reduction in the mitochondrial membrane potential and a decreased activity of several enzyme complexes of the mitochondrial electron transport chain (ETC). The cellular ATP level was also decreased, which was associated with phosphorylation of AMPK, dephosphorylation and nuclear translocation of FoxO3a as well as increased mRNA expression of atrogin-1. Markers of apoptosis were increased in simvastatin-treated H9c2 cells. Treatment of mice with 5 mg/kg/day simvastatin for 21 days was associated with a 5 % drop in heart weight as well as impaired activity of several enzyme complexes of the ETC and increased mRNA expression of atrogin-1 and of markers of apoptosis in cardiac tissue. Cardiomyocytes exposed to simvastatin in vitro or in vivo sustain mitochondrial damage, which causes AMPK activation, dephosphorylation and nuclear transformation of FoxO3a as well as increased expression of atrogin-1. Mitochondrial damage and increased atrogin-1 expression are associated with apoptosis and increased protein breakdown, which may cause myocardial atrophy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Statins are among the most often prescribed drugs in Western countries. They are used frequently in patients with cardiovascular diseases because they reduce morbidity and mortality from coronary heart disease (Group 1994; LaRosa et al. 2005; Ridker et al. 2008), reduce the risk of stroke (Amarenco et al. 2006) and improve the walking distance in patients with peripheral arterial disease (Mohler et al. 2003). Their major site of action is the liver, where they inhibit HMG-CoA (hydroxyl-methyl-glutaryl-coenzyme A) reductase, the rate-limiting step in cholesterol biosynthesis. Inhibition of this pathway also impairs other processes, such as ubiquinone production and the isoprenylation and N-linked glycosylation of proteins (Matzno et al. 2005). Altering these processes can reduce inflammation, oxidative stress and platelet adhesion (Davignon 2004), actions known as pleiotropic effects of statins.
Statins are generally well tolerated, but there are exposure-dependent adverse reactions, particularly on skeletal muscle. Myopathy is observed in 1.5–5 % of patients treated with statins (Joy and Hegele 2009), whereas rhabdomyolysis is much rarer (Graham et al. 2004). The most important risk factor for myotoxicity of statins is an increased exposure, for instance, due to impaired transport of statins into hepatocytes by OATP1B1 (Link et al. 2008) or due to impaired hepatocellular metabolism of statins caused by interactions with other drugs (Ratz Bravo et al. 2005; Roten et al. 2004). The molecular mechanisms how statins damage skeletal muscle are not completely clear. Frequently mentioned possibilities include impaired prenylation of critical proteins (Cao et al. 2009; Sakamoto et al. 2011), impaired mitochondrial function (Bouitbir et al. 2012; Kaufmann et al. 2006; Kwak et al. 2012; Larsen et al. 2013; Mullen et al. 2011; Sirvent et al. 2005) or impaired mitochondrial proliferation (Schick et al. 2007), accelerated skeletal muscle breakdown due to increased expression of atrogin-1 (Cao et al. 2009; Hanai et al. 2007) and impaired skeletal muscle protein synthesis (Tuckow et al. 2011).
Adverse reactions of statins on cardiac muscle have been much less intensively studied, but could also be clinically relevant. In two publications, cardiac muscle was described to be less sensitive to mitochondrial adverse reactions of statins than skeletal muscle (Bouitbir et al. 2012; Sirvent et al. 2005), findings which are compatible with the clinical experience that the heart is usually not affected in patients with statin-induced rhabdomyolysis (Joy and Hegele 2009; Roten et al. 2004). On the other hand, in vitro studies have clearly demonstrated that lipophilic statins such as simvastatin and lovastatin can be toxic on cardiac myocytes and can induce apoptosis (Demyanets et al. 2006; Kong and Rabkin 2004; Rabkin and Kong 2003; Rabkin et al. 2007). In addition, Bouitbir et al. demonstrated that cardiac mitochondria exposed to simvastatin in vivo react to a possible insult by increasing antioxidative mechanism and by proliferation (Bouitbir et al. 2012).
As mentioned above, mitochondrial damage and increased expression of atrogin-1 appear to be key findings for skeletal muscle injury associated with statins. Previous studies have shown that atrogin-1 is expressed in cardiac muscle and that it is upregulated during experimental heart failure (Adams et al. 2007; Mearini et al. 2010). Since there are so far no studies available about the effect of statins on mitochondrial function and the expression of atrogin-1 in cardiomyocytes, we decided to study the effect of simvastatin on these parameters on rodent cardiomyocytes in vitro and in mice in vivo. Furthermore, these studies allowed us to investigate possible links between mitochondrial damage and increased expression of atrogin-1.
Materials and methods
Chemicals
Simvastatin lactone (Sigma-Aldrich, St. Louis, MO, USA) was converted into the active acid following the protocol of Bogman et al. (2001). We prepared stock solutions of 10 and 100 mmol/L simvastatin in dimethylsulfoxide (DMSO) and stored them at −20 °C. All chemicals were supplied by Sigma-Aldrich (St. Louis, MO, USA), except where indicated.
Cell culture
H9c2 cardiomyocytes were provided by Dr Pfister (University Hospital Basel, Switzerland). They were grown in Dulbecco modified Eagle’s medium (DMEM) containing 4.5 g/l glucose with Glutamax from Invitrogen (Basel, Switzerland) and supplemented with 10 % FBS, 5 mmol/L HEPES, 1 mmol/L sodium pyruvate and 500 μg/ml penicillin–streptomycin. Cells were kept at 37 °C in a humidified 5 % CO2 cell culture incubator and were passaged using trypsin. The cell number was determined using a Neubauer hemacytometer and viability using the trypan blue exclusion method.
Myoblasts were seeded at 150,000 cells/well of a six-well plate (or equivalent) and grown in a humidified incubator with 5 % CO2 at 37 °C for 2 days before drug treatment. We then treated them with simvastatin at 10 and 100 μmol/L. All incubations contained 0.1 % DMSO (including control incubations).
Animals
The experiments were performed in agreement with the Declaration of Helsinki and were approved by the cantonal veterinary authority (License 2659). Male C57BL/6 mice were purchased from Charles River Laboratories (Sutzfeld, Germany) and were adult age-matched (7 weeks old) housed in a standard facility with 12 h light–dark cycles and controlled temperature (21–22 °C). The mice were fed with a standard pellet chow and water ad libitum. After 7 days of acclimatization, the mice were divided into two groups: (1) eight mice (SMV group) treated with simvastatin by oral gavage dissolved in water at 5 mg/kg body weight/day for 21 days group and (2) eight mice (CTL group) treated with the same amount of water and served as a control. Animals and food intakes were weighted every 2 days.
Sample collection
After the 21 days of treatment, the mice were anaesthetized with an intraperitoneal application of ketamine (100 mg/kg) and xylazine (10 mg/kg). The left ventricles were excised and conserved in ice-cold BIOPS buffer (10 mmol/L Ca-EGTA buffer, 0.1 μmol/L free calcium, 5.77 mmol/L ATP, 6.56 mmol/L MgCl2, 20 mmol/L taurine, 15 mmol/L phosphocreatine, 0.5 mmol/L dithiothreitol and 50 mmol/L MES, pH 7.1) until analysis.
Heart samples were frozen in liquid nitrogen immediately after excision. Since the mice were anesthetized, tissues were obtained from living animals, and the time between sampling and freezing was only a few seconds. Samples were kept at −80 °C until analysis.
Cytotoxicity assay
In vitro cytotoxicity was assessed by the release of adenylate kinase (AK), which results from the loss of cell membrane integrity. AK was quantified using the ToxiLight assay kit supplied from Lonza (Basel, Switzerland). After incubation with simvastatin for 6 and 24 h, we used the protocol outlined in Mullen et al. (2010).
Intracellular ATP content
The cellular ATP content, a marker for metabolic cell activity and cell viability, was determined using a CellTiter Glo kit from Promega (Madison, USA) following the producer’s instructions. In brief, 100-μL assay buffer was added to each 96-well containing 100-μL culture medium. After incubation in the dark for 30 min, luminescence was measured using a Tecan M200 Pro Infinity plate reader (Männedorf, Switzerland).
Mitochondrial membrane potential
To detect changes in mitochondrial membrane potential (Δψm), we used tetramethylrhodamine ethyl ester (TMRE), which distributes across the inner mitochondrial membrane in accordance with the Nernst equation (Bursac et al. 1999). Cells were incubated with simvastatin for 24 h and then detached and incubated with 100 nmol/L TMRE for 30 min at 37 °C and 5 % CO2. We used a Live/Dead® Near-IR Dead Cell stain kit from Invitrogen to exclude dead cells. Viable cells were analyzed with a DAKO Cyan cytometer. The mitochondrial uncoupler carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) was used as a control for depletion of the mitochondrial membrane potential.
Oxygen consumption
We measured intact cellular respiration with a Seahorse XF24 analyzer (Seahorse Biosciences, North Billerica, MA, USA). H9c2 myoblasts were seeded in Seahorse XF24-well cell culture microplates at 5,000 cells per well in 250-μL growth medium. We treated cells with simvastatin for 24 h and then replaced the medium with 750-μL medium containing Dulbecco modified Eagle’s medium (DMEM) high-glucose (4.5 g/l) medium with Glutamax (Invitrogen, Basel, Switzerland) supplemented with 1 mmol/L sodium pyruvate, 25 mmol/L glucose and 15 mg phenol red. Cells were equilibrated to the medium for 45 min at 37 °C in a CO2-free incubator and then transferred to the XF24 analyzer. We first measured the basal oxygen consumption rate (OCR). Then, we determined leak-state respiration, a measure for uncoupling of oxidative phosphorylation. For that, we added oligomycin (final concentration 2.5 μmol/L), an inhibitor of F0F1ATPase. An OCR higher than in control incubations is compatible with uncoupling by the toxicant investigated (Felser et al. 2013). This was followed by the addition of the chemical uncoupler FCCP (final concentration 1 μmol/L) to obtain maximum mitochondrial respiration. Finally, the complex I inhibitor rotenone (final concentration 1 μmol/L) was added for the determination of the residual respiration. Respiration rates were calculated as the mean of all determinations at a certain state (basal, leak and FCCP-stimulated respiration) after subtraction of residual respiration in the presence of rotenone.
Activity of enzyme complexes of the mitochondrial electron transport chain
Mitochondrial respiration was analyzed in permeabilized H9c2 myoblasts and in permeabilized myocardial fibers. Mitochondrial measurements were performed at 37 °C with an Oxygraph-2k system equipped with DatLab software (Oroboros Instruments, Austria). Multisubstrate and multiinhibitor protocols were used simulating the operation of the mitochondrial electron transport chain (ETC). H9c2 myoblasts were first permeabilized with digitonin (8.1 µg/million cells) before the activities of complexes I, II, III and IV were determined.
In a first incubation, we determined the activity of complex I and of complex III (see supplementary data for a more detailed description of the substrates and inhibitors used). First, we assessed the activity of complex I using glutamate and malate (final concentrations 10 and 5 mmol/L, respectively) as substrates in the presence of ADP (final concentration 2.5 mmol/L). Then, we added oligomycin (final concentration 2.5 μmol/L) for the determination of the leak-state respiration. This was followed by the addition of FCCP (final concentration 1 μmol/L) to reach a full stimulation of the ETC and of cytochrome c (final concentration 10 μmol/L) to assess mitochondrial integrity. After that, we added rotenone (final concentration 1 μmol/L), an inhibitor of complex I. Now, we added duroquinol (0.5 mmol/L), an artificial substrate of complex III, and finally antimycin A (final concentration 2.5 μmol/L), an inhibitor of complex III.
In a second run, we assessed the activity of complex II and of complex IV (see supplementary data for a more detailed description of the substrates and inhibitors used). We first added succinate (final concentration 10 mmol/L), a substrate of complex II. Then, we added rotenone (final concentration 1 μmol/L) and ADP (final concentration 2.5 mmol/L). Then, we added oligomycin (final concentration 2.5 μmol/L) for the determination of the leak-state respiration. This was followed by the addition of FCCP (final concentration 1 μmol/L) to reach a full stimulation of the ETC and of cytochrome c (final concentration 10 μmol/L) to assess mitochondrial integrity. Then, we added antimycin A (final concentration 2.5 μmol/L) in order to block the electron transport between complex II and IV and finally N,N,N′,N′-tetramethyl-p-phenylenediamine dihydrochloride (TMPD) and ascorbate (final concentrations 0.5 and 2 mmol/L, respectively) as artificial substrates of complex IV. Respiration rates are expressed in picomoles O2 per second per one million cells.
For the determination of the respiratory capacity of heart muscle fibers, the left ventricle of each heart was removed and transferred into 2 ml of ice-cold BIOPS buffer. After dissection, fiber bundles were permeabilized by gentle agitation for 30 min in ice-cold BIOPS solution supplemented with 50 μg/mL saponin. Permeabilized fibers were then washed in ice-cold BIOPS buffer for 10 min under shaking followed by washing two times in ice-cold mitochondrial respiration medium MiR05 (0.5 mmol/L EGTA, 1 g/l fatty acid free bovine serum albumin, 3 mmol/L MgCl2, 20 mmol/L taurine, 10 mmol/L KH2PO4, 110 mmol/L sucrose, 60 mmol/L K-lactobionate and 20 mmol/L HEPES, pH 7.1). Prior to respirometry measurements, 2–3 mg of permeabilized muscle bundles were blotted, weighed and immediately used for respirometry measurements. In these preparations, activity of complex I, II and IV was analyzed (see supplementary data for a more detailed description of the substrates and inhibitors used). In order to determine the activity of complex I, we added glutamate and malate (final concentration 10 and 5 mmol/L, respectively) as substrates, followed by ADP (final concentration 2.5 mmol/L). Then, we added rotenone (final concentration 1 μmol/L) as an inhibitor of complex I. After that, we added succinate (final concentration 10 mmol/L) as a substrate of complex II, followed by antimycin A (final concentration 2.5 μmol/L) as an inhibitor of complex III. Finally, we added the artificial substrates ascorbate and TMPD (final concentration 2 and 0.5 mmol/L, respectively) for complex IV, which was followed by the addition of cytochrome c (final concentration 10 μmol/L) to assess mitochondrial integrity. Respiration rates are expressed in picomoles O2 per second per gram wet weight.
Confocal microscopy
H9c2 cardiomyocytes were grown in Lab-Tek microscopy chambers (Nunc, NY, USA) and treated with simvastatin for 24 h. The cells were fixed with 4 % PFA, permeabilized with 0.2 % triton-X and then labeled with the appropriate antibody. We used DAPI to stain nuclei. We used a Zeiss LSM 710 confocal microscope to take the images, and Zen software to process them (Carl Zeiss, Switzerland).
Western blot analysis
After incubation with 0.1 % DMSO (control) or simvastatin (10 and 100 μmol/L) for 6 or 24 h, we lysed the H9c2 myoblasts with Phosphosafe extraction buffer (EMD Millipore, USA). We centrifuged the samples at 1,600g at 4 °C. Lysates were then resolved by electrophoresis on 4–12 % bis–tris polyacrylamide gels (Invitrogen, Carlsbad, USA) under reducing conditions and transferred to a polyvinylidendifluoride membrane (EMD Millipore, USA). We incubated the membranes with anti-phosphopeptide antibodies provided by Cell Signaling Technology (Danvers, USA). After washing, membranes were exposed to secondary antibodies (Santa Cruz Biotechnology, Dallas, USA).
Immunoblots were developed using enhanced chemiluminescence (GE Healthcare, Little Chalfont, UK). Results obtained using phosphorylation-specific antibodies were corrected for total protein expression and are presented as the ratio to control incubations. Band intensities of the scanned images were quantified using the National Institutes of Health Image J program, version 1.41.
Annexin/PI staining
Apoptosis and necrosis were investigated using Annexin V and propidium iodide (PI) staining (Vybrant TM Apoptosis Assay Kit #2, from Invitrogen). After 24-h incubation with simvastatin, we stained detached cells and stained them with 5 μL Annexin V-AlexaFluor 488 and 1 μL propidium iodide (final concentration 1 μg/ml) in Annexin V binding buffer (10 mmol/L HEPES, 140 mmol/L NaCl, 2.5 mmol/L CaCl2 in H2O, pH 7.4). Cells were incubated for 15 min at room temperature and analyzed by flow cytometry using a DAKO Cyan cytometer (Beckman Coulter, Marseille, France). Data were analyzed using FlowJo 9.3.2 software (Tree Star, Ashland, OR, USA).
Caspase 3/7 assay
Caspase 3/7 activity was determined using the luminescent Caspase-Glo 3/7 assay (Promega, Wallisellen, Switzerland). The assay was conducted according to the manufacturer’s protocol.
Atrogin-1 mRNA expression
We treated H9c2 cardiomyocytes with simvastatin for 24 h. RNA was extracted and purified using the Qiagen RNeasy mini extraction kit, with a DNA digest step to ensure pure RNA.
Moreover, we extracted mRNA from the heart of C57BL/6 mice with Trizol reagent™ (Invitrogen, Basel, Switzerland) following the instructions of the producer. We synthesized cDNA using the Qiagen omniscript system and used 10 ng cDNA for quantitative RT-PCR. We used SYBR green with primers specific for atrogin-1 (forward primer: 5′-GAAGACCGCTACTGTGGAA-3′ and reverse primer: 5′-ATCAATCGCTTGCGGATCT-3′ for in H9c2 myoblasts and forward primer: 5′AGTGAGGACCGGCTACTGTG3′; reverse primer: 5′GATCAAACGCTTGCGAATCT3′ for heart murine muscle fibers), Bcl-2 (forward primer: 5′GGTGGGCCCGGAACATCT3′ and reverse primer: 5′GGGCCCTACCGTCCTACTAAT3′) and Bax (forward primer: 5′-GGC TGG ACA CTG GAC TTC CT3′ and reverse primer 5′GGTGAGGACTCCAGCCACAA3′).
Relative quantities of specifically amplified cDNA were calculated with the comparative-threshold cycle method. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) acted as endogenous reference (forward primer: 5′-CATGGCCTTCCGTGTTCCTA-3′; reverse primer: 5′-CCTGCTTCACCACCTTCTTGA-3′), and no-template and no reverse transcription controls ensured nonspecific amplification could be excluded.
Statistical analysis
Statistical analysis was completed using the GraphPad Prism 5 program (GraphPad Software, San Diego, CA, USA). All results are expressed as mean ± SD and evaluated with one-way ANOVA followed by the comparisons between incubations containing toxicants and the control group using Dunnett’s posttest procedure. p values <0.05 (*) were considered significant.
Results
Simvastatin induces cytotoxicity in H9c2 cardiomyocytes
To firstly investigate whether simvastatin is directly toxic to cardiomyocytes, we performed AK release treating the H9c2 myoblasts for 6 and 24 h with 10 and 100 μmol/L simvastatin. We observed a significant AK release with 10 μmol/L simvastatin only after 24 h, whereas 100 μmol/L simvastatin was already toxic after 6 h (Fig. 1a). These results suggested that simvastatin could be associated with cardiac toxicity and led us to investigate in deep on the mechanisms of this toxicity using in vitro and in vivo models.

Toxicity of simvastatin (SMV) on cardiomyocytes. H9c2 cardiomyocytes were incubated with DMSO, 10 or 100 μmol/L simvastatin for 6 and 24 h. We measured the release of adenylate kinase (AK) into the medium (a) and the cellular ATP content (b). DMSO-treated cells were used as a control. Results are expressed as ratios to the DMSO control. Each bar represents the mean of four independent experiments carried out in triplicate. ***p < 0.001 versus control
Simvastatin decreases the ATP content in H9c2 cardiomyocytes
Previous work showed an impairment in ATP after simvastatin treatment (Alfazari et al. 2013). To understand whether the key event of simvastatin-induced cardiotoxicity is an energetic impairment, we analyzed the cellular ATP content. The intracellular ATP content started to decrease already after 6 h for 100 μmol/L simvastatin, whereas 10 μmol/L simvastatin induced a decrease in ATP content only after 24 h (Fig. 1b). These results suggested that the energetic drop could be involved in the induction of cytotoxicity.
Simvastatin reduces mitochondrial membrane potential in H9c2 cardiomyocytes
It has been shown by our group and others that statins impair the mitochondrial function in other cell types (Bouitbir et al. 2012; Kaufmann et al. 2006; Sirvent et al. 2012). To investigate whether the reduction in the cellular ATP content is a consequence of mitochondrial impairment also in cardiomyocytes, we assayed for changes in the mitochondrial membrane potential (Δψm) with the dye TMRE. A compromised mitochondrial Δψm is an indicator of impaired mitochondrial function (Felser et al. 2013). We pretreated the cells for 24 h with DMSO, 10 or 100 μmol/L simvastatin (Fig. 2a). H9c2 mitochondrial membrane potential was significantly reduced after treatment with both 10 and 100 μmol/L simvastatin. The positive control, FCCP, showed the expected marked reduction in mitochondrial membrane potential.

Effect of simvastatin (SMV) on cellular oxygen consumption and mitochondrial function. a Membrane potential. Cells were incubated with DMSO, 10 or 100 μmol/L simvastatin for 24 h. TMRE was used to determine mitochondrial membrane potential, and near-IR dye from Invitrogen to exclude dead cells. The uncoupler FCCP was used as positive control. b Effect of simvastatin on cellular OCR. Cells were incubated with DMSO (red lines), 10 µM (blue lines) or 100 µM (black lines) simvastatin. OCRs are shown for 24-h incubation. 1 µM oligomycin, 15 µM FCCP and 1 µM rotenone (final concentrations) were added at the indicated points. c Effect of simvastatin on enzymes complexes of the ETC. The activity of complexes I to IV was determined as described in “Methods” section. Results are means of four independent experiments carried out in triplicate. *p < 0.05, **p < 0.01 and ***p < 0.001 versus control. Glut glutamate, Mal malate, Olig oligomycin, FCCP carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone, Cyt c cytochrome c, Rot rotenone, Duro duroquinol, Succ succinate, Ant antimycin a, TMPD N,N,N′,N′-tetramethyl-p-phenylenediamine, Asc ascorbate (color figure online)
Simvastatin reduces oxygen consumption in intact H9c2 myocytes
To delve deeper into how simvastatin affects H9c2 mitochondria, we used a XF24 analyzer to determine changes in OCR in simvastatin-treated intact H9c2 cardiomyocytes. Cellular consumption of oxygen mainly reflects mitochondrial metabolism (Felser et al. 2013). As shown in Fig. 2b, after 24-h treatment with 10 μmol/L simvastatin, baseline OCR values were significantly reduced (mean ± SD, n = 4; 179 ± 25 vs. 229 ± 21 pmol/min/5,000 cells in simvastatin-treated vs. control incubations, p < 0.05). After the addition of oligomycin, which blocks ATP production, the OCR was not different between simvastatin 10 μmol/L and control values (109 ± 18 vs. 122 ± 14 pmol/min/5,000 cells in simvastatin treated vs. control incubations), excluding uncoupling, suggesting that the baseline reduction is due impaired oxidative phosphorylation. After the addition of FCCP to determine maximum respiration, the inhibitory effect of simvastatin was confirmed (308 ± 46 vs. 414 ± 68 pmol/min/5,000 cells in simvastatin treated vs. control incubations, p < 0.05). Treatment with 100 μmol/L simvastatin resulted in a complete abolition of oxygen consumption, suggesting a complete inhibition of oxidative phosphorylation at this concentration.
Simvastatin impairs the function of the mitochondrial electron transport chain in vitro
In order to investigate on the decrease in oxygen consumption, we determined the function of the enzyme complexes of the ETC after treatment with vehicle (DMSO 0.1 %) or simvastatin (10 and 100 μmol/L) for 24 h with a high-resolution respirometry system (Oxygraph-2k). After exposure to simvastatin for 24 h, the respiratory capacities through complexes I and IV were significantly decreased for both concentrations in a dose-dependent way, whereas the function of complex III was significantly impaired only at 100 μmol/L simvastatin.
Moreover, the respiratory leak after the addition of oligomycin was not increased by the simvastatin-treated samples, indicating that simvastatin had no uncoupling effect in H9c2 myoblasts exposed for 24 h (Fig. 2c).
Simvastatin leads to AMPK activation, and subsequent FoxO3a nuclear translocation and upregulation of atrogin-1
The observed decrease in the cellular ATP content in H9c2 myoblasts exposed to simvastatin suggested that exposure to simvastatin could activate AMPK (5′ adenosine monophosphate-activated protein kinase; Towler and Hardie 2007a). AMPK is a metabolic master switch that regulates several metabolic processes including cellular uptake of glucose and metabolism of glucose and fatty acids (Mihaylova and Shaw 2011). Once activated, AMPK promotes ATP production by increasing catabolic activity while conserving ATP by switching off biosynthetic pathways (Hardie et al. 2012). As shown in Fig. 3a, both 10 and 100 μmol/L simvastatin remarkably triggered the activation of AMPK at both time points (6 and 24 h).

Effect of simvastatin (SMV) on AMP-activated protein kinase (AMPK) activation, FoxO3a translocation and atrogin-1 expression in H9c2 cardiomyocytes. a Simvastatin induces AMPK phosphorylation in a concentration- and time-dependent fashion. b Treatment with simvastatin is associated with translocation of FoxO3a into the nucleus. c Numerical expression of the results (three independent experiments). d Atrogin-1 mRNA levels in H9c2 cardiomyocytes treated with 10 µM simvastatin for 6 or 24 h, relative to DMSO control (three independent experiments in triplicate). **p < 0.01 and ***p < 0.001 versus control (CTL)
Activation of AMPK is associated with dephosphorylation of the nuclear transcription factor FoxO, which then can migrate into the nucleus (Chiacchiera and Simone 2010). As shown in Fig. 3b, we could observe a nuclear translocation of FoxO3a using confocal microscopy at 10 μmol/L. Once in the nucleus, FoxO3a acts as a transcription factor of different genes like, for instance atrogin-1, a key player in muscle protein degradation and therefore associated with muscle atrophy (Sandri et al. 2004). As shown in Fig. 3c, as expected, nuclear translocation of FoxO3a was associated with upregulation of atrogin-1 mRNA levels (Fig. 3d).
Simvastatin triggers apoptosis as the major form of cell death
We used Annexin V/PI staining to measure the type of cell death seen in H9c2 cardiomyocytes treated with simvastatin for 24 h. H9c2 cardiomyocytes treated with 10 μmol/L simvastatin showed a significant increase in early apoptotic cells when compared to control (6.3 vs. 3.0 %). This increase was concentration dependent; indeed, 100 μmol/L simvastatin showed a larger increase in early apoptotic cells (9.2 %; Fig. 4a). An increase in the late apoptotic fraction was only seen in cells treated with 100 μmol/L simvastatin (18.2 vs. 11.6 %; data not shown). In addition, we also analyzed the activation of caspase 3 by the caspase 3/7 glo kit and Western blots (Fig. 4c, d). In both assays, we could confirm an activation of caspase after simvastatin treatment. The stimulation was dependent on the concentration of simvastatin and on the incubation time.

Mechanisms of cell death after simvastatin (SMV) treatment in cardiomyocytes. a H9c2 myoblasts exposed for 24 h to DMSO, 10 or 100 μM simvastatin were labeled with Annexin V and PI and analyzed via flow cytometry. b Representative blot of cleaved caspase 3. c Caspase 3/7 activity after drug exposure for 6 and 24 h, expressed as ratio compared with DMSO control. d Representative blot of Bax and Bcl-2. Data in (a) and (c) represent the mean ± SEM of three independent experiments. **p < 0.01 and ***p < 0.001 versus control
Bax and Bcl-2 are two mitochondrial proteins involved in the regulation of apoptosis (Bagci et al. 2006), and an increased Bax/Bcl-2 ratio is associated with apoptosis (Perlman et al. 1999). Figure 4b shows a strong increase in the expression of Bax in cells incubated with simvastatin for 24 h and a reduced expression of Bcl-2 after exposure to simvastatin for 6 h and for 24 h. The Bax/Bcl-2 ratio was increased in a concentration- and time-dependent manner.
Simvastatin impairs mitochondrial functions in vivo
To confirm the role of simvastatin as a cardiac mitochondrial toxicant also in vivo, we treated mice orally with simvastatin 5 mg/kg body weight/day for 21 days. The heart weight was decreased by 5 % in simvastatin treated compared to control mice, but this decrease did not reach statistical significance (Fig. 5a). Similar to the in vitro findings, oxygen uptake by heart tissue was reduced in simvastatin-treated mice, indicating impaired mitochondrial function (Fig. 5b). Impaired oxygen uptake resulted from reduced activities of complex I, II and IV of the ETC, agreeing well with the in vitro data. Identical to the in vitro results, there was an upregulation of atrogin-1 mRNA (Fig. 5c), and an increase in Bax mRNA as well as a decrease in Bcl-2 RNA, indicating increased apoptosis (Fig. 5d). These findings demonstrated that the results obtained in vitro could be reproduced in mice in vivo.

Effect of simvastatin (SMV) on heart weight and cardiac atrogin-1 expression in C57BL/6 mice. Male C57BL/6 mice received water (CTL) or simvastatin (5 mg/kg/day) for 21 days. a Heart weight. Each data point represents the mean of eight hearts. b Activity of enzyme complexes of the ETC. c Atrogin-1 mRNA levels in seven simvastatin-treated mice relative to seven water control mice. d Bax/Bcl-2 mRNA levels in seven simvastatin-treated mice, relative to seven water control mice. *p < 0.05, **p < 0.01 and ***p < 0.001 versus control. Glut glutamate, Mal malate, ADP adenosine diphosphate, Rot rotenone, Ant a antimycin a, TMPD N,N,N′,N′-tetramethyl-p-phenylenediamine, Asc ascorbate, Cyt c cytochrome c
Discussion
This study presents several novel findings about the effects of the HMG-CoA reductase inhibitor simvastatin on cultured cardiomyocytes and the myocardium of mice. First, it demonstrates that exposure to simvastatin is cytotoxic for H9c2 cells in a dose- and time-dependent manner and that simvastatin impairs the function of enzyme complexes I and IV (and also of complex II after long-term exposure in vivo) of the ETC. Second, exposure of H9c2 cells to simvastatin is associated with the translocation of FoxO3a into the nucleus, which increases atrogin-1 mRNA expression. Third, treatment of mice with simvastatin at 5 mg/kg/day for 21 days inhibits the activity of the mitochondrial ETC and increases the expression of atrogin-1 mRNA in the heart.
In H9c2 cells, we started to observe cytotoxicity at a simvastatin concentration of 10 μmol/L after 24-h incubation. These data are in agreement with cytotoxicity experiments with cultured myotubes (Bouitbir et al. 2012; Kaufmann et al. 2006; Rabkin and Kong 2003). This simvastatin concentration is higher than the concentrations normally reached in vivo, which are below 5 μmol/L (Kwak et al. 2012). Nevertheless, the data are in agreement with the clinical findings in humans, showing that exposure is a strong risk factor for statin-associated myopathy (Link et al. 2008; Ratz Bravo et al. 2005; Roten et al. 2004). Based on the current and our previous data (Kaufmann et al. 2006), we assume that systemic concentrations in the low µmolar range are necessary for inducing myopathy in humans. Such concentrations can obviously be reached in patients with drug–drug interactions or impaired activity of OATP1B1.
As a vital organ rich in mitochondria and with a high oxygen need, the heart is susceptible for mitochondrial damage. In the presence of simvastatin, we initially observed a decrease in the electrical potential across the inner mitochondrial membrane (Δψm), which represents an indicator of mitochondrial function (Felser et al. 2013). We believe that this change is not the result of cholesterol lowering by simvastatin, since the cholesterol precursor squalene does not reverse the effects of statins on cells (Thorpe et al. 2004), and the cellular cholesterol of cultured myotubes is maintained at cytotoxic simvastatin concentrations (Mullen et al. 2010). To confirm a toxic effect of simvastatin on mitochondria, we assayed for changes in oxygen consumption by H9c2 cells. Importantly, oxygen consumption by cardiomyocytes was reduced already after 6-h treatment with 10 μmol/L simvastatin, a time point when we did not observe cytotoxicity. This suggests that the changes in the mitochondrial function are a cause, rather than a consequence, of simvastatin-induced toxicity in H9c2 cells. This finding is new for cardiomyocytes and is in agreement with results reported from myotubes (Kaufmann et al. 2006; Liantonio et al. 2007).
In order to explore the effect of simvastatin on cardiac mitochondria in more detail, we measured the activity of the complexes of the ETC in permeabilized H9c2 cells. We could show that the exposure to simvastatin mainly inhibited the activity of the enzyme complexes I and IV of the ETC after 24 h of exposure. This is most probably a direct effect on the enzyme complexes and not an indirect effect due to a decrease in the mitochondrial ubiquinol pool. We have shown previously that exposure of myotubes to 10 μmol/L simvastatin for 24 h does not decrease the mitochondrial ubiquinone content (Mullen et al. 2010). More specific studies are needed to find out the precise mechanism how simvastatin impairs the function of the enzyme complexes of the respiratory chain.
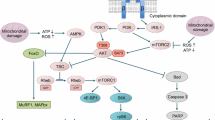
Impairment of mitochondrial function can be associated with a decrease in the cellular ATP content, what we observed in cardiomyocytes exposed to 10 μmol/L simvastatin for 24 h (Fig. 1b). A drop in cellular ATP is associated with phosphorylation of AMPK, which activates and/or increases the expression of enzymes involved in catabolic processes for replenishing the cellular ATP stores (Mihaylova and Shaw 2011; Towler and Hardie 2007b). As explained in Fig. 6, activation of AMPK has several consequences. One of them is dephosphorylation of FoxO3, a nuclear transcription factor, which can travel into the nucleus and increase the expression of atrogin-1 (Sandri et al. 2004). As it has been shown previously for skeletal muscle (Cao et al. 2009; Hanai et al. 2007), we could demonstrate activation of AMPK followed by dephosphorylation and nuclear translocation of FoxO3 and finally increased expression of atrogin-1 also in cardiomyocytes.

Effect simvastatin on cardiomyocytes. Simvastatin (SMV) impairs the function of several enzyme complexes in mitochondria of cardiomyocytes. One consequence is a drop in cellular ATP, which is associated with activation of AMPK, nuclear translocation of FoxO3a and increased expression of atrogin-1. Atrogin-1 stimulates protein degradation. Another consequence is increased expression of Bax and decreased expression of Bcl-2, favoring apoptosis as shown by activation of caspases. Both apoptosis and increased protein degradation can be associated with cardiac atrophy
In order to investigate whether these findings can be reproduced in vivo, we treated mice with simvastatin at 5 mg/kg/day for 21 days. This dose was calculated as described by Reagan-Shaw et al. (2008) and corresponds to approximately 0.4 mg/kg/day in humans, which is a normally used dose. Importantly, we could demonstrate mitochondrial dysfunction and increased atrogin-1 mRNA expression also in the myocardium of mice treated with simvastatin. Interestingly, the heart weight decreased numerically (by 5 %) in the simvastatin-treated mice, suggesting that the toxic effect of simvastatin on the heart could be associated with cardiac atrophy. On the one hand, atrophy-inducing effects of simvastatin could account for some of its cardiovascular benefits by counterbalancing cardiac hypertrophy and/or cardiac remodeling (Kang et al. 2009; Singh and Krishan 2010). On the other hand, statin-associated cardiac atrophy could have a negative effect on cardiac contractility, particularly in patients with heart failure.
Increased expression of atrogin-1 is associated with accelerated proteolysis and is therefore considered to be a major contributing factor to statin-induced myopathy (Hanai et al. 2007). Our previous (Kaufmann et al. 2006) and current data suggest that also increased apoptosis may play a role in muscle and/or cardiac toxicity associated with exposure to high systemic statin concentrations. Since in the current studies, mitochondrial damage was observed already after 6-h exposure to 10 μmol/L simvastatin, a time point when apoptosis was absent, apoptosis may have been triggered by a mitochondrial mechanism (Kaufmann et al. 2006). Importantly, the observed increase in the Bax/Bcl-2 ratio in the myocardium of mice treated with simvastatin suggests that statin-associated apoptosis also takes place in vivo. An increase in the Bax/Bcl-2 ratio indicates a high susceptibility to apoptotic stimuli (Vander Heiden and Thompson 1999).
Our study was not designed to answer the question why the heart is usually not affected in patients with rhabdomyolysis. Assuming that exposure is most probably not different between skeletal muscle and the myocardium, the most likely reason is a different susceptibility of myocytes and cardiomyocytes. In their study, Bouitbir et al. (2012) showed that ROS accumulation, which is associated with damage to the ETC (Felser et al. 2013), is much less in cardiomyocytes than in myocytes. This may be due to better defense mechanisms in cardiomyocytes such as, for instance, upregulation of SOD2, which metabolizes O −·2 .
In conclusion, our data show that cardiomyocytes exposed to simvastatin in vitro or in vivo sustain mitochondrial damage, causing AMPK activation, dephosphorylation and nuclear transformation of FoxO3 as well as increased expression of atrogin-1. Mitochondrial damage is associated with increased apoptosis. Increased protein degradation associated with atrogin-1 and increased apoptosis can lead to myocardial atrophy. Since AMPK is an important master switch for energy metabolism, further studies are necessary to study the effect of statins on signal transduction associated with metabolic processes.
References
Adams V, Linke A, Wisloff U et al (2007) Myocardial expression of Murf-1 and MAFbx after induction of chronic heart failure: effect on myocardial contractility. Cardiovasc Res 73(1):120–129. doi:10.1016/j.cardiores.2006.10.026
Alfazari AS, Al-Dabbagh B, Almarzooqi S, Albawardi A, Souid AK (2013) Bioenergetic study of murine hepatic tissue treated in vitro with atorvastatin. BMC Pharmacol Toxicol 14:15. doi:10.1186/2050-6511-14-15
Amarenco P, Bogousslavsky J, Callahan A III et al (2006) High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med 355(6):549–559. doi:10.1056/NEJMoa061894
Bagci EZ, Vodovotz Y, Billiar TR, Ermentrout GB, Bahar I (2006) Bistability in apoptosis: roles of Bax, Bcl-2, and mitochondrial permeability transition pores. Biophys J 90(5):1546–1559. doi:10.1529/Biophysj.105.068122
Bogman K, Peyer AK, Torok M, Kusters E, Drewe J (2001) HMG-CoA reductase inhibitors and P-glycoprotein modulation. Br J Pharmacol 132(6):1183–1192. doi:10.1038/sj.bjp.0703920
Bouitbir J, Charles AL, Echaniz-Laguna A et al (2012) Opposite effects of statins on mitochondria of cardiac and skeletal muscles: a ‘mitohormesis’ mechanism involving reactive oxygen species and PGC-1. Eur Heart J 33(11):1397–1407. doi:10.1093/eurheartj/ehr224
Bursac N, Papadaki M, Cohen RJ et al (1999) Cardiac muscle tissue engineering: toward an in vitro model for electrophysiological studies. Am J Physiol 277(2 Pt 2):H433–H444
Cao P, Hanai J, Tanksale P, Imamura S, Sukhatme VP, Lecker SH (2009) Statin-induced muscle damage and atrogin-1 induction is the result of a geranylgeranylation defect. FASEB J 23(9):2844–2854. doi:10.1096/fj.08-128843
Chiacchiera F, Simone C (2010) The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle 9(6):1091–1096
Davignon J (2004) Beneficial cardiovascular pleiotropic effects of statins. Circulation 109(23 Suppl 1):III39–III43. doi:10.1161/01.CIR.0000131517.20177.5a
Demyanets S, Kaun C, Pfaffenberger S et al (2006) Hydroxymethylglutaryl-coenzyme A reductase inhibitors induce apoptosis in human cardiac myocytes in vitro. Biochem Pharmacol 71(9):1324–1330. doi:10.1016/j.bcp.2006.01.016
Felser A, Blum K, Lindinger PW, Bouitbir J, Krahenbuhl S (2013) Mechanisms of hepatocellular toxicity associated with dronedarone—a comparison to amiodarone. Toxicol Sci 131(2):480–490. doi:10.1093/toxsci/kfs298
Graham DJ, Staffa JA, Shatin D et al (2004) Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA 292(21):2585–2590. doi:10.1001/jama.292.21.2585
Group SS (1994) Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 344(8934):1383–1389
Hanai J, Cao P, Tanksale P et al (2007) The muscle-specific ubiquitin ligase atrogin-1/MAFbx mediates statin-induced muscle toxicity. J Clin Invest 117(12):3940–3951. doi:10.1172/jci32741
Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13(4):251–262. doi:10.1038/Nrm3311
Joy TR, Hegele RA (2009) Narrative review: statin-related myopathy. Ann Intern Med 150(12):858–868
Kang BY, Wang W, Palade P, Sharma SG, Mehta JL (2009) Cardiac hypertrophy during hypercholesterolemia and its amelioration with rosuvastatin and amlodipine. J Cardiovasc Pharmacol 54(4):327–334. doi:10.1097/FJC.0b013e3181b76713
Kaufmann P, Torok M, Zahno A, Waldhauser KM, Brecht K, Krahenbuhl S (2006) Toxicity of statins on rat skeletal muscle mitochondria. Cell Mol Life Sci 63(19–20):2415–2425. doi:10.1007/s00018-006-6235-z
Kong JY, Rabkin SW (2004) Cytoskeletal actin degradation induced by lovastatin in cardiomyocytes is mediated through caspase-2. Cell Biol Int 28(11):781–790. doi:10.1016/j.cellbi.2004.07.012
Kwak HB, Thalacker-Mercer A, Anderson EJ et al (2012) Simvastatin impairs ADP-stimulated respiration and increases mitochondrial oxidative stress in primary human skeletal myotubes. Free Radic Biol Med 52(1):198–207. doi:10.1016/j.freeradbiomed.2011.10.449
LaRosa JC, Grundy SM, Waters DD et al (2005) Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 352(14):1425–1435. doi:10.1056/NEJMoa050461
Larsen S, Stride N, Hey-Mogensen M et al (2013) Simvastatin effects on skeletal muscle: relation to decreased mitochondrial function and glucose intolerance. J Am Coll Cardiol 61(1):44–53. doi:10.1016/j.jacc.2012.09.036
Liantonio A, Giannuzzi V, Cippone V, Camerino GM, Pierno S, Camerino DC (2007) Fluvastatin and atorvastatin affect calcium homeostasis of rat skeletal muscle fibers in vivo and in vitro by impairing the sarcoplasmic reticulum/mitochondria Ca2+-release system. J Pharmacol Exp Ther 321(2):626–634. doi:10.1124/jpet.106.118331
Link E, Parish S, Armitage J et al (2008) SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 359(8):789–799. doi:10.1056/NEJMoa0801936
Matzno S, Yasuda S, Juman S et al (2005) Statin-induced apoptosis linked with membrane farnesylated Ras small G protein depletion, rather than geranylated Rho protein. J Pharm Pharmacol 57(11):1475–1484. doi:10.1211/jpp.57.11.0014
Mearini G, Gedicke C, Schlossarek S et al (2010) Atrogin-1 and MuRF1 regulate cardiac MyBP-C levels via different mechanisms. Cardiovasc Res 85(2):357–366. doi:10.1093/cvr/cvp348
Mihaylova MM, Shaw RJ (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13(9):1016–1023. doi:10.1038/ncb2329
Mohler ER III, Hiatt WR, Creager MA (2003) Cholesterol reduction with atorvastatin improves walking distance in patients with peripheral arterial disease. Circulation 108(12):1481–1486. doi:10.1161/01.cir.0000090686.57897.f5
Mullen PJ, Luscher B, Scharnagl H, Krahenbuhl S, Brecht K (2010) Effect of simvastatin on cholesterol metabolism in C2C12 myotubes and HepG2 cells, and consequences for statin-induced myopathy. Biochem Pharmacol 79(8):1200–1209. doi:10.1016/j.bcp.2009.12.007
Mullen PJ, Zahno A, Lindinger P et al (2011) Susceptibility to simvastatin-induced toxicity is partly determined by mitochondrial respiration and phosphorylation state of Akt. Biochim Biophys Acta 1813(12):2079–2087. doi:10.1016/j.bbamcr.2011.07.019
Perlman H, Zhang XJ, Chen MW, Walsh K, Buttyan R (1999) An elevated bax/bcl-2 ratio corresponds with the onset of prostate epithelial cell apoptosis. Cell Death Differ 6(1):48–54. doi:10.1038/Sj.Cdd.4400453
Rabkin SW, Kong JY (2003) Lovastatin-induced cardiac toxicity involves both oncotic and apoptotic cell death with the apoptotic component blunted by both caspase-2 and caspase-3 inhibitors. Toxicol Appl Pharmacol 193(3):346–355
Rabkin SW, Lodha P, Kong JY (2007) Reduction of protein synthesis and statin-induced cardiomyocyte cell death. Cardiovasc Toxicol 7(1):1–9. doi:10.1007/s12012-007-0003-7
Ratz Bravo AE, Tchambaz L, Krahenbuhl-Melcher A, Hess L, Schlienger RG, Krahenbuhl S (2005) Prevalence of potentially severe drug–drug interactions in ambulatory patients with dyslipidaemia receiving HMG-CoA reductase inhibitor therapy. Drug Saf 28(3):263–275
Reagan-Shaw S, Nihal M, Ahmad N (2008) Dose translation from animal to human studies revisited. FASEB J 22(3):659–661. doi:10.1096/fj.07-9574LSF
Ridker PM, Danielson E, Fonseca FA et al (2008) Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 359(21):2195–2207. doi:10.1056/NEJMoa0807646
Roten L, Schoenenberger RA, Krahenbuhl S, Schlienger RG (2004) Rhabdomyolysis in association with simvastatin and amiodarone. Ann Pharmacother 38(6):978–981. doi:10.1345/aph.1D498
Sakamoto K, Wada I, Kimura J (2011) Inhibition of Rab1 GTPase and endoplasmic reticulum-to-Golgi trafficking underlies statin’s toxicity in rat skeletal myofibers. J Pharmacol Exp Ther 338(1):62–69. doi:10.1124/jpet.111.179762
Sandri M, Sandri C, Gilbert A et al (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117(3):399–412
Schick BA, Laaksonen R, Frohlich JJ et al (2007) Decreased skeletal muscle mitochondrial DNA in patients treated with high-dose simvastatin. Clin Pharmacol Ther 81(5):650–653. doi:10.1038/sj.clpt.6100124
Singh R, Krishan P (2010) Modulation of impact of high fat diet in pathological and physiological left ventricular cardiac hypertrophy by fluvastatin. Biomed Pharmacother 64(3):147–153. doi:10.1016/j.biopha.2009.06.016
Sirvent P, Bordenave S, Vermaelen M et al (2005) Simvastatin induces impairment in skeletal muscle while heart is protected. Biochem Biophys Res Commun 338(3):1426–1434. doi:10.1016/j.bbrc.2005.10.108
Sirvent P, Fabre O, Bordenave S et al (2012) Muscle mitochondrial metabolism and calcium signaling impairment in patients treated with statins. Toxicol Appl Pharmacol 259(2):263–268. doi:10.1016/j.taap.2012.01.008
Thorpe JL, Doitsidou M, Ho SY, Raz E, Farber SA (2004) Germ cell migration in zebrafish is dependent on HMGCoA reductase activity and prenylation. Dev Cell 6(2):295–302
Towler MC, Hardie DG (2007) AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res 100(3):328–341. doi:10.1161/01.Res.0000256090.42690.05
Tuckow AP, Jefferson SJ, Kimball SR, Jefferson LS (2011) Simvastatin represses protein synthesis in the muscle-derived C(2)C(1)(2) cell line with a concomitant reduction in eukaryotic initiation factor 2B expression. Am J Physiol Endocrinol Metab 300(3):E564–E570. doi:10.1152/ajpendo.00383.2010
Vander Heiden MG, Thompson CB (1999) Bcl-2 proteins: regulators of apoptosis or of mitochondrial homeostasis? Nat Cell Biol 1(8):E209–E216. doi:10.1038/70237
Acknowledgments
The authors thank David Paterson for proof-reading and Vreni Jäggin for aid with the flow cytometry. This study was supported by a Grant to Stephan Krähenbühl from the Swiss National Science Foundation (PDFMP3_132477).
Conflict of interest
None of the authors has any conflict of interest regarding this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Annalisa Bonifacio and Peter J. Mullen have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bonifacio, A., Mullen, P.J., Mityko, I.S. et al. Simvastatin induces mitochondrial dysfunction and increased atrogin-1 expression in H9c2 cardiomyocytes and mice in vivo. Arch Toxicol 90, 203–215 (2016). https://doi.org/10.1007/s00204-014-1378-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-014-1378-4