
Abstract
In addition to its well-characterized role in the regulation of drug metabolism and transport by xenobiotics, pregnane X receptor (PXR) critically impacts on lipid homeostasis. In mice, both ligand-dependent activation and knockout of PXR were previously shown to promote hepatic steatosis. To elucidate the respective pathways in human liver, we generated clones of human hepatoma HepG2 cells exhibiting different PXR protein levels, and analyzed effects of PXR activation and knockdown on steatosis and expression of lipogenic genes. Ligand-dependent activation as well as knockdown of PXR resulted in increased steatosis in HepG2 cells. Activation of PXR induced the sterol regulatory element-binding protein (SREBP) 1-dependent lipogenic pathway via PXR-dependent induction of SREBP1a, which was confirmed in primary human hepatocytes. Inhibiting SREBP1 activity by blocking the cleavage-dependent maturation of SREBP1 protein impaired the induction of lipogenic SREBP1 target genes and triglyceride accumulation by PXR activation. On the other hand, PXR knockdown resulted in up-regulation of aldo–keto reductase (AKR) 1B10, which enhanced the acetyl-CoA carboxylase (ACC)-catalyzed reaction step of de novo lipogenesis. In a cohort of human liver samples histologically classified for non-alcoholic fatty liver disease, AKR1B10, SREBP1a and SREBP1 lipogenic target genes proved to be up-regulated in steatohepatitis, while PXR protein was reduced. In summary, our data suggest that activation and knockdown of PXR in human hepatic cells promote de novo lipogenesis and steatosis by induction of the SREBP1 pathway and AKR1B10-mediated increase of ACC activity, respectively, thus providing mechanistic explanations for a putative dual role of PXR in the pathogenesis of steatohepatitis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a spectrum of diseases ranging from non-alcoholic fatty liver (or simple steatosis) to non-alcoholic steatohepatitis (NASH) with potential risk to progression toward cirrhosis. It is part of the metabolic syndrome and as such associated with obesity, insulin resistance and type 2 diabetes (Abdelmalek and Diehl 2007). NAFLD is characterized by the excess storage of triglycerides in hepatocytes, resulting from the dysregulation of glucose and lipid homeostasis (Anderson and Borlak 2008). However, the precise molecular mechanisms of pathogenesis and disease progression are only partially understood. While excess release of fatty acids by lipolysis in adipose tissue due to insulin resistance is generally accepted as contributing to the etiology of hepatic steatosis (Sanyal 2005), a significant role of increased hepatic de novo lipogenesis (DNL) was only very recently proven (Lambert et al. 2014). By acting as key metabolic sensors, nuclear receptors control hepatic glucose and lipid metabolism. Thus, their activation and/or ablation have been shown to induce or protect from steatosis (George and Liddle 2008; Wagner et al. 2011). As ligand-activated transcription factors, nuclear receptors offer therapeutic options in the treatment of NAFLD and associated diseases through pharmacological modulation of their respective activities (Trauner and Halilbasic 2011).
Pregnane X receptor (PXR, NR1I2) mainly protects the body from potential hazardous and toxic compounds by inducing xenobiotic metabolism and transport, in an adaptive response to chemical assault (Tolson and Wang 2010). More recently, PXR was shown to participate in the regulation of glucose and lipid metabolism. Activation of PXR in mice inhibits gluconeogenesis and β-oxidation of fatty acids and induces fatty acid uptake, via up-regulation of the fatty acid translocase Cd36, and lipogenesis, consequently resulting in the induction of hepatic steatosis (Zhou et al. 2006; Nakamura et al. 2007). PXR-mediated lipid accumulation in mice was claimed to be independent of the sterol-regulatory element-binding protein (SREBP) 1, which is the major transcriptional regulator of DNL, as neither Srebp1c nor its target genes encoding acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS) were induced (Zhou et al. 2006). On the other hand, ablation of PXR has been shown to result in increased basal hepatic steatosis by as yet unknown mechanisms (Nakamura et al. 2007; Spruiell et al. 2014). However, effects of PXR on liver metabolism appear to be species-specific in part, as has been recently demonstrated, when comparing the response to high-fat diet of PXR-humanized transgenic and wild-type mice (Spruiell et al. 2014). Ligand-dependent activation of PXR in primary human hepatocytes was shown to induce thyroid hormone responsive SPOT 14 homolog (THRSP), thereby up-regulating FAS expression, whereas CD36 was not affected, indicating that steatosis due to human PXR activation does not involve increased fatty acid uptake (Moreau et al. 2009). Furthermore, PXR may play a role in the pathogenesis of human NAFLD, since PXR single nucleotide polymorphisms are associated with NAFLD disease severity (Sookoian et al. 2010).
Here, we show that ligand-dependent activation and knockdown of PXR both resulted in enhanced steatosis of HepG2 cells. Our data suggest novel PXR-dependent mechanisms of DNL in human liver cells: Activation of PXR resulted in induction of the SREBP1-lipogenic pathway by increased SREBP1a expression, whereas knockdown of PXR led to the up-regulation of aldo–keto reductase (AKR) 1B10, which enhanced the ACC-catalyzed reaction step of DNL. A role of PXR in human NAFLD was further corroborated by demonstrating reduced PXR protein and subsequent expression changes of regulated genes in histologically classified NASH liver specimens.
Materials and methods
Chemicals
DMSO, rifampicin, bisphenol A, sulforaphane, Nile red and bisbenzimide H33258 were purchased from Sigma-Aldrich (Taufkirchen, Germany). PK11195 was obtained from Enzo Life Sciences (Plymouth Meeting, PA, USA). MG132 was provided by Santa Cruz (Dallas, TX, USA) and PF-429242 was obtained from Tocris Bioscience (Bristol, UK). TOFA was purchased from Cayman Chemical (Ann Arbor, MI, USA). 14C-labeled acetic acid (sodium salt of [2-14C] acetic acid, specific activity 56 mCi/mmol) was provided by American Radiolabeled Chemicals (St. Louis, MO, USA).
Plasmids
The eukaryotic expression plasmid pcDhPXR, encoding human PXR, has been described previously (Geick et al. 2001). Annealed complimentary oligonucleotides, designed using the BLOCK-iT RNAi Designer (Invitrogen, Carlsbad, CA, USA) to target human PXR 5′-GAC ACT ACC TTC TCC CAT TTC-3′ (position 2,326–2,346 of NM_003889) or non-mammalian sequence 5′-CAA CAA GAT GAA GAG CAC CAA-3′, were cloned into pENTR/U6 vector and inserts sequenced. The expression cassettes were shuttled to pLenti6/BLOCK-iT-DEST vector using the LR clonase II enzyme mix (Invitrogen), thereby generating pLenti6-shP3, encoding shRNA targeting human PXR, and pLenti6-shCTR-EH, encoding the non-mammalian negative control shRNA.
Cell culture and generation of stably transfected cell clones
The origin and culture of HepG2 cells were described previously (Hoffart et al. 2012). The cells were transfected with pcDhPXR, pLenti6-shCTR-EH or pLenti6-shP3. Stable transfected clones were selected by treatment with appropriate antibiotics and analyzed for PXR protein expression by immunoblotting.
Protein analysis
For immunodetection of proteins, cells were lysed as described previously (Sundqvist et al. 2005). To avoid proteasomal degradation of SREBP1, cells were treated with 25 µM MG132 2 h before harvesting. Total protein lysates were quantified using the bicinchoninic acid method and further used for standard protein gel electrophoresis and Western blotting. For detection of PXR in human primary hepatocytes and liver, cells and tissue samples, the latter of which were ground in liquid nitrogen, were lysed directly with Laemmli protein sample buffer. Nitrocellulose membranes were incubated with the following specific primary antibodies: PXR (H-160), SREBP1 (2A4) and AKR1B10 (D-8) from Santa Cruz; FAS (C20G5) and ACC (C83B10) from Cell Signaling Technology (Danvers, MA, USA); β-actin (AC-15) from Sigma-Aldrich. Fluorescence intensity of appropriate infrared-labeled secondary antibodies (LI-COR, Lincoln, NE, USA) was analyzed and quantified using the Odyssey Infrared Imaging System (LI-COR). Equal protein loading was verified by comparing to β-actin levels.
Fluorescence quantification and imaging of neutral lipids
7 × 104 cells/well were seeded in black poly-d-lysine-coated 96-well microplates (PerkinElmer, Waltham, MA, USA). After 72 h of culture, cells were treated with 3 % formaldehyde in medium for 30 min at 37 °C, washed with phosphate-buffered saline (PBS) and incubated with 70 µl of 15 µg/ml Nile red in PBS for 20 min at 37 °C to stain neutral lipids, which were quantified by fluorescence spectroscopy using the Wallac 1420 VICTOR microplate reader (PerkinElmer), with excitation and emission wavelengths of 535 and 580 nm, respectively. Subsequently, DNA was stained by adding 40 µl of 110 µg/ml bisbenzimide H33258 in PBS for 40 min at 37 °C. The supernatant was aspirated and DNA quantified by fluorescence spectroscopy with excitation and emission wavelengths of 360 and 460 nm, respectively. Nile red fluorescence was normalized to cell numbers. To record pictures of intracellular lipid accumulation, cells were fixed in 3 % formaldehyde/PBS, washed twice with PBS and stained with Nile red as described above. The CKX41 inverse fluorescent microscope (Olympus, Hamburg, Germany) with integrated digital camera was used to take the pictures.
Quantification of total triglycerides
2 × 107 cells were seeded in 10 cm dishes and supplied with fresh medium every 24 h. After 72 h of culture, cells were harvested, resuspended at 1 × 107 cells/300 µl in PBS and lysed by sonication in the Bioruptor ultrasonic water bath (Diagenode, Liege, Belgium). An aliquot was used for DNA quantification. Lipids were extracted by the method of Bligh and Dyer (1959). Triglycerides were quantified using the Serum Triglyceride Determination Kit (Sigma-Aldrich), according to the recommendations of the manufacturer.
Quantitative real-time RT-PCR analysis
Total RNA and first-strand cDNA were prepared as described previously (Burk et al. 2002). The integrity of RNA samples was either confirmed by formaldehyde agarose gel electrophoresis or by Agilent2100 Bioanalyzer analysis (Agilent Technologies, Santa Clara, CA, USA).
For relative quantification analyses, cDNA samples (25 ng each) were pre-amplified for 14 cycles using a mix of primer/probe sets of the respective genes (omitting the set of 18S rRNA) and TaqMan PreAmp Master Mix (Applied Biosystems, Foster City, CA, USA), according to the Fluidigm specific target amplification protocol (Fluidigm, South San Francisco, CA, USA) and finally diluted 1:5 with nuclease-free H2O. 48.48 or 96.96 Dynamic Array Integrated Fluidic Circuits (Fluidigm) were loaded with these diluted pre-amplified samples and primer/probe sets of the respective genes, added into TaqMan Gene Expression Master Mix (Applied Biosystems). TaqMan real-time quantitative PCR was performed using the BioMark HD system (Fluidigm) according to the manufacturer’s protocol. Assays were done in triplicate. Oligonucleotides used for SREBP1c were as follows: 900 nM each of primers 5′-CGG AGC CAT GGA TTG CA-3′ (exon 1c) and 5′-GGA AGT CAC TGT CTT GGT TGT TGA-3′ (exon 2); 250 nM of 6-carboxyfluorescein-labeled probe 5′-CTT TCG AAG ACA TGC TTC A-3′ (exon 1c/2). The 18S rRNA assay has been described (Hoffart et al. 2012). The following commercial TaqMan gene expression assays (Applied Biosystems), consisting of pre-designed primer/probe sets, were used to quantify the other genes: Hs01046047_m1 (ACACA), Hs00153715_m1 (ACACB), Hs00982738_m1 (ACLY), Hs01546975_gH (AKR1B10), Hs00604506_m1 (CYP3A4), Hs00225412_m1 (ELOVL6), Hs01005622_m1 (FASN), Hs02758991_g1 (GAPDH), Hs01114267_m1 (PXR), Hs01682761_m1 (SCD), Hs00231674_m1 (SREBP1a), Hs00930058_m1 (THRSP). Data were analyzed using the BioMark real-time PCR analysis software and further processed by applying the ΔΔCt method. 18S rRNA levels were used to normalize gene expression levels, if not indicated otherwise.
For absolute quantification analyses of ACACA and ACACB, the 7900 Real-Time PCR system (Applied Biosystems) and qPCR MasterMix (Eurogentec, Liege, Belgium) were used. Serial dilutions of a linearized plasmid, which contains cDNAs of both ACACA and ACACB, were used to create the respective calibration curves, ranging from 30 to 3 × 107 copies. Assays were done in triplicate and normalized to 18S rRNA levels to calculate copy numbers per ng of total RNA.
Preparation and culture of primary human hepatocytes
Tissue samples from human liver resections were obtained from 25 patients, who underwent partial hepatectomy because of primary or secondary liver tumors at the Department of General, Visceral and Transplantation Surgery (University Medical Center Charité, Berlin, Germany). Donor data are shown in Supplementary Table S1. Human hepatocytes were isolated as described previously (Nussler et al. 2009). For induction experiments, the isolated cells were seeded at 1.5 × 106 cells/well into collagen type I-coated 6-well plates and treated with chemicals as described previously (Hoffart et al. 2012).
siRNA transfection of cultured cells
Primary human hepatocytes were transfected with nontargeting negative control siRNA (Silencer Select negative control 1) or gene-specific Silencer Select siRNA (Ambion/Life Technologies, Carlsbad, CA, USA) at a final concentration of 20 nM using Lipofectamine RNAiMAX (Invitrogen), according to manufacturer’s protocol, 6 h after plating of 4 × 105 cells/well into collagen type-I-coated 12-well plates. Similarly, 3.5 × 105 HepG2 cells/well of a 6-well plate were reverse-transfected with siRNA. Supplementary Table S2 shows siRNA sequences. Culture medium was renewed daily and cells were harvested for RNA and/or protein analysis 72 h after transfection. Transfected cells of HepG2 clone H–S1 were harvested for protein analysis or used in metabolic labeling experiments 72 h after transfection.
Quantification of de novo lipogenesis
De novo lipogenesis was analyzed by quantifying the incorporation of 14C-labeled acetic acid into lipids. For this purpose, siRNA-transfected H–S1 cells, grown in 6-well plates, were labeled by adding 2 µCi/well of 14C-acetic acid for 3 h. Cells were harvested and lipids were extracted as previously described (Ma et al. 2008). Radioactivity was measured using the Hidex 300 SL liquid scintillation counter (Hidex, Turku, Finland).
Human liver biobank and NAFLD grading
The human liver biobank, which was built up with histologically normal, non-tumorous liver tissue samples, collected from patients undergoing liver surgery at the Department of General, Visceral and Transplantation Surgery (University Medical Center Charité, Berlin, Germany) between 1999 and 2001 was described previously (Nies et al. 2009). Samples from patients with hepatitis, cirrhosis or chronic alcohol abuse were excluded. None of the patients received pre-surgical chemotherapy within 6 weeks before surgery. NAFLD livers were identified retrospectively by histological analysis of hematoxylin/eosin- and Masson’s trichrome-stained sections of paraffin-embedded tissue, performed by a board certified pathologist (P.R.). Samples were graded as non-NASH (N = 23), mild-NASH (N = 22) and moderate to severe NASH (N = 20), taking into account the variable degrees of steatosis, hepatocellular ballooning, lobular and portal inflammation (Supplementary Table S3), according to the classification by Brunt et al. (1999). Significant differences in serum liver parameters were not observed between groups.
Data analysis
Data are presented as mean ± SD of at least three independent experiments, if not otherwise indicated. Multiple comparisons were performed using one-way or two-way ANOVA with Dunnett’s multiple comparisons test, if not otherwise identified. Pairwise comparisons were performed using unpaired or paired t test. One sample t test or Wilcoxon signed-rank test were used for comparisons with a hypothetical mean. All calculations were done using GraphPad Prism 6.03 (GraphPad Software, La Jolla, CA, USA).
Results
Ligand-dependent activation and knockdown of PXR result in increased steatosis in HepG2
To investigate the effects of PXR expression levels and activation on human hepatic steatosis, we established a set of HepG2 cell clones by stable over-expression (clone H–P) or shRNA-mediated knockdown (clone H–S1) of PXR. Figure 1a shows that these clones differ in PXR protein expression by a factor of 10. Control clone H–C expressed the same amount of PXR protein as parental HepG2 cells (Supplementary Fig. S1a). Analysis of cytochrome P450 (CYP) 24A1, which is directly regulated by PXR (Pascussi et al. 2005), demonstrated the functional effect of variable PXR expression levels on basal and ligand-induced target gene expression (Fig. 1b). PXR activation by the prototypical ligand rifampicin resulted in increased intracellular lipid accumulation, as shown by fluorescent staining of neutral lipids (Fig. 1c, d) and triglyceride quantification (Fig. 1e). Irrespective of the tenfold difference in PXR protein, H–P and H–S1 cells showed similar dose-dependent induction of lipid accumulation (Fig. 1d). However, basal lipid content was already elevated in PXR knockdown H–S1 cells (Fig. 1c), which accordingly exhibited significantly elevated basal triglyceride levels (Fig. 1f). The increased basal steatosis was not a clonal trait of H–S1, since a second PXR knockdown clone H–S2, exhibiting equally low PXR protein expression as H–S1 (Supplementary Fig. S1b), showed a similar increase in basal triglycerides (Fig. 1f).

Ligand-dependent activation and knockdown of PXR increase lipid accumulation in HepG2. a PXR protein expression in HepG2 clones, generated by stable transfection of PXR expression plasmid (H–P), shRNA targeting PXR (H–S1) and non-mammalian negative control shRNA (H–C). b Basal expression (inset) and time course of induction by 30 µM rifampicin (RIF) of CYP24A1 mRNA. c Nile red fluorescence images (magnification 400×) of H–P (i, ii) and H–S1 cells (iii, iiii), treated for 72 h with 0.1 % DMSO (i, iii) or 30 µM rifampicin (ii, iiii). d Nile red fluorescence quantification of cells treated for 72 h with the indicated concentrations of rifampicin or 0.1 % DMSO (−). Columns show mean ± SD of three independent experiments, each performed in replicates of ten. e Triglyceride accumulation in cells treated for 72 h with 30 µM rifampicin (+) or 0.1 % DMSO (−). f Basal triglyceride content of HepG2 clones. Columns show mean ± SD (n = 5). H–S2, second HepG2 clone generated by stable transfection of shRNA targeting PXR. Significant difference to respective cells treated with DMSO (d, e) or to control H–C cells (a, b inset, f): *P < 0.05; ***P < 0.001
Activation of PXR induces the expression of SREBP1a and lipogenic SREBP1-target genes in HepG2 and primary human hepatocytes
Next, we analyzed whether changes in the expression of transcriptional regulators of lipogenic genes may be responsible for the rifampicin-induced steatosis in HepG2 cells. In accordance with the induction of steatosis, treatment with rifampicin resulted in the induction of SREBP1a mRNA expression in both H–P and H–S1 cells (Fig. 2a). However, PXR knockdown H–S1 cells showed lower basal and rifampicin-induced SREBP1a expression levels, which was confirmed by transient siRNA-mediated knockdown of PXR in parental HepG2 cells (Fig. 2b), thereby indicating PXR-dependent regulation of SREBP1a. Induction by further PXR ligands bisphenol A and PK11195 in H–P and inhibition of rifampicin-dependent induction in H–S1 by co-treatment with the PXR antagonist sulforaphane (Fig. 2c) further proved that SREBP1a was up-regulated in a PXR-dependent way. Due to cytotoxicity, sulforaphane could only be used at 6 µM, which was below the reported IC50 of 12 to 16 µM for PXR antagonism (Zhou et al. 2007). In contrast to H–S1, this dose did not inhibit rifampicin-dependent induction of SREBP1a in H–P cells (Supplementary Fig. S2), which demonstrate tenfold higher PXR protein levels than H–S1. As H–P cells showed stable induction of SREBP1a over time (Supplementary Fig. S3), they were used to analyze the effects of PXR ligand-dependent activation on lipogenesis. In agreement with the induction of SREBP1a mRNA, precursor and mature SREBP1 protein expression were induced by rifampicin and bisphenol A (Fig. 2d). However, PK11195 solely resulted in induction of precursor protein, without a corresponding increase in the level of mature protein, indicating that the compound blocked processing of precursor SREBP1 in H–P cells by an as yet unknown mechanism. As only pan-SREBP1-specific antibodies, recognizing both the 1a and 1c isoform, were available, we used isoform-specific siRNA-mediated knockdown to demonstrate that the increased SREBP1 protein levels resulted from induced expression of SREBP1a mRNA (Fig. 2e). These data further confirmed that SREBP1a represents the major SREBP1 protein isoform in HepG2 cells. Lipogenic SREBP1 target genes ACACA and FASN, encoding the key lipogenic enzymes ACC1 and FAS, were induced by rifampicin (Fig. 2f), which further resulted in elevated protein levels (Fig. 2g). Lack of induction of ACACB (Fig. 2f), encoding ACC2, confirmed that the increase in ACC protein was exclusively derived from the induction of ACACA, which has to be noted, as the antibody recognized both protein isoforms.

Activation of PXR induces the expression of SREBP1a and of key lipogenic enzymes ACC and FAS. a SREBP1a expression in HepG2 clones, treated for 24 h with 0.1 % DMSO or 30 µM rifampicin (RIF). b PXR (left) and SREBP1a expression (right) in parental HepG2 cells transfected with negative control (siCtr) or PXR-specific siRNA (siPXR) for 72 h and treated with 0.1 % DMSO or 30 µM rifampicin during the last 24 h. c Induction of SREBP1a in H–P cells, treated with 30 µM of PXR ligands (left), and in H–S1 cells, co-treated with 30 µM rifampicin and with or without 6 µM sulforaphane (SFN) (right), for 24 h. Expression in DMSO-treated cells was designated as 1. BPA, bisphenol A; PK, PK11195. d Induction of SREBP1 protein expression in H–P cells, treated with 30 µM of the indicated PXR ligands or 0.1 % DMSO for 24 h. P- and m- indicate precursor and mature protein, respectively. e Down-regulation of mature SREBP1 protein by 48 h silencing of SREBP1a using three isoform-specific siRNAs (#1, #2 and #3) in H–P cells, treated with 30 µM rifampicin (+) or 0.1 % DMSO (−) during the last 24 h. siCtr, negative control siRNA. f Induction of the indicated genes in H–P cells by treatment with 30 µM rifampicin or 0.1 % DMSO for 72 h. g ACC and FAS protein expression in H–P cells treated as described in (f). Data were analyzed as described in Materials and methods except that in (a) and (b) one-way ANOVA with Tukey’s multiple comparisons test was used. Asterisks indicate significant differences to respective DMSO-treated groups, daggers denote significant differences between connected groups: *,† P < 0.05; **, †† P < 0.01; ***P < 0.001
In primary human hepatocytes, rifampicin also induced the expression of SREBP1a and of selected lipogenic direct SREBP1 target genes (Reed et al. 2008), including ACACA and FASN (Fig. 3a). SREBP1c, the major hepatic SREBP1 transcript (Shimomura et al. 1997), which was previously shown to be auto-regulated by SREBP1 protein (Dif et al. 2006), was not significantly induced by rifampicin in this cohort. Nevertheless, single donors demonstrated induction of 1.5-fold or more. To further demonstrate that PXR is mediating the induction of SREBP1a and lipogenic gene expression by rifampicin, siRNA-mediated knockdown of PXR was performed additionally in primary human hepatocytes. Transfection of PXR-specific siRNA resulted in 80 % PXR mRNA knockdown, thereby preventing the induction of SREBP1a and key lipogenic target genes by rifampicin (Fig. 3b). The same effect was achieved by co-treatment with the PXR antagonist sulforaphane (Fig. 3c).

PXR-dependent induction of SREBP1a and lipogenic target genes in primary human hepatocytes. a Primary human hepatocyte cultures of 23 donors were treated for 48 h with 30 µM rifampicin (RIF). Expression of indicated genes is shown as fold induction by rifampicin in scatter plots, with medians indicated by lines. Respective expression levels by treatment with DMSO only were set as 1. Wilcoxon signed-rank test was used to calculate P values. b Relative PXR expression (left) and induction of SREBP1a and lipogenic target genes (right) in siRNA-transfected (72 h) primary human hepatocytes (donor RH17) treated with 5 µM rifampicin or 0.1 % DMSO during the last 24 h. c Induction of SREBP1a and its lipogenic target genes in primary human hepatocytes (donor RH17) by treatment with 5 µM rifampicin for 24 h in the absence (−SFN) or presence (+SFN) of 10 µM sulforaphane. mRNA data are shown as fold induction by rifampicin in siPXR and negative control siRNA (siCtr) transfected cells (b) and in −SFN and +SFN treated cells (c), compared to respective cells treated with DMSO
Induction of lipogenic gene expression and steatosis by PXR activation is mediated by SREBP1a
Next, we analyzed the possible intermediary role of SREBP1a in PXR activation-dependent lipogenesis and steatosis. Linear regression analysis of the fold change by rifampicin of SREBP1a and respective lipogenic target genes showed that the extent of SREBP1a induction largely determined the extent of induction of lipogenic SREBP1 target genes, except THRSP (Supplementary Fig. S4). Interestingly, SREBP1a induction did also not determine induction of CYP3A4 (Supplementary Fig. S4).
Precursor SREBP1 protein has to be processed by successive proteolytic cleavage, involving site-1 and site-2 proteases, to result in the transcriptionally active mature nuclear protein (Horton et al. 2002). Co-treatment of primary human hepatocytes with the site-1 protease inhibitor PF-429242 (Hawkins et al. 2008) prevented the induction of SREBP1c and stearoyl-CoA desaturase (SCD) by rifampicin, whereas induction of FASN was halved (Fig. 4a). Expression of THRSP was largely down-regulated by PF-429242; however, fold induction by rifampicin was even increased. Similarly, induction of CYP3A4 and SREBP1a by rifampicin persisted (Fig. 4a), indicating that PF-429242 was not inhibiting ligand-dependent PXR activation per se. Accordingly, the compound neither activated nor inhibited PXR in transient reporter gene assays (Supplementary Fig. S5). In the following, H–P cells were used to analyze the contribution of SREBP1a to PXR activation-dependent intracellular lipid accumulation, as this could not be performed in primary human hepatocytes due to their variable and mostly pronounced basal steatosis. PF-429242 completely blocked processing of SREBP1 protein in H–P cells, resulting in the loss of mature protein, without compromising the induction of the precursor by rifampicin (Fig. 4b). The compound strongly reduced the dose-dependent induction of intracellular lipid accumulation in H–P cells by rifampicin (Fig. 4c). In agreement with not inducing mature SREBP1 protein levels (see Fig. 2d), PK11195 did not induce the accumulation of intracellular lipids (Fig. 4d). In conclusion, these data clearly indicate that the SREBP1 lipogenic pathway is largely contributing to the steatogenic effect of ligand-dependent activation of PXR in human hepatic cells.

Induction of lipogenic gene expression and steatosis by PXR activation is mediated by SREBP1. a SREBP1 target gene, SREBP1a and CYP3A4 mRNA expression in primary human hepatocytes (donor BH61) treated for 48 h with 5 µM rifampicin (RIF) and/or 10 µM PF-429242, as indicated. b Induction of SREBP1 protein expression in H–P cells, treated with 0.1 % DMSO, 30 µM rifampicin or co-treated with 3 µM PF-429242 for 24 h. P- and m- indicating precursor and mature SREBP1 protein, respectively. NC not calculable. c, d Quantification of lipid accumulation by Nile red fluorescence in H–P cells co-treated for 72 h with the indicated concentrations of rifampicin and with or without 3 µM PF-429242 (c) or treated with the indicated concentrations of PK11195 (PK) (d). Columns show mean ± SD of two independent experiments, each performed in quadruplicates. Significant differences to cells treated with vehicle DMSO only (−): *P < 0.05; ***P < 0.001
PXR knockdown results in up-regulation of AKR1B10, which promotes DNL
Next, we aimed to elucidate the mechanism of the elevated basal lipid accumulation in PXR knockdown H–S1 cells (see Fig. 1c, f). The mRNA expression of SREBP1a and lipogenic genes was even lower in these cells than in H–C controls (Supplementary Fig. S6). The elevated basal steatosis of H–S1 cells may thus be a consequence of the 1.6-fold increase in ACC protein expression (Fig. 5a), which was not reflected on mRNA level. On the contrary, ACACA mRNA was reduced in H–S1 compared to H–C. Expression differences in ACACB do not account for the difference in ACC protein, as ACACB expression was six–ninefold lower than ACACA and not different between the cell clones (Fig. 5a). Recently, it was shown that ACC1 protein is protected from degradation by interaction with AKR1B10 protein, thereby stabilizing the former enzyme and inducing DNL (Ma et al. 2008). To investigate whether this mechanism may cause the elevated ACC protein level and basal steatosis of H–S1, we determined respective AKR1B10 mRNA and protein amounts. Figure 5b shows that expression of AKR1B10 mRNA and protein was higher in H–S1 than in H–C. The siRNA-mediated knockdown of PXR in primary human hepatocytes resulted in similarly elevated mRNA and protein levels of AKR1B10 (Fig. 5c), thereby excluding clonal cell line effects. To analyze, whether AKR1B10 levels play an ACC-dependent role in DNL, H–S1 cells were transfected with AKR1B10-specific siRNA and DNL was quantified in the absence or presence of the ACC inhibitor TOFA. Knockdown of AKR1B10 resulted in 80 % decrease of protein expression and consequently reduced DNL by 20 %, if ACC was not inhibited by TOFA (Fig. 5d). In conclusion, these data suggest that increased AKR1B10 levels contribute to the elevated basal steatosis of PXR knockdown H–S1 cells.

PXR knockdown dependent up-regulation of AKR1B10 promotes DNL. a Relative ACC protein expression (left) and absolute quantification of mRNA of ACC encoding genes (right) in H–C and H–S1 cells. b Relative AKR1B10 mRNA (left) and protein (right) expression in H–C and H–S1 cells. c PXR and AKR1B10 mRNA (top) and protein expression (bottom) of siRNA-transfected primary human hepatocytes (phh). mRNA data, normalized to GAPDH, are shown as fold change by transfection of siPXR as compared to negative control siRNA (siCtr), expression levels of which were designated as 1, and presented as scatter plots, with medians indicated by lines. Western Blot analysis of a single experiment (donor RH16) is shown. Numbers indicate relative expression of AKR1B10 protein. d Relative de novo lipogenesis, as measured by 14C-acetate incorporation into lipids, in siAKR1B10 (siAKR) and negative control siRNA (siCtr) transfected H–S1 cells, treated with or without 20 µM ACC inhibitor TOFA. Columns show mean ± SD of two independent experiments, each performed in triplicate. Western blot of AKR1B10 knockdown is shown on top. Significant differences to H–C or between groups: *P < 0.05; **P < 0.01; ***P < 0.001
PXR protein expression declines in NASH, whereas AKR1B10, SREBP1a and SREBP1-dependent lipogenic gene expression increase
To investigate whether the here identified PXR-regulated lipogenic pathways are of pathophysiological relevance, we analyzed expression of PXR and of the respective genes in histologically classified NAFLD liver specimens (Supplementary Table S3). Linear regression analysis of log2-transformed mRNA and protein expression of PXR showed that hepatic PXR mRNA amounts did not predict protein levels (R 2 = 0.0037; P = 0.6959; N = 43). PXR protein was more than halved in moderate to severe NASH compared to non-NASH (Fig. 6a). Consistently, AKR1B10 showed threefold higher median level in moderate to severe NASH (Fig. 6b). Figure 6c shows that SREBP1a and the SREBP1-dependent lipogenic genes demonstrated increased expression in NASH.

Decreased PXR protein levels and increased expression of AKR1B10, SREBP1a and its lipogenic target genes in human NASH. a PXR protein expression was analyzed by Western blot of tissue lysates of histopathologically diagnosed human liver samples and normalized to β-actin levels. Relative expression was calculated by referring to an unrelated liver sample, which was co-analyzed on each individual blot. Representative Western blot is shown on top. mRNA expression of AKR1B10 (b), SREBP1a and lipogenic target genes (c) in histologically diagnosed human liver samples. Numbers in groups correspond to Supplementary Table S3, except that in (b) two non-NASH and one moderate to severe NASH sample were identified as outliers by the ROUT method and thus removed. Expression data are shown in scatter plots, lines indicating medians. Median gene and PXR protein expression of the non-NASH group was designated as 1. P values were calculated by Mann–Whitney test (a, b) or Kruskal–Wallis test with Dunn’s multiple comparisons test (c)
Discussion
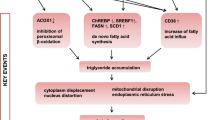
Previous data from mouse models indicate that the modulation of PXR activity or expression may be a therapeutic option in the treatment of NAFLD. However, information on the molecular mechanisms underlying PXR-dependent steatosis in human liver is limited. Here, we have shown that ligand-dependent activation of human PXR results in induction of the SREBP1-dependent lipogenic pathway, whereas knockdown enhances the ACC-catalyzed reaction step of DNL via up-regulation of AKR1B10. By increasing DNL, both mechanisms independently result in elevated hepatic steatosis (Fig. 7).

Scheme of the dual role of PXR in the development of human hepatic steatosis. On the one hand, PXR, which is activated by the binding of ligands, results in increased DNL and steatosis via induction of the SREBP1-lipogenic pathway, mediated by PXR-induced SREBP1a. On the other hand, down-regulation of PXR expression enhances AKR1B10 expression. The AKR1B10 protein stabilizes acetyl-CoA carboxylase, which subsequently promotes DNL and steatosis
Our data indicate that PXR- and SREBP1-dependent pathways are integrated by PXR inducing the expression of SREBP1a. Transient as well as stable knockdown of PXR in HepG2 cells resulted in reduced basal and rifampicin-induced SREBP1a mRNA expression levels, clearly arguing for a role of PXR in the regulation of SREBP1a expression. However, the residual 30–40 % of PXR was still sufficient for induction of SREBP1a by PXR activation, which also explains the induction of steatosis by rifampicin in PXR knockdown H–S1 cells. Attempts to further reduce PXR expression in HepG2 (e.g. by transfection of PXR-specific siRNA in H–S1 cells) failed, which may be related to the fact that PXR participates in the proliferation control of hepatocytes (Dai et al. 2008) and may thus be essential for the proliferating HepG2 cells. In contrast, siRNA-mediated knockdown succeeded to reduce PXR expression to 10–20 % of control levels in the nonproliferating primary hepatocytes, which was no longer sufficient to maintain rifampicin-dependent induction of SREBP1a. Co-treatment of H–S1 cells and primary human hepatocytes with the PXR antagonist sulforaphane further confirmed that PXR is necessary for rifampicin-dependent induction of SREBP1a and lipogenic SREBP1 target genes. Altogether these data unequivocally demonstrate that the induction of SREBP1a by rifampicin depends on the activation of PXR.
The molecular mechanism of SREBP1a induction by PXR is currently unknown. We hypothesize that it most likely will differ from the classical direct transcriptional activation by binding of PXR to response elements in regulatory regions. First, time course of induction markedly differed between SREBP1a and the directly regulated CYP24A1; while induction of CYP24A1 steadily increased over time in H–P cells, SREBP1a induction reached its maximal level after 12 h and stayed constant, thereafter. This further indicates that the mechanism was rapidly saturated, implicating that a limited amount of PXR protein may be sufficient. Second, the extent of induction of SREBP1a and of direct PXR target genes as CYP3A4 or THRSP was not correlated in primary human hepatocytes. Third, using a promoter reporter gene construct encompassing 2,553 bp of the 5′ upstream region of SREBP1a (Fernández-Alvarez et al. 2008), which comprises several putative PXR binding sites of DR4-type, we did not observe any PXR-dependent induction of promoter activity in HepG2 cells (data not shown). Up to now, DNA-binding independent mechanisms have only been demonstrated for ligand-dependent gene repression by PXR, wherein the receptor was shown to inhibit the activity of other transcription factors, as, e.g., cAMP-response element-binding protein, and forkhead box proteins A2 and O1, by protein–protein interaction (Kodama and Negishi 2013). However, overlay of genome-wide PXR chromatin binding and ligand-dependent gene expression data in mice revealed the existence of a large group of up-regulated genes without apparent PXR binding sites in their chromatin (Cui et al. 2010).
We clearly have shown here that steatosis by activation of PXR in human hepatic cells largely depends on the induction of the SREBP1-dependent lipogenic pathway, as blocking the generation of the mature, transcriptionally active SREBP1 protein strongly reduced rifampicin-induced lipogenic gene expression and intracellular lipid accumulation. Induction of this pathway by PXR was not reported in previous studies using mice (Zhou et al. 2006) and human hepatocytes (Moreau et al. 2009). However, these studies did not analyze SREBP1a expression. Although SREBP1a is expressed at significantly lower levels in liver, it is a much stronger transcriptional activator of SREBP1 target genes than SREBP1c (Amemiya-Kudo et al. 2002). Thus, even a moderate increase of its expression can be expected to result in disproportionately high effects. Very recently, ob/ob VP−PXR mice, expressing activated PXR, also demonstrated induction of SREBP1-regulated genes encoding ACC, FAS and of Srebp1c (He et al. 2013), further corroborating the capacity of PXR to activate this pathway and demonstrating that the effects of PXR activation differ between mouse strains. It has yet to be established, whether murine SREBP1a is activated by PXR. In primary human hepatocytes, Moreau and colleagues previously described a PXR-dependent lipogenic mechanism, involving THRSP-mediated up-regulation of FASN (Moreau et al. 2009), which may explain that blocking the maturation of SREBP1 in these cells resulted in only 50 % inhibition of FASN induction by PXR. However, this mechanism cannot be operative in HepG2 cells, as THRSP was not induced here by rifampicin (data not shown).
We successfully mimicked in a human cell-based PXR knockdown model the steatogenic effect of PXR knock-out in mice. Elevated basal expression of Scd1 (Nakamura et al. 2007), may contribute to the steatogenic effect of PXR knockout in mice. However, SCD did not show increased expression in PXR knockdown H–S1 cells. Instead, reduced PXR protein expression, as generated by knockdown in HepG2 cells and primary human hepatocytes, and further observed in NASH, resulted in elevated AKR1B10 levels. AKR1B10 was previously shown to protect ACC from degradation by direct protein–protein interaction (Ma et al. 2008), thereby raising lipid synthesis (Wang et al. 2009), which may contribute to DNL being permanently up-regulated in NAFLD (Donnelly et al. 2005). Notably, increased expression of AKR1B10 has recently been shown to be associated with hepatic steatosis and steatohepatitis (Tsuzura et al. 2014; Starmann et al. 2012). Furthermore, the resulting increase in ACC protein may contribute to the sustained induction of steatosis by rifampicin in PXR low expressing H–S1 cells. A higher basal synthesis rate of malonyl-CoA, due to the AKR1B10-dependent increase in ACC activity, may compensate for the lower level of rifampicin-induced SREBP1a expression in these cells, as malonyl-CoA represents the building block of fatty acid synthesis. The molecular mechanism of AKR1B10 up-regulation by PXR knockdown most likely relies on the release of PXR-dependent repression of a transcription factor, which activates AKR1B10 gene expression, as PXR is well known for repressive protein–protein interactions with several other transcription factors (Kodama and Negishi 2013). Akr1b8, the mouse ortholog of human AKR1B10, was shown to exert a similar effect on ACC stability and DNL as its human counterpart (Joshi et al. 2010). Whether it is regulated similarly by PXR and may contribute to the steatogenic effect of PXR knock-out in mice deserves further study.
A role of PXR in human NAFLD has been previously suggested by a genetic association study (Sookoian et al. 2010). Most likely due to the small sample sizes of our NAFLD groups, we could not confirm the reported association of the respective PXR single nucleotide polymorphisms with the disease. However, we here provide further evidence for the putative role of PXR in human NAFLD, by showing for the first time that PXR protein is significantly reduced in human NASH. In mice, hepatic PXR mRNA and protein expression are reduced by feeding a high-fat diet, which simultaneously increased the expression of pro-inflammatory cytokines (Ghose et al. 2011). The mechanism by which PXR protein is down-regulated in human NASH is expected to be different, because we did not observe any corresponding decrease of mRNA. Since human PXR protein expression is subject to regulation by miRNA (Takagi et al. 2008) and miRNAs are increasingly recognized as important players in NASH (Cheung et al. 2008), an inflammatory epigenetic mechanism may be involved in the down-regulation of PXR protein in human steatohepatitis.
The expression of SREBP1a, -1c and a battery of SREBP1-regulated genes encoding lipogenic enzymes was shown here for the first time to be consistently up-regulated in steatohepatitis. To link PXR to this increase in expression, we have to hypothesize that the receptor is activated in NAFLD, implicating that its activation promotes human hepatic steatosis. Although a case report demonstrated hyperlipidemia as a consequence of rifampicin treatment in a patient with tuberculosis (Khogali et al. 1974), thereby indicating that PXR activation may result in clinically relevant effects on lipid homeostasis, it has yet to be shown unequivocally in a controlled study that xenobiotic activation of human PXR can cause hepatic steatosis. In further support of a respective role of PXR, it was shown that treatment of volunteers with rifampicin resulted in elevated postprandial insulin and glucose levels (Rysä et al. 2013), which are highly associated with NAFLD.
Even if PXR activation and knockdown both stimulate DNL, the resulting steatosis most likely differs due to the different underlying molecular mechanisms. In accordance with this assumption, PXR knockout and PXR activation-dependent steatosis differed largely in mice, with knockout resulting in macrovesicular lipid droplets and activation in microvesicular lipid droplets (Nakamura et al. 2007). By increasing the expression of SREBP1a, PXR activation induces all steps of fatty acid synthesis (Fig. 7). Thus, even if the total amount of lipids is increased, the composition of the free fatty acid pool should still be balanced. In contrast, PXR knockdown specifically results in the increased activity of ACC, without any inducing effect on lipogenic gene expression (Fig. 7). It may thus be hypothesized that the composition of the cellular-free fatty acid pool is changed toward a higher share of saturated long-chain fatty acids, as, e.g., palmitate, because expression of the desaturating enzyme SCD was not concomitantly induced. As especially saturated fatty acids have been shown to damage liver cells and promote steatohepatitis (Li et al. 2009), steatosis by PXR knockdown may be regarded as a pathophysiological feature. On the other hand, steatosis by PXR activation may represent an adaptive reaction. Lipid droplets have been discussed to possibly function as buffers for lipophilic compounds that may be harmful if present in excess (Farese and Walther 2009). Increasing such a buffer capacity may thus be the cellular objective of the induction of DNL by PXR activation. Otherwise, it is unintelligible why PXR activation induces DNL, which consumes 14 NADPH molecules for the synthesis of a single palmitate, and thereby strongly competes for NADPH with cytochrome P450-dependent detoxification. With respect to steatosis, it is currently unclear whether inhibition of PXR is functionally equivalent to its knockdown, thus PXR antagonists cannot yet be considered as novel therapeutic agents for the treatment of NAFLD. Respective further investigations are clearly warranted.
In conclusion, we have identified here novel PXR-dependent pathways in human DNL, which may participate in hepatic steatosis and pathogenesis of steatohepatitis. The influence of PXR on multiple metabolic pathways represents an obstacle for use of its ligands in the therapy of metabolic diseases. Our study has identified SREBP1a and AKR1B10 as novel PXR-regulated genes in lipogenic pathways. Elucidation of the molecular mechanisms of their regulation by PXR, which is in progress, may further aid in the future development of appropriate selective PXR modulators.
References
Abdelmalek MF, Diehl AM (2007) Nonalcoholic fatty liver disease as a complication of insulin resistance. Med Clin North Am 91:1125–1149
Amemiya-Kudo M, Shimano H, Hasty AH et al (2002) Transcriptional activities of nuclear SREBP-1a, −1c, and −2 to different target promoters of lipogenic and cholesterogenic genes. J Lipid Res 43:1220–1235
Anderson N, Borlak J (2008) Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol Rev 60:311–357
Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917
Brunt EM, Janney CG, Di Bisceglie AM et al (1999) Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 94:2467–2474
Burk O, Tegude H, Koch I et al (2002) Molecular mechanisms of polymorphic CYP3A7 expression in adult human liver and intestine. J Biol Chem 277:24280–24288
Cheung O, Puri P, Eicken C et al (2008) Nonalcoholic steatohepatitis is associated with altered hepatic microRNA expression. Hepatology 48:1810–1820
Cui JY, Gunewardena SS, Rockwell CE, Klaassen CD (2010) ChIPing the cistrome of PXR in mouse liver. Nucleic Acids Res 38:7943–7963
Dai G, He L, Bu P, Wan YJ (2008) Pregnane X receptor is essential for normal progression of liver regeneration. Hepatology 47:1277–1287
Dif N, Euthine V, Gonnet E et al (2006) Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem J 400:179–188
Donnelly KL, Smith CI, Schwarzenberg SJ et al (2005) Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 115:1343–1351
Farese RV, Walther TC (2009) Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 139:855–860
Fernández-Alvarez A, Tur G, López-Rodas G, Casado M (2008) Reciprocal regulation of the human sterol regulatory element binding protein (SREBP)-1a promoter by Sp1 and EGR-1 transcription factors. FEBS Lett 582:177–184
Geick A, Eichelbaum M, Burk O (2001) Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampicin. J Biol Chem 276:14581–14587
George J, Liddle C (2008) Nonalcoholic fatty liver disease: pathogenesis and potential for nuclear receptors as therapeutic targets. Mol Pharm 5:49–59
Ghose R, Omoluabi O, Gandhi A et al (2011) Role of high-fat diet in regulation of gene expression of drug metabolizing enzymes and transporters. Life Sci 89:57–64
Hawkins JL, Robbins MD, Warren LC et al (2008) Pharmacologic inhibition of site 1 protease activity inhibits sterol regulatory element-binding protein processing and reduces lipogenic enzyme gene expression and lipid synthesis in cultured cells and experimental animals. J Pharmacol Exp Ther 326:801–808
He J, Gao J, Xu M et al (2013) PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice. Diabetes 62:1876–1887
Hoffart E, Ghebreghiorghis L, Nussler AK et al (2012) Effects of atorvastatin metabolites on induction of drug-metabolizing enzymes and membrane transporters through human pregnane X receptor. Br J Pharmacol 165:1595–1608
Horton JD, Goldstein JL, Brown MS (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109:1125–1131
Joshi A, Rajput S, Wang C et al (2010) Murine aldo–keto reductase family 1 subfamily B: identification of AKR1B8 as an ortholog of human AKR1B10. Biol Chem 391:1371–1378
Khogali AM, Chazan BI, Metcalf VJ, Ramsay JH (1974) Hyperlipidaemia as a complication of rifampicin treatment. Tubercle 55:231–233
Kodama S, Negishi M (2013) PXR cross-talks with internal and external signals in physiological and pathophysiological responses. Drug Metab Rev 45:300–310
Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ (2014) Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 146:726–735
Li ZZ, Berk M, McIntyre TM, Feldstein AE (2009) Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl-CoA desaturase. J Biol Chem 284:5637–5644
Ma J, Yan R, Zu X et al (2008) Aldo–keto reductase family 1 B10 affects fatty acid synthesis by regulating the stability of acetyl-CoA carboxylase-alpha in breast cancer cells. J Biol Chem 283:3418–3423
Moreau A, Téruel C, Beylot M et al (2009) A novel pregnane X receptor and S14-mediated lipogenic pathway in human hepatocyte. Hepatology 49:2068–2079
Nakamura K, Moore R, Negishi M, Sueyoshi T (2007) Nuclear pregnane X receptor cross-talk with FoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver. J Biol Chem 282:9768–9776
Nies AT, Koepsell H, Winter S et al (2009) Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology 50:1227–1240
Nussler AK, Nussler NC, Merk V et al (2009) The holy grail of hepatocyte culturing and therapeutic use. In: Santin M (ed) Strategies in regenerative medicine. Springer, New York, pp 283–320
Pascussi JM, Robert A, Nguyen M et al (2005) Possible involvement of pregnane X receptor-enhanced CYP24 expression in drug-induced osteomalacia. J Clin Invest 115:177–186
Reed BD, Charos AE, Szekely AM et al (2008) Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLoS Genet 4:e1000133
Rysä J, Buler M, Savolainen MJ et al (2013) Pregnane X receptor agonists impair postprandial glucose tolerance. Clin Pharmacol Ther 93:556–563
Sanyal AJ (2005) Mechanisms of disease: pathogenesis of nonalcoholic fatty liver disease. Nat Clin Pract Gastroenterol Hepatol 2:46–53
Shimomura I, Shimano H, Horton JD et al (1997) Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest 99:838–845
Sookoian S, Castaño GO, Burgueño AL et al (2010) The nuclear receptor PXR gene variants are associated with liver injury in nonalcoholic fatty liver disease. Pharmacogenet Genomics 20:1–8
Spruiell K, Richardson RM, Cullen JM et al (2014) Role of pregnane X receptor in obesity and glucose homeostasis in male mice. J Biol Chem 289:3244–3261
Starmann J, Fälth M, Spindelböck W et al (2012) Gene expression profiling unravels cancer-related hepatic molecular signatures in steatohepatitis but not in steatosis. PLoS One 7:e46584
Sundqvist A, Bengoechea-Alonso MT, Ye X et al (2005) Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab 1:379–391
Takagi S, Nakajima M, Mohri T, Yokoi T (2008) Post-transcriptional regulation of human pregnane X receptor by micro-RNA affects the expression of cytochrome P450 3A4. J Biol Chem 283:9674–9680
Tolson AH, Wang H (2010) Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv Drug Deliv Rev 62:1238–1249
Trauner M, Halilbasic E (2011) Nuclear receptors as new perspective for the management of liver diseases. Gastroenterology 140:1120–1125
Tsuzura H, Genda T, Sato S et al (2014) Expression of aldo–keto reductase family 1 member B10 in the early stages of human hepatocarcinogenesis. Int J Mol Sci 15:6556–6568
Wagner M, Zollner G, Trauner M (2011) Nuclear receptors in liver disease. Hepatology 53:1023–1034
Wang C, Yan R, Luo D et al (2009) Aldo–keto reductase family 1 member B10 promotes cell survival by regulating lipid synthesis and eliminating carbonyls. J Biol Chem 284:26742–26748
Zhou J, Zhai Y, Mu Y et al (2006) A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J Biol Chem 281:15013–15020
Zhou C, Poulton EJ, Grün F et al (2007) The dietary isothiocyanate sulforaphane is an antagonist of the human steroid and xenobiotic nuclear receptor. Mol Pharmacol 71:220–229
Acknowledgments
We appreciate the expert technical assistance of K. Abuazi-Rincones. The nonprofit foundation Human Tissue and Cell Research (Regensburg, Germany) kindly provided human hepatocytes from four additional donors. This work contains parts of the doctoral thesis of A.B. It was supported, in whole or in part, by the Robert Bosch Foundation, Stuttgart, Germany, by the Interfaculty Center for Pharmacogenomics and Drug Research of the University of Tübingen, Germany, Grant 3-0-0 (to O.B.), by the German Federal Ministry of Education and Research, HepatoSys Grants 0313080I (to O.B.) and 0313081B (to A.K.N.) and Virtual Liver Network Grant 0315755 (to U.M.Z., and M.S.), and by Grant F3008 from the Austrian Science Foundation (to M.T.).
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standard
Liver resections of tumor patients, which were used for the preparation of primary human hepatocytes, were performed according to the respective institutional guidelines including the patient’s written informed consent, which were approved by the local ethical committee of the Charité, Humboldt University Berlin, Germany. The human liver biobank study was approved by the local ethical committees of the Charité, Humboldt University, Berlin, and University of Tübingen and conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from each patient.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bitter, A., Rümmele, P., Klein, K. et al. Pregnane X receptor activation and silencing promote steatosis of human hepatic cells by distinct lipogenic mechanisms. Arch Toxicol 89, 2089–2103 (2015). https://doi.org/10.1007/s00204-014-1348-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-014-1348-x