
Abstract
Trichothecenes are a large family of structurally related toxins mainly produced by Fusarium genus. Among the trichothecenes, T-2 toxin and deoxynivalenol (DON) cause the most concern due to their wide distribution and highly toxic nature. Trichothecenes are known for their inhibitory effect on eukaryotic protein synthesis, and oxidative stress is one of their most important underlying toxic mechanisms. They are able to generate free radicals, including reactive oxygen species, which induce lipid peroxidation leading to changes in membrane integrity, cellular redox signaling, and in the antioxidant status of the cells. The mitogen-activated protein kinases signaling pathway is induced by oxidative stress, which also induces caspase-mediated cellular apoptosis pathways. Several new metabolites and novel metabolic pathways of T-2 toxin have been discovered very recently. In human cell lines, HT-2 and neosolaniol (NEO) are the major metabolites of T-2 toxin. Hydroxylation on C-7 and C-9 are two novel metabolic pathways of T-2 toxin in rats. The metabolizing enzymes CYP3A22, CYP3A29, and CYP3A46 in pigs, as well as the enzymes CYP1A5 and CYP3A37 in chickens, are able to catalyze T-2 toxin and HT-2 toxin to form the C-3′–OH metabolites. Similarly to carboxylesterase, CYP3A29 possesses the hydrolytic ability in pigs to convert T-2 toxin to NEO. T-2 toxin is able to down- or upregulate cytochrome P-450 enzymes in different species. The metabolism of DON in humans is region-dependent. Free DON and DON-glucuronide are considered to be the biomarkers for humans. The masked mycotoxin DON-3-β-d-glucoside can be hydrolyzed to free DON in the body. This review will provide useful information on the progress of oxidative stress as well as on the metabolism and the metabolizing enzymes of T-2 toxin and DON. Moreover, the literature will throw light on the blind spots of metabolism and toxicological studies in trichothecenes that have to be explored in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
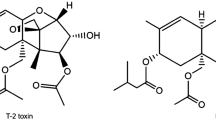
The trichothecenes are a large family of metabolites that are mainly produced by the fungi of Fusarium genus (Bennett and Klich 2003). The consumption of these toxins by farm animals and humans has a wide variety of toxic effects: hemorrhage, diarrhea, emesis, leucopenia, immunosuppression, decreased reproductive capacity, bone marrow damage, and radiomimetic injury to tissues are the common clinical signs (Wu et al. 2010). According to their characteristic functional groups, the trichothecenes are subdivided into four types (A–D) (Li et al. 2011; Wu et al. 2013). T-2 toxin and DON, belonging to type A and type B group, respectively, are considered to be the most important trichothecenes due to their great toxicity and widespread dissemination (Fig. 1).

Chemical structure of T-2 toxin and deoxynivalenol (DON)
T-2 toxin with the highest toxicity cause sublethal and even lethal toxicosis in humans and farm animals (Dohnal et al. 2008; Wu et al. 2012; Bennett and Klich 2003). Alimentary toxic aleukia (ATA), a disease that affects only humans, has been associated primarily with the ingestion of musty cereal infected with T-2 toxin (Joffe 1974). Due to its great toxicity and stability, T-2 toxin has been classified as a biological weapon (Kuca and Pohanka 2010). Recently, it is reported that T-2 toxin enters the brain via the blood–brain barrier (Weidner et al. 2013a) and that it reaches the fetus through the placenta (Wang et al. 2014). In China, high contamination of T-2 toxin in drinking water and grains has been reported in the provinces of Qinghai and Sichuan (Sun et al. 2012; Wang et al. 2012). In Europe, especially in the Nordic countries, the contamination of cereals with T-2 and HT-2 toxins is also a serious problem (Beyer et al. 2009). The WHO/FAO Joint Expert Committee on Food Additives (JECFA 2001) as well as other international bodies, such as the European Union (SCF 2001, 2002), have carried out risk assessment and management of T-2 toxin in human foods.
Deoxynivalenol is less toxic than T-2 toxin and many others, but it is the most widely distributed trichothecene and is commonly found in barley, corn, rye, safflower seed, wheat, and mixed feed (Bennett and Klich 2003; Wu et al. 2013). For humans, dizziness, throat irritation, nausea, vomiting, diarrhea, and blood in the stool are mentioned as symptoms of DON intoxication (SCF 1999). The European Commission (2006) has established the maximum levels for DON (1,250 μg/kg in cereals; 200 μg/kg in processed cereal-based foods; and baby foods). The temporary tolerable daily intake (TDI) has been set at 1.0 μg/kg b.w. Furthermore, in crops including rice, wheat, and barley, DON can be transformed to the glucose-conjugated product, DON-3-O-glucoside (DON-3-Glc) (Berthiller et al. 2005, 2013). The conjugated mycotoxins, which are generally referred to as “masked” or “hidden” mycotoxins, have become an increasing concern for scientists over the last few decades. The chemical behavior of masked mycotoxins is fundamentally different from that of their unmasked counterparts. Thus, their vast majority is not detected during routine analyses. These compounds can be totally or partially hydrolyzed after ingestion to release the parent aglycone, thus leading to an increased exposure (Galaverna et al. 2009). Normally, DON-3-Glc has lower toxicity, and the free DON that is released in the digestive tract will cause the toxic effects on animals and humans (Berthiller et al. 2013).
Trichothecenes have the ability to induce apoptosis in macrophages (Zhou and Pestka 2003). The most prominent molecular target of trichothecenes is the 60S ribosomal subunit. Trichothecenes binding to ribosomes can rapidly activate mitogen-activated protein kinases (MAPKs) and induce apoptosis in a process known as “ribotoxic stress response” (Pestka 2007). Numerous studies have reported that oxidative stress is also an important underlying mechanism in the toxicity of trichothecenes (El Golli et al. 2006; Krishnaswamy et al. 2010; Chaudhary and Rao 2010; Wu et al. 2011a, b; Fang et al. 2012). Trichothecenes significantly increase the levels of reactive oxygen species (ROS) and deplete intracellular reduced glutathione (GSH). Moreover, they increase lipid peroxidation which leads to single-strand breaks in DNA (Chaudhari et al. 2009a, b). Subsequently, some signaling pathways, including MAPK, are induced by which cell apoptosis is triggered (Sehata et al. 2005). However, some other researchers have shown that oxidative damage is not the major contributor to trichothecene toxicity, since the indexes of the oxidative stress, such as the generation of ROS, lipid peroxidation, or the induction of Heat Shock Protein-70 (HSP-70), have not been registered in these studies, although the DNA damage and apoptosis have been observed (Frankic et al. 2006; Rezar et al. 2007; Bensassi et al. 2009; Awad et al. 2012). Thus, it seems that a discrepancy with regard to the effect of oxidative stress on trichothecenes may exist, albeit some researchers have explained that the different doses used in the various studies are the potential reason of this mismatch (Frankic et al. 2006; Sahu et al. 2010). Therefore, a detailed and systematical discussion of trichothecene-induced oxidative stress is necessary for getting a thorough understanding of the toxic mechanism of trichothecenes.
During the last decades, the metabolism of trichothecenes has been accurately studied and many metabolites have been identified (Li et al. 2011). The major metabolic pathways of T-2 toxin in animals are hydrolysis, hydroxylation, deepoxidation, and conjugation. The common metabolites of T-2 toxin in animals and humans are HT-2 toxin, neosolaniol (NEO), 3′-OH-T-2, 3′-OH-HT-2, T-2 triol, T-2 tetraol, and some C12,13-deepoxy products (Wu et al. 2010, 2013). Today, it seems to be difficult to find new metabolites of T-2 toxin, and most studies are currently focusing on the toxic mechanism of the major metabolites or on controlling strategies. However, several new T-2 toxin metabolites and some novel metabolic pathways are reported recently, as for example, nine novel metabolites of T-2 toxin have been reported in rats in vivo. Moreover, hydroxylations at C-7 and C-9 have been identified as two novel metabolic pathways (Yang et al. 2013). Also in pigs, a new phase II metabolite of T-2 toxin, HT-2-4-glucuronide, has been identified (Welsch and Humpf 2012). In addition, Wu et al. (2011a, b) have reported on the metabolism of T-2 in carp for the first time. The metabolizing enzymes play important roles in the biotransformation of the T-2 toxin. Furthermore, the enzymes are responsible for different metabolic fates in species. However, the metabolizing enzymes have rarely been reported before, which hinders our in-depth knowledge of the metabolism of trichothecenes. In recent years, as it has been reported that CYP3A22, CYP3A29, and CYP3A46 in pigs, as well as CYP1A5 and CYP3A37 in chickens, are critical metabolizing enzymes of T-2 toxin and HT-2 toxin (Ge et al. 2010; Wu et al. 2011a, b; Wang et al. 2011; Shang et al. 2013; Yuan et al. 2013). These studies cast a new light on metabolizing enzymes and toxicities of trichothecenes, which could lead to further studies dedicated to this topic.
Deoxynivalenol can be transformed to C12,13-deepoxy metabolite, DOM-1, in rodents, pigs, chickens, and ruminants. The conjugated products, DON-glucuronide, DON-sulfate, and DOM-l-glucuronide, have also been identified in ruminants (Wu et al. 2010). Some new metabolites of DON have been detected recently in rats and chicken (Wan et al. 2014). In earlier studies, liver is considered not to be able to metabolize DON to any phase I of phase II products (Cote et al. 1987). Recently, several studies have proven that DON can be metabolized to a variety of phase II metabolites by liver enzymes (Maul et al. 2012; Uhlig et al. 2013). Free DON and DON-GlcA are considered to be the biomarkers for humans exposed to DON. The metabolism of DON in humans is region-dependent (Turner et al. 2011; Warth et al. 2011, 2012; Gratz et al. 2013). Thus, it is necessary to provide a new summary of the metabolism of the most important trichothecenes T-2 toxin and DON in animals and humans.
In this review, we aim at discussing the toxic mechanism of trichothecenes from the vantage point of oxidative stress. Furthermore, we would like to find an explanation to the differences between the effects that have been ascribed to the oxidative stress caused by trichothecenes in different studies. Our main goal, however, consists in summarizing the most recent findings on the metabolism of T-2 toxin and DON as well as on their metabolizing enzymes. In the present review, we will mainly focus on the reports on the metabolism of T-2 toxin and DON that have been published in the recent years. Additionally, the conjugated metabolites of DON as well as the fate of “masked” DON in animals and humans will be addressed. We believe that this review will provide a comprehensive overview of the knowledge on the toxic mechanism and metabolism of trichothecenes. Moreover, it will cast some light on the metabolism and toxicological studies in trichothecenes.
Oxidative stress and trichothecenes
The toxic mechanism of trichothecenes was well discussed by Pestka (2007). Trichothecenes bind to ribosome and rapidly activate MAPKs via a mechanism known as the “ribotoxic stress response.” MAPKs are important transducers of downstream signaling events related to immune response and apoptosis. Currently, increasing studies have proven that oxidative stress is an important underlying cytotoxic mechanism of trichothecenes (Doi and Uetsuka 2011).
Oxidative stress induced by the generation of ROS is the mechanism of toxicity for a variety of chemicals (Stacey and Klaassen 1981; Sahu et al. 2010; Sinha et al. 2013). Oxidative stress may occur either due to the overproduction of ROS or due to a decrease in cellular antioxidant levels. Overproduction of ROS may induce cell oxidative injury, such as DNA damage, oxidation of proteins, and lipid peroxidation (Mates 2000; Chaudhari et al. 2009a). The primary antioxidant enzymes responsible for protection from oxidative stress are superoxidase dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) (Hou et al. 2013).
Several studies have shown that oxidative stress is involved in the toxicities of trichothecene mycotoxins (El Golli et al. 2006). Trichothecenes are able to generate free radicals, including ROS that induce lipid peroxidation, which leads to changes in membrane integrity, cellular redox signaling, and changes in the antioxidant status of the cells (Krishnaswamy et al. 2010). Elucidating trichothecene-induced oxidative stress is of importance for the understanding of the toxic mechanism of trichothecenes as well as for improving the strategies for controlling toxin contamination.
Trichothecenes are able to induce lipid peroxidation in different species. In rats, trichothecenes have induced free radical-mediated lipid peroxidation (Rizzo et al. 1994). In chickens, the concentration of hepatic malondialdehyde (MDA), a typical biomarker for lipid peroxidation, has been considerably increased (128 %) after 7 days of treatment with T-2 toxin (1.5 mg/kg BW) (Leal et al. 1999). Similarly, after treatment with T-2 toxin (1–6.25 mg/kg BW) for 24–48 h, an increase of lipid peroxidation in the liver as measured by MDA production is detected in mice (Vilà et al. 2002).
Pregnant rats were treated orally with 2 mg/kg of T-2 toxin. An increased expression of oxidative stress- and apoptosis-related genes was detected in the liver before the mice were killed after 12 h (Sehata et al. 2005). Decreased expression of lipid metabolism- and metabolizing enzyme-related genes were also detected. Their study also shows that MAPK pathway might be involved in apoptosis. In particular, c-jun gene seems to play an important role in the process of apoptosis. Thus, T-2 toxin induces oxidative stress, followed by the activation of the MAPK pathway, finally inducing apoptosis. Caspase-2 is essential for T-2 toxin-induced apoptosis, and apoptotic signals are mainly transmitted via caspase-8 and caspase-3 rather than through mitochondrial pathway (Huang et al. 2007).
Chaudhari et al. (2009a) have shown that oxidative stress is one of the mechanisms of T-2 toxin-mediated toxicity. The oxidative stress was determined after mice were treated intraperitoneally with T-2 toxin (5.61 or 11.22 mg/kg BW) for 24 h. A threefold increase in MDA levels was noticed at the time of death. Additionally, at the higher dose (11.22 mg/kg BW), the activity of both antioxidant enzyme glutathione-S-transferase (GST) and SOD was increased after 4 h. HSP-70 was induced by the oxidative stress in response to T-2 toxin. In another study (Chaudhari et al. 2009b), T-2 toxin (10 ng/mL) caused generation of ROS as early as 30 min and reached its peak after 4 h. Finally, it was followed by increased lipid peroxidation in human cervical cancer cells (HeLa cells). T-2 toxin-induced single-strand DNA damage was detectable at 2 and 4 h. This working group also showed that T-2 toxin could induce significant oxidative damage in mice brain in terms of depletion of hepatic glutathione, increased ROS and lipid peroxidation, alternations in activity of antioxidant enzymes as well as protein oxidation in a time-dependent manner (Chaudhary and Rao 2010). Oxidative stress is also one potential mechanism of the skin inflammation and cutaneous injury caused by T-2 toxin. Significant increase of ROS in mice skin could be observed as early as 3 h post-exposure of T-2 toxin (Agrawal et al. 2012). Finally, it is demonstrated that T-2 toxin-induced oxidative stress contributes to the pathological process of cartilage damage in Kashin-Beck disease (Chen et al. 2012).
The ROS-mediated mitochondrial pathway plays an important role in the process of T-2 toxin-induced apoptosis. Ovarian granulosa cells of rats were treated with T-2 toxin (1-100 nM) for 24 h, ROS accumulation was induced, and the reduction of mitochondrial transmembrane potential could be detectable (Wu et al. 2011a). The mitochondrial dysfunction by T-2 toxin in rat cardiomyocytes is largely associated with oxidative stress (Ngampongsa et al. 2013). ROS generation and GSH depletion, together with lipid peroxidation in differentiated murine embryonic stem cells, have confirmed that oxidative stress is an underlying mechanism of T-2 toxin cytotoxicity (Fang et al. 2012). In the studies of Wu et al. (2011a) as well as those of Fang et al. (2012), T-2 toxin-induced apoptosis was not completely blocked by the inhibitor, thus the authors have suggested that other pathways may also be involved in the regulation of apoptosis. Indeed, another caspase-independent apoptosis-inducing factor (AIF) pathway is also involved in T-2 toxin-induced apoptosis (Chaudhari et al. 2009b). The toxicity of T-2 toxin in chicken primary hepatocytes was investigated by proteomic analysis (Mu et al. 2013). The proportion of ROS-positive cells and intensity of the ROS signal in 0.05 μg/mL T-2 toxin-treated cells increased over time. The mitochondrial mass and cellular ATP levels were also increased. The induced oxidative stress and mitochondrial enhancement induced by T-2 toxin are very possibly caused by the changes in the expression of proteins related to cell oxidative stress and the mitochondrial proteome.
Contrary to the studies mentioned above, however, several studies have not been caused by the oxidative stress induced by T-2 toxin. Schuster et al. (1987) studied the effects of T-2 toxin on the lipid peroxidation in rats, but no increase of lipid peroxidation was observed. Therefore, they have suggested that lipid peroxidation does not play a major role in T-2 toxin toxicity. Chickens received feed, which contained T-2 toxin (10 mg/kg feed) or DON (10 mg/kg feed) for 17 days. Although DNA fragmentation was induced significantly in spleen leukocytes, no induction of the lipid peroxidation was observed (Frankic et al. 2006). Similarly, no oxidative stress was detectable when chickens were treated with T-2 toxin (0.5–13.5 mg/kg of feed) for 17 days (Rezar et al. 2007). Possibly, the different doses of the toxin are responsible for the different results, since a much higher dose of mycotoxins was used in the studies, in which the oxidative stress effect was registered.
Deoxynivalenol as the important type B trichothecenes induces oxidative stress. In the human intestinal cell line Caco-2 cells, DON (1–150 μM) significantly increased the production of MDA starting at the concentration of 10 μM (Kouadio et al. 2005). In U937 and HT-29 cells, DON induced a significant increase of ROS levels (49–65 %) (Costa et al. 2009; Krishnaswamy et al. 2010). The formation of free radicals and ROS are involved in the DNA damage. Zhang et al. (2009) used human hepatoma HepG2 cells to assess the role of oxidative stress in DON-induced DNA damage. A remarkable increase in ROS occurred when a concentration of DON 60 μM was used, whereas lower concentrations (15 or 30 μM) were not able to trigger a significant induction of ROS. DON increases the DNA migration in a dose-dependent manner. A notable increase in lipid peroxidation is also detectable at a relatively higher dose of DON (15 μM). The non-cytotoxic concentrations of DON had not lead to a higher oxidation rate in HepG2 cells (Sugiyama et al. 2012). In rat liver clone-9 cells, however, DON induced a significant concentration-dependent increase in oxidative stress at concentrations from 10 μg/mL onward. The mitochondrial function decreased with the increasing concentration of DON, but it was not statistically different from that of the control value (Sahu et al. 2008). Very recently, the human lymphocytes were cultured in different doses of DON during 6, 12 and 24 h (Yang et al. 2014). The damage to the membrane, the chromosomes, or the DNA was observed at all times of culture. The levels of lipid peroxidation and the 8-hydroxydeoxyguanosine and ROS were also increased.
Naturally contained mycotoxins are able to induce the oxidative stress in mice (Hou et al. 2013). Maize contaminated with mycotoxins (DON 3.1 mg/kg, zearalenone 0.729 mg/kg, aflatoxins 0.597 mg/kg) was incorporated into the mouse’s diet, and blood and tissue samples were collected to examine the oxidative stress-related indexes. The treatment increased the GPx activity and MDA level in the serum and liver. The activity of CAT and SOD was also markedly decreased. Moreover, the multiple mycotoxin-contaminated diet may cause more serious damage than the single or pure mycotoxin.
Similarly to T-2 toxin, several studies have shown that the oxidative damage is not the major contributor to DON toxicity. Bensassi et al. (2009) studied the cytotoxicity of DON on human colon carcinoma cells, but they observed neither the generation of ROS nor the induction of HSP-70. Thus, they have suggested that DON directly induces DNA lesions, but not via the release of ROS. Furthermore, this toxin seems to act as a direct genotoxic agent, which causes DNA fragmentation and leads to p53- and caspase-dependent apoptosis. Broilers were fed with a diet artificially contaminated with 10 mg of feed-grade DON/kg of diet (Awad et al. 2012). DNA damage in chicken lymphocytes was observed; however, the concentration of mitochondrial thiobarbituric acid substance (TBARS), which acts as an indicator of lipid peroxidation, was not different from that of the control group. A similar assumption has also been proposed that DNA damage is mostly induced by the direct action of DON or through different epigenetic mechanisms, such as the formation of DNA adducts rather than via the formation of free radicals. Sahu et al. (2010) assessed the hepatotoxicity of DON (0–25 μg/mL) in rat (Clone9 and MH1C1), mouse (NBL CL2) and human liver (WRL68 and HepG2) cells. They have not observed any oxidative stress in any of these cell lines. Similar result was also reported by Frankic et al. (2008), who have studied the effect of DON (4 mg/kg feed) and T-2 toxin (3 mg/kg feed) on lipid peroxidation, lymphocyte DNA fragmentation in weaned pigs. DON and T-2 toxin significantly increased the amount of DNA damage in pig lymphocytes. However, MDA excretion rate in plasma and 24-h urine as well as erythrocyte GPx levels were not changed significantly.
In summary, trichothecenes are able to induce oxidative stress in cells. Normally, trichothecenes damage the normal function of mitochondria and release the free radicals, including ROS, induce the lipid peroxidation, and change the antioxidant status of the cells, thereby reducing the activity of the antioxidant enzymes, such as GST, SOD and CAT in the body. DNA damage is an earlier event correlating with the generation of ROS and lipid peroxidaion. Some signaling pathways, including MAPK, are subsequently induced by oxidative stress, and caspase-mediated apoptosis pathways are activated. Some caspase-independent pathways, such as the AIF pathway, are also involved in cell apoptosis. However, there exists a discrepancy regarding the involvement of oxidative stress in trichothecene toxicity. The different doses that have been used in the different studies seem to be the reason for these contradictory results. The speculated mechanism of oxidative stress-mediated trichothecene toxicity in animals is presented in Fig. 2.

Speculated mechanism of oxidative stress-mediated toxicity of trichothecene in animals
Metabolism of T-2 toxin and DON
T-2 toxin
Metabolic pathways
The metabolism of T-2 toxin has already been studied since 1980s, and a variety of metabolites are identified over the last decades. The metabolism of T-2 toxin can notably occur in the liver and the digestive tract. The major concerned metabolic pathways of T-2 toxin are hydrolysis, hydroxylation, deepoxidation, and conjugation (Dohnal et al. 2008). The detailed information on the metabolic pathways in different species has already been summarized in earlier reviews (Dohnal et al. 2008; Wu et al. 2010; Li et al. 2011). In recent years, numerous studies on biotransformation of T-2 toxin in animals and humans have been carried out and have given us a new understanding of T-2 metabolism.
In order to get a global metabolic profile of T-2 toxin in food-producing animals, a comparison of hepatic in vitro metabolism of T-2 toxin in pigs, chicken, carp (grass carp and common carp), and rats was made (Wu et al. 2011b). No new metabolites were detectable, but some qualitative similarities and interesting differences across the species were observed. The metabolites including HT-2 toxin, NEO, 3′-OH-T-2, 3′-OH-HT-2, and T-2 triol were detected in these animals. Hydrolysis for the formation of HT-2 toxin was the major metabolic pathway in these land animals (pigs, chickens, and rats). Moreover, contrary to the three land animals, the relative content of HT-2 toxin was lower or even undetectable in the carp liver microsomes. However, a higher percentage of 3′-OH-T-2 could be detected. These results suggest that ester hydrolysis in a carp may not be as an important route as it is in land animals. Although the authors of this study have demonstrated a different major metabolic pathway of T-2 toxin in carp, the metabolic fate of T-2 toxin in more aquatic species needs to be established to confirm their conclusion. In order to reveal an important aspect of the bioavailability of T-2 toxin, Wu et al. (2012) studied the degradation and the metabolism of T-2 toxin in the pig intestine. A large variation in the intestinal degradation of T-2 toxin was visible for individual pigs. Moreover, HT-2 toxin was the only detectable metabolite. Therefore, it is highly plausible that the toxicity of T-2 toxin for pigs is caused by the combination of T-2 and HT-2 toxins.
Recently, the research group led by Prof. Dr. Humpf (Muenster University, Germany) has contributed a series of publications on the metabolism of T-2 toxin in human cells (2009–2013). A new metabolite of T-2 toxin, namely HT-2-4-glucuronide, was identified in pig liver microsomes as well as in pig urine (Welsch and Humpf 2012) (Fig. 3). In this study, glucuronidation of T-2 and HT-2 toxin in liver microsomes of rat, mouse, pig, and human was compared. HT-2-3-glucuronide accounted for half of the quantified metabolites in human liver microsomes. Thus, both HT-2 toxin glucuronide isomers should be taken into account when studying the metabolism of T-2 toxin.

Glucuronide metabolites when incubating T-2 toxin with pig liver microsomes in the presence of NAPDH (cited from Welsch and Humpf 2012)
After the incubation of T-2 toxin with human renal proximal tubule epithelial cells (RPTEC) and normal human lung fibroblasts (NHLF) in primary culture for 24 and 48 h, HT-2 toxin was detected as the major metabolite (Königs et al. 2009). Further metabolic activity could only be observed in RPTECs, which formed NEO as a second metabolite. Moreover, the cleavage of the acetyl group at the C-4 position does not affect the toxicity seriously, but once the C-8-isovaleryl group is lost, the cytotoxicity reduces greatly. In order to study the metabolism of T-2 toxin close to the human situation in vivo, the metabolism of T-2 toxin in a cell line derived from human colon carcinoma cells (HT-29) and RPTECs was investigated (Weidner et al. 2012). Besides HT-2 toxin and NEO, other metabolites, such as 3′-OH-T-2, 4-deacetylneosolaniol, T-2-glucuronide and HT-2-glucuronide, were also identified in RPTECs after an incubation of 48 h. Nevertheless, HT-2 toxin and NEO were still the major metabolites, while the others only accounted for less than approximately 1–5 % of the total amount of T-2 toxin metabolites. In HT-29 cells, besides these metabolites, T-2 triol was also detected, while HT-2 toxin represented the main metabolite. Importantly, the metabolic fate of T-2 toxin in human primary cells does not necessary lead to a detoxification (Weidner et al. 2012). Furthermore, both T-2 and HT-2 toxins are able to enter the brain via the blood–brain barrier to induce neurotoxic effects (Weidner et al. 2013a). The uptake and metabolism of T-2 toxin in normal human astrocytes in primary culture (NHA) was also studied by the same research group (Weidner et al. 2013b). They have also analyzed the uptake of T-2 toxin by human cell line HT-29 cells. Normal human astrocytes exhibited a fast uptake of T-2 toxin (within 15 min) and could transform T-2 toxin to HT-2 toxin rapidly. Therefore, it is very possible that the strong cytotoxic effects of T-2 toxin on NHA are most likely facilitated by the high cellular accumulation and fast uptake of T-2 toxin. However, these cells are unable to completely eliminate the absorbed toxin. Notably, this formation from T-2 to HT-2 toxin seems to occur within the cells since no degradation of T-2 toxin is visible when incubating T-2 toxin with cell-depleted culture medium. The glucuronic acid conjugate of T-2 toxin is not detectable in NHA but occurs in the media of HT-S9 cells. Therefore, the human cell line, but not human astrocytes, seems to possess the ability to form phase II conjugates.
Most new metabolites of T-2 toxin that have been identified recently are phase II metabolites. Actually, after more than 30 years of study, it is difficult to find new phase I T-2 toxin metabolites. However, a very recent study has reported some novel phase I metabolites of T-2 toxin (Yang et al. 2013). In this study, biological transformation of T-2 toxin in the in vitro (liver S9 fraction and microsomes) and in vivo system of Wistar rats was analyzed. A total of 19 metabolites were identified where 9 of them are novel. These 9 metabolites are the following: 15-deacetyl-T-2 (1), 3′-OH-15-deacetyl-T-2 (2), 3′,7-dihyroxy-T-2 (3), isomer of 3′,7-dihyroxy-T-2 (4), 7-OH-HT-2 (5), isomer of 7-OH-HT-2 (6), deepoxy-3′,7-dihyroxy-HT-2 (7), 9-OH-T-2 (8), and 3′,9-dihydroxy-T-2 (9) (Fig. 4). In the in vivo system, HT-2 toxin was the metabolite with the highest detection, followed by NEO. 15-acetyl-T-2 and 3′-OH-15-acetyl-T-2 were first detected in rats in vivo, but they were undetectable in the in vitro systems. One novel metabolic pathway of T-2 toxin, the hydroxylation at C-9 (9-OH-T-2 and 3′,9-dihydroxy-T-2), was found for the first time in both the in vivo and the in vitro systems of rats. Another new metabolic pathway, the hydroxylation at C-7 (3′,7-dihyroxy-T-2 and 7-OH-HT-2), was detectable only in vivo. Moreover, in this study, the relative amount of 3′,7-dihydroxy-HT-2 toxin was higher than HT-2 toxin. Additionally, hydroxylation is the major metabolic pathway in vivo, whereas hydrolysis is the main metabolic fate of T-2 in the liver. However, some issues from this study still need to be elucidated. (1) Do these new metabolites and new metabolic pathways also exist in other species, such as pigs, chickens, and fish? (2) Why does there exist such a big difference of T-2 metabolism between the in vivo and the in vitro system? (3) What are the potential toxic profiles of these new metabolites? (4) Although the authors have used HPLC–MS/MS with very high sensitivity and resolution to identify the new products, these products still need to be synthesized and confirmed by nuclear magnetic resonance (NMR).

Some novel metabolites of T-2 toxin in rats. (1): 15-deacetyl-T-2, (2): 3′-OH-15-deacetyl-T-2, (3): 3′,7-dihyroxy-T-2, (4): isomer of 3′,7-dihyroxy-T-2, (5): 7-OH-HT-2, (6): isomer of 7-OH-HT-2, (7): deepoxy-3′,7-dihyroxy-HT-2, (8): 9-OH-T-2, and (9): 3′,9-dihydroxy-T-2
In summary, the metabolic pathway of T-2 toxin seems to be highly similar in different land animals. However, the major metabolic pathways of T-2 toxin in aquatic species seem to be fundamentally different from those of land animals. In many human cell lines, HT-2 and NEO are the main metabolites, but some other products, such as 3′-OH-T-2, 4-deacetylneosolaniol, T-2-glucuronide, and HT-2-glucuronide, are also minor metabolites in human cells. The hydroxylation on C-7 and C-9 are two new metabolic pathways that have recently been found in rats. However, more studies are necessary to confirm these results.
Metabolizing enzymes
Currently, the study of T-2 metabolizing enzymes is taking two directions simultaneously: The first one consists of characterizing the involvement of metabolizing enzymes in the biotransformation of T-2 toxin in animals, whereas the second one consists of studying the toxic effect of T-2 toxin on the metabolizing enzymes. Specific inhibitors of the metabolizing enzymes are used, and the aimed enzymes are recombined in order to generate a direct incubation with T-2 toxin and to monitor the potential products. Therefore, we will summarize the findings on the metabolizing enzymes of T-2 toxin that were generated from these two directions.
In the 1980s, the study of the metabolism of T-2 toxin began to emerge and some researchers focused directly on the metabolic mechanism of T-2 toxin. However, several reports on the metabolizing enzymes of T-2 toxin were published at that time (Yoshizawa et al. 1984; Johnsen et al. 1986), and most studies still focused on the identification of new metabolites. Johnsen et al. (1986) have found out that T-2 toxin can be hydrolyzed rapidly to HT-2 toxin by rat liver microsomes. According to them, carboxylesterase (EC 3.1.1.1) plays an important role in hydrolyzing T-2 toxin to HT-2 toxin. Carboxylesterase activity from liver microsomes is separated into at least five different isoenzymes, but in this study, only the isoenzyme of pI 5.4 was able to catalyze T-2 toxin to HT-2 toxin. In another study (Johnsen et al. 1988), the hydrolysis of T-2 to HT-2 toxin in human white blood cells and T-2 to NEO in human red blood cells was all identified as carboxylesterases. Thus, the authors have suggested that the carboxylesterases present in the blood cells are of the same identity as liver carboxylesterases pI 5.4. An in vitro formation of 3′-OH-T-2 and 3′-OH-HT-2 from T-2 toxin by liver homogenates from mice and monkeys was observed (Yoshizawa et al. 1984), the formation of these hydroxyl products needs the presence of NADPH, implying that a cytochrome P-450 (CYP450) is catalyzing the hydroxylation at the C-3′ of T-2 and HT-2 toxins.
After the above-mentioned studies, the research on the metabolizing enzymes of T-2 toxin has almost been forgotten for nearly 30 years. Only recently, a variety of key T-2 toxin CYP450 metabolizing enzymes are reported. Based on the earlier investigations, it is only known that T-2 toxin can be converted to HT-2 toxin by CYP450 enzymes. However, it is still unclear which CYP subfamilies play the major main role. Ge et al. (2010) have incubated T-2 toxin with pig hepatocytes and found out that it significantly induced CYP3A22 expression. Recombinant pig CYP3A22 can convert T-2 and HT-2 toxin to 3′-OH-T-2 and 3′-OH-HT-2 toxins, respectively. Moreover, these metabolites are detected after the treatment of CYP3A-specific inhibitor. Thus, CYP3A22 is considered to be a key enzyme in the metabolism of T-2 and HT-2 toxin by 3′-hydroxylation of the C-8 isolaveryl group. The main function of CYP3A22 consists of facilitating the removal of T-2 or HT-2 toxin from the body by C-hydroxylation and excretion into the urine, without mediating the C-4 deacylation to T-2 toxin. Similarly, pig CYP3A29 can also convert T-2 and HT-2 toxin to their C-3′ hydroxyl products (Wu et al. 2011b). Moreover, CYP3A29 can also catalyze T-2 toxin to some amount of NEO. Thus, the cleavage of the C-8 ester group cannot be catalyzed only by carboxylesterase but also by CYP450 enzymes, at least by pig CYP3A29.
Wang et al. (2011) have studied the biotransformation of T-2 toxin by pig CYP 3A46 as well as the hydrolytic ability of carboxylesterase 1C5 (CES1C5) and the deepoxy ability of epoxide hydrolase (EPHX1) in T-2 toxin-treated primary hepatocytes from porcines. CYP3A46 is able to catalyze T-2 toxin to 3′-OH-T-2, which is further proved by purified CYP3A46 protein. However, recombinant porcine CES1C5 and EPHX1 have not enhanced hydrolysis and deepoxidation of T-2 toxin, which implies that CES1C5 and EPHX1 are not the dominant enzymes in the canalization of these reactions. The deepoxylation of T-2 toxin is a very important toxic-reducing pathway. Most studies demonstrate that the deepoxylation of T-2 toxin happens in the intestine but not in the liver (Wu et al. 2010). Thus, we may suspect whether the deepoxylation of T-2 toxin is catalyzed only by the bacteria in the intestine while not by the metabolizing enzymes. If this assumption is correct, the detailed mechanism still needs to be unraveled. It has also been suspected that CYP2C42 is the potential metabolizing enzyme of T-2 toxin in pigs (Wang et al. 2011).
T-2 toxin significantly induces CYP1A4 and CYP1A5 activity in chicken embryonic hepatocytes. However, only CYP1A5 can metabolize T-2 toxin to 3′-OH-T-2. The in vitro recombinant CYP1A5 also confirms that CYP1A5 is able to convert T-2 toxin to 3′-OH-T-2 (Shang et al. 2013). In addition to CYP1A5, CYP3A37 plays an important role in the hydroxylation of T-2 toxin (Yuan et al. 2013), which implies that, similar to pigs, multiple CYP450 enzymes are involved in the metabolism of T-2 toxin in chickens. Weidner et al. (2013a, b) have demonstrated that the metabolism of T-2 toxin in human astrocytes takes place after its uptake into the cell interior and is not conducted by secreted enzymes outside the cells.
Thus, CYP3A22, CYP3A29, and CYP3A46 in pigs, as well as CYP1A5 and CYP3A37 in chickens are able to catalyze T-2 toxin and HT-2 toxin to form the C-3′-OH products. Similar to carboxylesterase, pig CYP3A29 has the ability to convert T-2 toxin to NEO through hydrolysis. The enzymatic mechanism of deepoxidation of T-2 toxin is not yet known and needs to be further investigated. It is postulated that other underlying CYP450 subfamilies exist, which are able to catalyze T-2 toxin metabolism in these animals. Furthermore, since some researchers have found out that the metabolism of T-2 toxin in carp showed several different metabolic profiles (Wu et al. 2011b), the metabolizing enzymes of T-2 toxin in aquatic species have to be investigated in the future.
Effects on metabolizing enzymes
The metabolizing enzymes are crucial for maintaining body health. Some of these enzymes are biomarkers of the damages in vivo. For example, GST and GPx are important antioxidant enzymes. A change in the levels of these enzymes is a very good indicator for the occurrence of oxidative stress. The observation of the changes of the metabolizing enzymes that have been induced by trichothecenes is also a fast method for screening typical key metabolizing enzymes. On the other hand, understanding the effects of T-2 toxin on enzymes in vivo is very important for establishing the prevention of the toxins. Moreover, it will help us to understand the pharmacokinetics and withdrawal time of drugs that are substrates of these metabolizing enzymes (Goossens et al. 2013).
Suneja et al. (1984) have studied the effects of T-2 toxin on intestinal brush border enzymes (sucrase, lactase, Na+-K+-ATPase, and alkaline phosphatase) and lysosomal enzymes in rats which received T-2 toxin orally. The activity of brush border sucrase, lactase, and Na+-K+-ATPase was significantly decreased, whereas the alkaline phosphatase activity was not significantly altered. T-2 toxin did not significantly alter the release of the lysosomal enzymes, phosphatase, and acid ribonuclease. In addition to the intestine, T-2 toxin also induces changes in rat liver and serum enzymes (Suneja et al. 1987). At daily doses (0.75 mg/kg) and even with a single dose (2 mg/kg), T-2 toxin was able to significantly reduce activities of hepatic glutamate pyruvate transaminase (GPT) and alkaline and acid phosphatases. The levels of GPT increased, whereas the levels of acid and alkaline phosphatases decreased. This study indicates that the rat liver is damaged through the oral intake of feed that had been contaminated with T-2 toxin.
After 7 days of treatment of T-2 toxin to chickens, hepatic MDA concentration was highly increased (128 %). A significant consumption of endogenous antioxidant GSH as well as a marked increase in hepatic enzyme activities of GST (312 %), γ-glutamyltransferase (GGT) (187 %), and GPx (324 %) was induced. Interestingly, the addition of lycopene has led to a decline of some parameters. This finding suggests that, as an antioxidant agent, lycopene would be able to protect the cellular level of GSH (Leal et al. 1999). After having been fed with T-2 toxin (2.5 and 2.8 mg/kg body weight) for 24 h, the liver tissue enzymes, aspartate aminotransferase (ASAT), alanine aminotransferase (ALAT), and γ-glutamyl transferase (γ-GT) activities, highly increased in rats, whereas the protein level decreased importantly (Atroshi et al. 2000).
Pigs were offered diets that have been contaminated with 540, 1324, or 2,102 μg T-2 toxin/kg feed for 28 days. P450 1A-related activities (ethoxyresorufin O-deethylation and benzo-(a)-pyrene hydroxylation) were reduced for all pigs given T-2 toxin (Meissonnier et al. 2008). The exposure of T-2 toxin also led to a reduction of certain N-demethylase activities. The reduction of monooxygenase activities suggests possible deleterious interactions between T-2 toxin and these enzymes. Similarly, after feeding New Zealand white rabbits with T-2 toxin (0.25 mg/kg/day) for 5 days, the expression of P450 1A subfamilies (1A1 and 1A2) was also decreased. Additionally, CYP 2A1 and 2B4 have also decreased (Guerre et al. 2000). However, this has not been the case of P450 2C3 and 3A6. In another study, the effects of T-2 toxin on hepatic and intestinal drug-metabolizing enzymes (CYP450) in poultry were investigated by Osselaere et al. (2013). In their study, T-2 toxin downregulated CYP3A37, CYP1A4, CYP1A5 mRNA expression in the liver; however, the gene expression of these enzymes was not changed in the intestine. Moreover, CYP3A enzymatic activity was upregulated in the liver and ileum after 3 weeks of exposure to T-2 toxin at 752 μg/kg feed.
Different to the above-mentioned studies, in human intestinal epithelial cell line Caco-2, T-2 toxin can highly upregulate the P450 1A1 mRNA expression and lead to MAPK/p38-dependent interleukin-8 secretion (Kruber et al. 2011). After having been fed T-2 toxin (903 μg/kg/feed) for 14 days, a substantial inhibition of the CYP3A activity has been observed in the livers of pigs. The differences concerning the investigated species and the doses of T-2 toxin are possible reasons that have been observed in the enzymatic activates.
Furthermore, some in vitro experiments have been carried out to study the effect of T-2 toxin on metabolizing enzymes in hepatocytes. In pig hepatocytes, T-2 toxin (0.05 μg/mL) could upregulate the mRNA levels of CYP3A46, CYP3A39, CYP2C42, carboxylesterase CES1C4, and the microsomal EPHX1, but decreased CYP2D25 and CYP51 gene levels (Wang et al. 2011). In pig hepatocytes, T-2 toxin (0.1 μg/mL) significantly upregulated CYP3A22 gene expression, whereas other CYP450 genes (1A2, 2E1, 3A29, and 2C49) were weak or even undetectable (CYP2D25) (Ge et al. 2010). With the same dose (0.1 μg/mL T-2 toxin), CYP1A4 and CYP1A5 were upregulated 132-fold and 47-fold, respectively, in chicken embryonic hepatocytes. Furthermore, an increased gene expression of other CYP450 enzymes, such as CYP2C18 (5.3-fold), CYP2H1 (8.1-fold), CYP3A37 (5.7-fold), was also observed.
In summary, T-2 toxin down- or upregulates different CYP450 enzymes in different species. Differences among the analyzed species or between the administered doses of T-2 toxin may be the reasons for the differences among the regulated enzymatic profiles. Nevertheless, we can obtain the aimed enzymes from the information of the significant up- or downregulations and facilitate the fast screening of key metabolizing enzymes. The enzymatic catalyzing mechanism of the biotransformation of T-2 toxin and the effects of T-2 toxin on the enzymes are summarized in Fig. 5.

Enzymatic catalyzing mechanism of T-2 toxin in animals
Deoxynivalenol (DON)
Metabolic pathways
The C12,13-deepoxy product DOM-1 is a well-known metabolite of DON in different species (rodents, pigs, chicken, and ruminants). Since the deepoxy metabolism of DON has already been thoroughly reviewed (Wu et al. 2010), this pathway will not be discussed here. Recently, more conjugated metabolites of DON have been identified in animals and humans. The phase II metabolites have been considered to be the biomarkers of DON detection in humans.
Since DON is rapidly metabolized to its glucuronide metabolites, free DON combined with DON-glucuronide (DON-GlcA) is proposed as a suitable biomarker for the effective assessment of DON exposure in humans. Meky et al. (2003) have investigated the metabolism of DON in rats as a basis to establish a methodology for a candidate biomarker of human exposure to DON. In total, 37 % of the administered DON was excreted via urine and DON-GlcA was proposed as the major urinary metabolite. Based on the methods established in rat urine, they have demonstrated that the inhabitants in Linxian, China, an area with a potentially high DON exposure, are converting DON to DON-GlcA and excrete it through urine. However, it is never elucidated so far, which site of the DON structure is conjugated with the glucuronide. A first exposure biomarker survey for DON was established in a French population (Turner et al. 2010). The deepoxy metabolite DOM-1 was observed in the urine from 21/26 farm workers. In addition, human fecal microbiota could metabolize DON to DOM-1, which were detected in the urine from the same volunteers (Gratz et al. 2013). Interestingly, the formation of DON metabolites in humans is region-dependent: while DOM-1 is not detectable in British adults, DON-GlcA seems to be the major product of the inhabitants of the UK (Turner et al. 2011). In Austrian adults, DON-15-GlcA is identified as the major DON metabolite in urine and the ratio of DON-15-GlcA/DON-3-GlcA has been 3/1 (Warth et al. 2012).
In the previous studies, β-glucuronidase was usually utilized to hydrolyze the conjugated DON. Therefore, it is not possible so far to determine the exact ratio of DON/DON-GlcA in a sample in a one-step procedure. Thus, the standard of DON-GlcA was synthesized by Warth et al. (2012) and applied to the direct quantification of DON-GlcA in human urine as a biomarker of exposure of DON. The concentration of DON-3-GlcA was >30 μg/L, whereas DON was undetectable in the urine. This observation indicates that DON is quickly converted to the glucuronide-conjugated product in humans in order to reduce the toxicity, since DON-GlcA is proven to be less toxic than DON (Wu et al. 2007).
An in vivo study has provided detailed information on DON metabolism in humans over a period of 8 days (Warth et al. 2013). The average DON excretion and glucuronidation amounted to 68 and 76 %, respectively. In addition to DON-3-GlcA, another conjugated product, DON-15-GlcA has also been firstly identified as the main conjugated product. Meanwhile, DON-7-GlcA was assumed to be a new conjugate (Maul et al. 2012).
When DON or DOM-I was incubated with uridine 5′-diphospho-glucuronic acid (UDPGA) and liver microsomes from rats or pigs, no glucuronides were detectable with the TLC plates. Moreover, no glucuronide conjugates were observed in rat and pig urine (Cote et al. 1987). Thus, the liver was considered not to catalyze the metabolism of DON. However, a very recent study has provided a new insight: The liver is able to metabolize DON (Maul et al. 2012). The metabolism of DON was investigated using liver microsomes from humans, rats, bovines, porcine, chicken, carp, and trouts (Maul et al. 2012). DON-3-GlcA was detectable in human and animal liver microsomes. The 3-conjugation ability is as follows: bovine > rat > carp > trout > human. Only human liver microsomes could catalyze DON to DON-15-GlcA. A third conjugate product detected in rat, carp, and human liver was assumed to be DON-7-GlcA. DON-3-GlcA and DON-15-GlcA were also identified in rat and human microsomes in another study (Uhlig et al. 2013). Additionally, another conjugated product was identified as DON-8-GlcA. In the earlier studies, the sensitivity of the methods and equipments limited the detection of new metabolites. In the study of Cote et al. (1987), TCL and HPLC are the major analytical equipments for the identification of metabolites, whereas in the studies of Maul et al. (2012) and Uhlig et al. (2013), LC–MS/MS with high sensitivity and resolution are applied. This is possibly the potential reason why Cote et al. (1987) have not detected any conjugates.
The study of the metabolism of DON has been performed very rarely in chicken. Very recently, Wan et al. (2014) have studied the in vivo metabolism of [3H]-DON in chicken and rats. Metabolites were characterized by comparing their LC-γ.ARC retention times and mass spectra to authentic synthetic standards. Five metabolites were identified: DON-10-sulfonate, DON-3-GlcA, DON-3-sulfate, DOM-1, and DOM-1-10-sulfonate. DON-10-sulfonate and DOM-1-10-sulfonate were only detectable in rats, whereas DON-3-sulfate was merely formed in chickens. Besides in chicken, DON-3-sulfate was already reported in sheep urine (Prelusky et al. 1987). DON-sulfonate is prepared in vitro to assess the potential physicochemical characteristics in intestines. Schwartz et al. (2013) prepared three different DON-sulfonates termed DON-sulfonate 1, 2, and 3. DON-sulfonate 1 and 2 are stable in a pH ranging 2–8 at 37 °C, which suggests that the two compounds are stable during digestion and in plasma of pigs. DON-sulfonate 3 is likely to survive in the acidic environment of the stomach, but might partly degrade to DON-sulfonate 1 and 2 in the neutral to slightly alkaline ambience of small intestine and plasma.
Sulfonate conjugation of DON is also a toxic-reducing pathway in animals. DON-sulfonate lacks the emetic activity characteristic for DON when given to pigs (Young et al. 1987). DON-sulfonate shows no toxic effect on the viability of porcine peripheral blood mononuclear cells (PBMC) and the intestinal porcine epithelial cell lines IPEC-1 and IPEC-J2 (Dänicke et al. 2010). Similarly, DON–sulfate conjugate is a detoxification pathway (Wan et al. 2014).
In summary, free DON and DON-GlcA are considered to be the biomarker for humans who have been exposed to DON. Liver and intestine are both able to metabolize DON. Conjugation of DON to glucuronic acid and elimination via the urine is the major metabolic pathway. Other conjugated metabolites, i.e., DON-sulfate, DON-sulfonate, DON-8-GlcA, and DOM-1-sulfonate, are also identified; however, some metabolites, including DON-7-GlcA, are just tentatively identified and still need to be confirmed in the future study. All these conjugates are considered to be important toxic-reducing pathways. The metabolism of DON in humans is region-dependent, and more epidemiological study of DON, especially in Asian and American countries, has to be carried out.
Metabolic fate of masked mycotoxin DON
During the last decades, the so-called masked mycotoxins, plant metabolites of the parent mycotoxins, have caused increasing concern. Besides DON and DON-3-β-d-glucoside (DON-3-Glc), other conjugates, such as DON-glutathione (GSH), DON-S-cysteine, and DON-S-cysteinyl-glycine, have been detected in plants (Kluger et al. 2013). These masked mycotoxins have a very different chemical behavior; thus, they are often not detected during routine analyses. Moreover, these forms could be hydrolyzed to their precursors in the digestive tracts of animals or exert toxic effects comparable to those imputable to free mycotoxins. Therefore, understanding the metabolic fate of the masked DON in animals is of the most importance for animal health and food safety.
In order to mimic the different stages of the digestion of DON in humans and animals, in vitro model systems were used to investigate the stability of the masked toxin DON-3-Glc to acidic conditions, hydrolytic enzymes, and to intestinal bacteria (Berthiller et al. 2011). DON-3-Glc is not able to be degraded in the acidic conditions as well as by digestive enzymes in vitro, implying that DON-3-Glc cannot be hydrolyzed into DON in the stomach and the small intestine of mammals. However, several lactic acid bacteria including Enterococcus durans, Enterococcus mundtii, and Lactobacillus plantarum show a high capability to hydrolyze DON-3-Glc. Warth et al. (2013) studied the in vivo metabolism of 3-actyl-DON and DON-3-Glc in humans. After the administration of a low dose of these masked mycotoxins (DON-3-Glc: 7 μg/day, 3-actyl-DON: 20 μg/day) to an individual for 8 days, these masked forms of DON were not detected in urine, indicting that they were hydrolyzed to free DON in the body.
Hydrolysis of DON-3-Glc is a common feature of human fecal microbiota. The fecal microbiota from human could release DON from DON-3-Glc very rapidly and efficiently (Gratz et al. 2013). The rapid degradation of the masked mycotoxin suggests that the human micriobiota can completely hydrolyze DON-3-Glc within normal residence time in the human colon. The human colonic epithelium will be exposed to significant levels of free DON.
In rats, DON-3-Glc has a low bioavailability and is hydrolyzed to DON during digestion. Then, it is partially converted to DOM-1 and DON-GlcA prior to excretion. After the administration of DON-3-Glc (3.1 mg/kg BW) for 15 days, rat urine and feces were collected for 48 h. Only 3.7 ± 0.7 % of the given dose was found in the urine as DON-3-Glc. In feces, the majority of administered DON-3-Glc was recovered as DON and DOM-1 (Nagl et al. 2012). Therefore, DON-3-Glc present in food and feed seems to have significantly lower toxicity than DON.
In summary, DON-3-Glc can be hydrolyzed to free DON in animals’ body. The exposure of human to masked mycotoxins should be taken into account in risk assessment, since their toxic precursors can be released after hydrolysis in animals and present an additional potential risk. The conjugated metabolism of DON in different species and its effect on the metabolizing enzymes is presented in Fig. 6.

Proposed phase II biotransformation of DON in animals and humans
Metabolizing enzymes
Few reports exist on the metabolizing enzymes of DON. To date, it is still unclear which enzymes are catalyzing the C12,13-deepoxy pathway. Because no phase I metabolism of DON has been detected in the liver (Cote et al. 1987; Maul et al. 2012), CYP450 enzymes are not involved in the metabolism of DON, albeit a bacterial CYP450 isolated from lake water has been able to catalyze DON to 16-OH-DON (Ito et al. 2013). The precise enzymatic mechanism of sulfonation is not yet clear. One possibility is the generation from the DON-GSH adducts, another metabolic pathway may result from the cooperation of the liver or intestinal cells with the microorganism (Wan et al. 2014).
In vertebrates, the glucuronidation of small lipophilic agents is catalyzed by the endoplasmic reticulum UDP-glucuronosyltransferases (UGTs). The β-d-glucuronides can be formed through hydroxyl, carboxyl, sulfuryl, carbonyl, and amino linkages (Tukey and Strassburg 2000). As mentioned above, DON-GlcA is found mostly in liver (microsomes) and urine. Glucuronide conjugation of DON was found in pig serum samples after oral exposure (Goyarts and Dänicke 2006), but not after intravenous application, implying that DON is very possibly conjugated in the intestine before absorption. Besides in liver, UGTs are also widespread in intestine, kidney, and other tissues (Tukey and Strassburg 2000). When rats were fed with DON, the UGT levels were twice higher in the liver (Obol’skiĭ et al. 1998). Thus, it is highly possible that UGTs play an important role in the formation of DON-GlcA, but the exact subfamily of the UGTs has yet to be elucidated.
Effects on metabolizing enzymes
There exist only very few data on the effect of DON on the metabolizing enzymes. When rats were treated with DON for 90 days, the activities of CYP450 enzymes and GST remained unchanged (Morrissey and Norred 1985). Gouze et al. (2006) have studied the effect of the low dose of DON (0.014–1.774 mg/kg BW) on liver xenobiotic metabolizing enzymes in mice. After an exposure of 28 days, the activities of two liver microsomal mixed-function oxidase ethoxyresorufin O-deethylase (EROD) and methoxyresorufin O-demethylase (MROD) were unaffected, whereas pentoxyresorufin O-depentylase (PROD) activity was increased (by 38–53 %). Protein level of CYP2B, GSTα and GSTπ was increased, whereas CYP1A protein level was unchanged. Additionally, DON is considered to be a putative substrate for GST. Rats were fed with a diet supplemented with DON (3,500 ± 5 % μg/kg) for 28 days. The activity of GST was not affected by DON. Although the activities of alanine transaminase (ALT) and aspartate transaminase (AST) were relatively increased, the AST/ALT, known as the index of liver damage, was normal (Sobrova et al. 2012). More research on the toxic effects of DON on the metabolizing enzymes will be necessary in the future.
Conclusion
Oxidative stress is an important underlying toxic mechanism of trichothecenes. Trichothecenes generate ROS that induce lipid peroxidation which in turn leads to changes in membrane integrity, cellular redox signaling and the antioxidant status of the cells. DNA damage correlates with but precedes the generation of ROS and lipid peroxidaion. The MAPK signaling pathway is possibly induced after the occurrence of oxidative stress through the caspase pathway to induce cell apoptosis. The caspase-independent AIF pathway is also involved in T-2 toxin-induced apoptosis. The mitochondrial dysfunction by T-2 toxin is largely associated with oxidative stress.
However, it is demonstrated that the oxidative damage is not the major contributor to trichothecene toxicity, especially in the studies of DON. The different results are possibly due to the different doses of toxin that have been used in the different studies. But it should be investigated whether some other reasons are involved in the different results. Furthermore, the relationship between oxidative stress and several important signaling pathways is still unclear, although some studies have shown that MAPK is induced by the oxidative stress in response to T-2 toxin, but very few studies on the link between oxidative stress and MAPK or Janus kinase/signal transducers and activators of transcription (JAK/STAT) singling pathways have become available. Therefore, more work on the oxidative stress induced by trichothecenes is necessary.
As for the metabolism of T-2 toxin in animals and humans, hydroxylation on C-7 and C-9 are identified as two novel metabolic pathways in rats. HT-2 toxin is the major metabolite of T-2 toxin in animals and humans. The toxicity of T-2 toxin for animals is possibly caused by the combination of T-2 and HT-2 toxins. In addition, some new conjugates including HT-2-4-glucuronide are also identified. Metabolism of T-2 toxin in aquatic species is possibly not similar to that of land animals, but more work is needed to confirm this assumption.
In comparison with T-2 toxin, relatively few metabolites of DON are identified so far. Contrary to the structure of T-2 toxin, there is almost no subsidence in the structure of DON, which may be the one explanation for the limited detection of metabolites. DON-GlcA is proposed as a suitable biomarker for an effective assessment of the exposure to DON by humans. Furthermore, DON-15-GlcA is the major conjugated product in humans. DON-sulfate, DON-sulfonate, DON-8-GlcA, and DOM-1-sulfonate are also identified as metabolites. Masked mycotoxins could be hydrolyzed to their precursors in animals and exert toxic effects. Nevertheless, some problems still exist in the metabolism of DON: DON-7-GlcA, a potential metabolite, has just been tentatively identified and still needs to be confirmed in future studies. The formation of DON metabolites in humans is region-dependent. Therefore, more epidemiological studies of DON have to be carried out in the future.
The investigation of the metabolizing enzyme of trichothecenes is crucial for elucidating the mechanism of the biotransformation of these toxins. Currently, we already know that CYP3A22, CYP3A29, and CYP3A46 in pigs, as well as CYP1A5 and CYP3A37, in chicken are able to catalyze T-2 and HT-2 toxins to form the C-3′-OH products. Similarly, together with carboxylesterase, pig CYP3A29 also possesses the ability to hydrolyze and to convert T-2 toxin to NEO. More CYP450 enzymes seem to be involved in the biotransformation of T-2 toxin. Therefore, more studies need to be carried out in the future, especially to find the potential important metabolizing enzymes of trichothecenes in the food-producing animals. Several reports have shown that important differences exist between the individual species concerning the metabolism of T-2 toxin, but the underlying mechanism that is responsible for these differences is not yet clear. It is highly possible that some important metabolizing enzymes are different among individual animals and lead to the different metabolic profiles of T-2 toxin in different species. However, almost no data are available to support this assumption. Therefore, the elucidation of the potential metabolic mechanism of trichothecens, together with the differences among the individual species, is an important point that has to be worked on in the future.
Contrary to T-2 toxin, CYP450 enzymes are not involved in the metabolism of DON. The enzymatic catalyzing mechanism of the C12,13-deepoxydation of DON has not yet been found. It seems that this process occurs in the intestines and is carried out by bacteria. UGTs are involved in the formation of DON-GlcA, but which subfamily of UGTs is exactly involved in this process should be elucidated. Similar to T-2 toxin, the metabolism of DON in different species has to be observed, but the precise mechanism which is responsible for these differences should also be elucidated. The metabolizing enzymes may be the key player in this process.
Abbreviations
- ALAT:
-
Alanine aminotransferase
- ALT:
-
Alanine transaminase
- ASAT:
-
Aspartate aminotransferase
- AST:
-
Aspartate transaminase
- ATA:
-
Alimentary toxic aleukia
- BBB:
-
Blood–brain barrier
- CAT:
-
Catalase
- CES1C5:
-
Carboxylesterase 1C5
- CYP450:
-
Cytochrome P-450
- DOM-1:
-
C12,13-deepoxy-DON
- DON:
-
Deoxynivalenol
- DON-3-Glc:
-
DON-3-O-glucoside
- DON-GlcA:
-
DON-glucuronide
- EPHX1:
-
Epoxide hydrolase
- EROD:
-
Ethoxyresorufin O-deethylase
- GGT:
-
γ-Glutamyltransferase
- GPT:
-
Glutamate pyruvate transaminase
- GPx:
-
Glutathione peroxidase
- GSH:
-
Glutathione
- HSP-70:
-
Heat-shock protein-70
- MAPK:
-
Mitogen-activated protein kinases
- MDA:
-
Malondialdehyde
- MROD:
-
Methoxyresorufin O-demethylase
- NEO:
-
Neosolaniol
- NHA:
-
Normal human astrocytes
- NHLF:
-
Normal human lung fibroblasts
- NMR:
-
Nuclear magnetic resonance
- PBMC:
-
Peripheral blood mononuclear cells
- PROD:
-
Pentoxyresorufin O-depentylase
- ROS:
-
Reactive oxygen species
- RPTEC:
-
Renal proximal tubule epithelial cells
- SOD:
-
Superoxidae dismutase
- TBARS:
-
Thiobarbituric acid substance
- TDI:
-
Tolerable daily intake
- UDPGA:
-
Uridine 5′-diphospho-glucuronic acid
- UGTs:
-
UDP-glucuronosyltransferases
- γ-GT:
-
γ-Glutamyl transferase
References
Agrawal M, Yadav P, Lomash V, Bhaskar ASB, Lakshmana Rao PV (2012) T-2 toxin induced skin inflammation and cutaneous injury in mice. Toxicology 302:255–265
Atroshi F, Rizzo A, Sankari S, Biese I, Westermarck T, Veijalainen P (2000) Liver enzyme actives of rats exposed to Ochratoxin A and T-2 toxin with antioxidants. Bull Environ Contam Toxicol 64:586–592
Awad WA, Ghareeb K, Dadak A, Gille L, Staniek K, Hess M, Böhm J (2012) Genotoxic effects of deoxynivalenol in broiler chickens fed low-protein feeds. Poult Sci 91(3):550–555
Bennett JW, Klich M (2003) Mycotoxins. Clin Microbiol Rev 16(3):497–516
Bensassi F, El Golli-Bennour E, Abid-Essefi S, Bouaziz C, Hajlaoui MR, Bacha H (2009) Pathway of deoxynivalenol-induced apoptosis in human colon carcinoma cells. Toxicology 264(1–2):104–109
Berthiller F, Dall’Asta C, Schumacher R, Lemmens M, Adam G, Krska R (2005) Masked mycotoxins: determination of a deoxynivalenol glucoside in artificially and naturally contaminated wheat by liquid chromatography-tandem mass spectrometry. J Agric Food Chem 53:3421–3425
Berthiller F, Krska R, Domig KJ, Kneifel W, Juge N, Schuhmacher R, Adam G (2011) Hydrolytic fate of deoxynivalenol-3-glucoside during digestion. Toxicol Lett 206:264–267
Berthiller F, Crews C, Dall’Asta C, Saeger SD, Haesaert G, Karlovsky P, Oswald IP, Seefelder W, Speijers G, Stroka J (2013) Masked mycotoxins: a review. Mol Nutr Food Res 57(1):165–186
Beyer M, Ferse I, Mulac D, Würthwein EU, Humpf HU (2009) Structural elucidation of T-2 toxin thermal degradation products and investigations toward their occurrence in retail food. J Agric Food Chem 57(5):1867–1875
Chaudhari M, Jayaraj R, Bhaskar ASB, Lakshmana Rao PV (2009a) Oxidative stress induction by T-2 toxin cause DNA damage and triggers apoptosis via caspase pathway in human cervical cancer cells. Toxicology 262:153–161
Chaudhari M, Jayaraj R, Santhosh SR, Lakshmana Rao PV (2009b) Oxidative damage and gene expression profile of antioxidant enzymes after T-2 toxin exposure in mice. J Biochem Mol Toxicol 23(3):212–221
Chaudhary M, Rao PV (2010) Brain oxidative stress after dermal and subcutaneous exposure of T-2 toxin in mice. Food Chem Toxicol 48:3436–3442
Chen JH, Xue S, Li S, Wang ZL, Yang H, Wang W, Song D, Zhou X, Chen C (2012) Oxidant damage in Kashin-Beck disease and a rat Kashin-Beck disease model by employing T-2 toxin treatment under selenium deficient conditions. J Orthop Res 30(8):1229–1237
Commission European (2006) Commission regulation (EC) No1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Off J Eur Union L364:5–24
Costa S, Schwaiger S, Cervellati R, Stuppner H, Speroni E, Guerra MC (2009) In vitro evaluation of the chemoprotective action mechanisms of leontopodic acid against aflatoxin B1 and deoxynivalenol-induced cell damage. J Appl Toxicol 29(1):7–14
Cote LM, Buck W, Jeffery E (1987) Lack of hepatic microsomal metabolism of deoxynivalenol and its metabolites DOM-1. Food Chem Toxicol 25:291–295
Dänicke S, Hegewald AK, Kahlert S, Kluess J, Rothkötter HJ, Breves G, Döll S (2010) Studies on the toxicity of deoxynivalenol (DON), sodium metabisulfite, DON-sulfonate (DONS) and de-epoxy-DON for porcine peripheral blood mononuclear cells and the Intestinal Porcine Epithelial Cell lines IPEC-1 and IPEC-J2, and on effects of DON and DONS on piglets. Food Chem Toxicol 48(8–9):2154–2162
Dohnal V, Jezkova A, Jun D, Kuca K (2008) Metabolic pathways of T-2 toxin. Curr Drug Metabol 9:77–82
Doi K, Uetsuka K (2011) Mechanisms of mycotoxin-induced neurotoxicity through oxidative stress-associated pathways. Int J Mol Sci 12:5213–5237
El Golli E, Hassen W, Bouslimi A, Bouaziz C, Ladjimi MM, Bacha H (2006) Induction of Hsp 70 in Vero cells in response to mycotoxins cytoprotection by sub-lethal heat shock and by Vitamin E. Toxicol Lett 166(2):122–130
Fang H, Wu Y, Guo J, Rong J, Ma L, Zhao Z, Zuo D, Peng S (2012) T-2 toxin induces apoptosis in differentiated murine embryonic stem cells through reactive oxygen species-mediated mitochondrial pathways. Apoptosis 17:895–907
Frankic T, Pajk T, Rezar V, Levart A, Salobir J (2006) The role of dietary nucleotides in reduction of DNA damage induced by T-2 toxin and deoxynivalenol in chicken leukocytes. Food Chem Toxicol 44(11):1838–1844
Frankic T, Salobir J, Rezar V (2008) The effect of vitamin E supplementation on reduction of lymphocyte DNA damage induced by T-2 toxin and deoxynivalenol in weaned pigs. Anim Feed Sci Tech 141:274–286
Galaverna G, Dall’asta C, Mangia M, Dossena A, Marchelli R (2009) Masked mycotoxins: an emerging issue for food safety. Czech J Food Sci 27:S89–S92
Ge X, Wang J, Liu J, Jiang J, Lin H, Wu J, Quyang M, Tang X, Zheng M, Liao M, Deng Y (2010) The catalytic activity of cytochrome P450 3A22 is crucial for the metabolism of T-2 toxin in procine reservoirs. Catal Comun 12:71–75
Goossens J, De Bock L, Osselaere A, Verbrugghe E, Devreese M, Boussery K, Van Bocxlaer J, De Backer P, Croubels S (2013) The mycotoxin T-2 inhibits hepatic cytochrome P4503A activity in pigs. Food Chem Toxicol 57:54–56
Gouze ME, Laffitte J, Rouimi P, Loiseau N, Oswald IP, Galtier P (2006) Effect of various doses of deoxynivalenol on liver xenobiotic metabolizing enzymes in mice. Food Chem Toxic 44:476–483
Goyarts T, Dänicke S (2006) Bioavailability of the Fusarium toxin deoxynivalenol (DON) from naturally contaminated wheat for the pig. Toxicol Lett 163:171–182
Gratz SW, Duncan G, Richardson AJ (2013) The human fecal microbiota metabolizes deoxynivalenol and deoxynivalenol-3-glucoside and may be responsible for urinary deepoxy-deoxynivalenol. Appl Environ Microbiol 79(6):1821–1825
Guerre P, Eeckhoutte C, Burgat V, Galtier P (2000) The effects of T-2 toxin exposure on liver drug metabolizing enzymes in rabbit. Food Addit Contam 17(12):1019–1026
Hou YJ, Zhao YY, Xiong B, Cui XS, Kim NH, Xu YX, Sun SC (2013) Mycotoxin-containing diet causes oxidative stress in the mouse. PLoS One 8(3):e60374
Huang P, Akagawa K, Yokoyama Y, Nohara K, Kano K, Morimoto K (2007) T-2 toxin initially activates caspase-2 and induces apoptosis in U937 cells. Toxicol Lett 170(1):1–10
Ito M, Sato I, Ishizaka M, Yoshida S, Koitabashi M, Yoshida S, Tsushima S (2013) Bacterial cytochrome P450 system categorizing the Fusarium Toxin deoxynivalenol. Appl Environ Microb 79(5):1619–1628
JECFA (2001) T-2 and HT-2. Joint FAO/WHO Expert Committee on Food additives, 56th report. Safety evaluation of certain mycotoxins in food. WHO Food additives series 47. WHO, Geneva, pp 419–556. http://www.inchem.org/documents/jecfa/jecmono/v47je01.htm. Accessed Feb 2014
Joffe AZ (1974) Toxicity of Fusarium poae and F. sporotrichoides and its relation to alimentary toxic aleukia. In: Purchase IFH (ed) Mycotoxins. Elsevier, Amsterdam, pp 229–262
Johnsen H, Odden E, Lie O, Johnsen BA, Fonnum F (1986) Metabolism of T-2 toxin by rat liver carboxylesterase. Biochem Pharmacol 35(9):1469–1473
Johnsen H, Odden E, Johnsen BA, Fonnum F (1988) Metabolism of T-2 toxin by blood cell carboxylesterases. Biochem Pharmacol 37(16):3193–3197
Kluger B, Bueschl C, Lemmens M, Berthiller F, Häubl G, Jaunecker G, Adam G, Krska R, Schuhmacher R (2013) Stable isotopic labeling-assisted untargeted metabolic profiling reveals novel conjugates of the mycotoxin deoxynivalenol in wheat. Anal Bioanal Chem 405:5031–5036
Königs M, Mulac D, Schwerdt G, Gekle M, Humpf HU (2009) Metabolism and cytotoxic effects of T-2 toxin and its metabolites on human cells in primary culture. Toxicology 258:106–115
Kouadio JH, Mobio TA, Baudrimont I, Moukha S, Dano SD, Creppy EE (2005) Comparative study of cytotoxicity and oxidative stress induced by deoxynivalenol, zearalenone or fumonisin B1 in human intestinal cell line Caco-2. Toxicology 213(1–2):56–65
Krishnaswamy R, Devaraj SN, Padma VV (2010) Lutein protects HT-29 cells against deoxynivalenol-induced oxidative stress and apoptosis: prevention of NF-kappaB nuclear localization and down regulation of NF-kappaB and Cyclo-Oxygenase-2 expression. Free Radic Biol Med 49(1):50–60
Kruber P, Trump S, Behrens J, Lehmann I (2011) T-2 toxin is a cytochrome P450 1A1 inducer and leads to MAPK/p38-but not arylhydrocarbon receptor-dependent interleukin-8 secretion in the human intestinal epithelial cell line Caco-2. Toxicology 284:34–41
Kuca K, Pohanka M (2010) Chemical warfare agents. Mol Clin Environ Toxicol 100:543–558
Leal M, Shimada A, Ruíz F, González de Mejía E (1999) Effect of lycopene on lipid peroxidation and glutathione-dependent enzymes induced by T-2 toxin in vivo. Toxicol Lett 109(1–2):1–10
Li Y, Wang Z, Beier RC, Shen J, De Smet D, De Saeger S, Zhang S (2011) T-2 toxin, a trichothecene mycotoxin: review of toxicity, metabolism, and analytical methods. J Agric Food Chem 59:3441–3453
Mates JM (2000) Effects of antioxidant enzymes in the molecular control of reactive oxygen species toxicology. Toxicology 153:83–104
Maul R, Warth B, Kant JS, Schebb NH, Krska R, Koch M, Sulyok M (2012) Investigation of the hepatic glucuronidation pattern of the Fusarium mycotoxin deoxynivalenol in various species. Chem Res Toxicol 25:2715–2717
Meissonnier GM, Laffitte J, Raymond I, Benoit E, Cossalter AM, Pinton P, Bertin G, Oswald IP, Galtier P (2008) Subclinical doses of T-2 toxin impair acquired immune response and liver cytochrome P450 in pigs. Toxicology 247(1):46–54
Meky FA, Turne PC, Ashcroft AE, Miller JD, Qiao YL, Roth MJ, Wild CP (2003) Development of a urinary biomarker of human exposure to deoxynivalenol. Food Chem Toxicol 41:265–273
Morrissey RE, Norred WP (1985) Subchronic toxicity of vomitoxin in Sprague-Dawley rats. Food Chem Toxicol 23(11):995–999
Mu P, Xu M, Zhang L, Wu K, Wu J, Jiang J, Chen Q, Wang L, Tang X, Deng Y (2013) Proteomic changes in chicken primary hepatocytes exposed to T-2 toxin are associated with oxidative stress and mitochondrial enhancement. Proteomics 13:3137–3188
Nagl V, Schwartz H, Krska R, Moll WD, Knasmüller S, Ritzmann M, Adam G, Berthiller F (2012) Metabolism of the masked mycotoxin deoxynivalenol-3-glucoside in rats. Toxicol Lett 213(3):367–373
Ngampongsa S, Hanafusa M, Ando K, Ito K, Kuwahara M, Yamamoto Y, Yamashita M, Tsuru Y, Tsubone H (2013) Toxic effects of T-2 toxin and deoxynivalenol on the mitochondrial electron transport system of cardiomyocytes in rats. J Toxicol Sci 38(3):495–502
Obol’skiĭ OL, Kravchenko LV, Avren’eva LI, Tutel’ian VA (1998) Effect of dietary selenium on the activity of UDP-glucuronosyltransferases and metabolism of mycotoxin deoxynivalenol in rats. Vopr Pitan 4:18–23
Osselaere A, Li SJ, De Bock L, Devreese M, Goossens J, Vandenbroucke V, Van Bocxlaer J, Boussery K, Pasmans F, Martel A, De Backer P, Croubels S (2013) Toxic effects of dietary exposure of T-2 toxin on intestinal and hepatic biotransformation enzymes and drug transporter systems in broiler chickens. Food Chem Toxicol 55:150–155
Pestka JJ (2007) Deoxynivalenol: toxicology, mechanisms and animal health risks. Anim Feed Sci Technol 137:283–298
Prelusky DB, Veira DM, Trenholm HL, Foster BC (1987) Metabolic fate and elimination in milk, urine and bile of deoxynivalenol following administration to lactating sheep. J Environ Sci Health B 22(2):125–148
Rezar V, Frankic T, Narat M, Levart A, Salobir J (2007) Dose-dependent effects of T-2 toxin on performance, lipid peroxidation, and genotoxicity in broiler chickens. Poult Sci 86(6):1155–1160
Rizzo AF, Atroshi F, Ahotupa M, Sankari S, Elovaara E (1994) Protective effect of antioxidants against free radical-mediated lipid peroxidation induced by DON or T-2 toxin. Zentralbl Veterinarmed A 41(2):81–90
Sahu SC, Garthoff LH, Robl MG, Chirtel SJ, Ruggles DI, Flynn TJ, Sobotka TJ (2008) Rat liver clone-9 cells in culture as a model for screening hepatotoxic potential of food-related products: hepatotoxicity of deoxynivalenol. J Appl Toxicol 28(6):765–772
Sahu SC, O’Donnell MW Jr, Wiesenfeld PL (2010) Comparative hepatotoxicity of deoxynivalenol in rat, mouse and human liver cells in culture. J Appl Toxicol 30(6):566–573
SCF (Scientific Committee on Food) (1999) Opinion of the scientific committee on Fusarium toxins. Part 1: Deoxynivalenol (DON). http://ec.europa.eu/food/fs/sc/scf/out44_en.pdf. Accessed 20 Feb 2014
SCF (Scientific Committee on Food) (2001) Opinion on Fusarium toxins. Part 5. T-2 toxin and HT-2 toxin). http://ec.europa.eu/food/fs/sc/scf/out123_en.pdf. Accessed 20 Feb 2014
SCF (Scientific Committee on Food) (2002) Opinion on Fusariumtoxins. Part 6. Group evaluation of T-2 toxin, HT-2 toxin, nivalenoland deoxynivalenol. http://europa.eu.int/comm/food/fs/sc/scf/out123_en.pdf. Accessed 20 Feb 2014
Schuster A, Hunder G, Fichtl B, Forth W (1987) Role of lipid peroxidation in the toxicity of T-2 toxin. Toxicon 25(12):1321–1328
Schwartz HE, Hametner C, Slavik V, Greitbauer O, Bichl G, Kunz-Vekiru E, Schatzmayr D, Berthiller F (2013) Characterization of three deoxynivalenol sulfonates formed by reaction of deoxynivalenol with sulfur reagents. J Agric Food Chem 61:8941–8948
Sehata S, Kiyosawa N, Atsumi F, Ito K, Yamoto T, Teranishi M, Uetsuka K, Uetsuka K, Nakayama H, Doi K (2005) Microarray analysis of T-2 toxin-induced liver, placenta and fetal liver lesions in pregnant rats. Exp Toxicol Pathol 57(1):15–28
Shang S, Jiang J, Deng Y (2013) Chicken cytochrome P450 is the key enzyme for metabolizing T-2 toxin to 3′OH-T-2. Int J Mol Sci 14:10809–10818
Sinha K, Das J, Pal PB, Sil PC (2013) Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol 87:1157–1180
Sobrova P, Vasatkova A, Skladanka J, Beklova M, Zeman L, Kizek R, Adam V (2012) Study of deoxynivalenol effect on metallothionein and glutathione levels, antioxidant capacity, and glutathione-S-transferase and liver enzymes activity in rats. Chem Pap 66(12):1092–1102
Stacey NH, Klaassen CD (1981) Inhibition of lipid peroxidation without prevention of cellular injury in isolated rat hepatocytes. Toxicol Appl Pharmacol 58:8–18
Sugiyama K, Kinoshita M, Kamata Y, Minai Y, Tani F, Sugita-Konishi Y (2012) Thioredoxin-1 contributes to protection against DON-induced oxidative damage in HepG2 cells. Mycotoxin Res 28(3):163–168
Sun LY, Li Q, Meng FG, Fu Y, Zhao ZJ, Wang LH (2012) T-2 Toxin Contamination in grains and selenium concentration in drinking water and grains in Kashin-Beck Disease endemic areas of Qinghai Province. Biol Trace Elem Res 150:371–375
Suneja SK, Ram GC, Wagle DS (1984) Effects of T-2 toxin on glucose and tryptophan uptaken and intestinal mucosal enzymes. Toxicon 22:39–43
Suneja SK, Wagle DS, Ram GC (1987) T-2 toxin induced changes in liver and serum enzymes of rats. Toxicon 25(7):793–796
Tukey RH, Strassburg CP (2000) Human UDP-Glucuronosyltransferases: metabolism, expression, and disease. Annu Rev PharmacolToxicol 40:581–616
Turner PC, Hopton RP, Lecluse Y, White KLM, Fisher J, Lebailly P (2010) J Agric Food Chem 58:5206–5212
Turner PC, Hopton RP, White KLM, Fisher J, Wild CP (2011) Assessment of deoxynivalenol metabolite profiles in UK adult. Food Chem Toxicol 49:132–135
Uhlig S, Ivanova L, Fæste CK (2013) Enzyme-assisted synthesis and structural characterization of the 3-, 8-, and 15-glucuronides of deoxynivalenol. J Agric Food Chem 61(8):2006–2012
Vilà B, Jaradat ZW, Marquardt RR, Frohlich AA (2002) Effect of T-2 toxin on in vivo lipid peroxidation and vitamin E status in mice. Food Chem Toxicol 40(4):479–486
Wan D, Huang L, Pan Y, Wu Q, Chen D, Tao Y, Wang X, Liu Z, Li J, Wang L, Yuan Z (2014) Metabolism, distribution, and excretion of deoxynivalenol with combined techniques of radiotracing, high-performance liquid chromatography ion trap time-of-flight mass spectrometry, and online radiometric detection. J Agric Food Chem 62(1):288–296
Wang J, Jiang J, Zhang H, Wang J, Cai H, Li C, Li K, Liu J, Guo X, Zou G, Wang D, Deng Y, Dai J (2011) Integrated transcriptional and proteomic analysis with in vitro biochemical assay reveal the important role of CYP3A46 in T-2 toxin hydroxylation in porcine primary hepatocytes. Mol Cell Proteomics 10(9):M111-008748
Wang XC, Liu XD, Liu JC, Wang G, Wan KY (2012) Contamination level of T-2 and HT-2 toxin in cereal crops from Aba Area in Sichuan Province, China. Bull Environ Contam Toxicol 88:396–400
Wang X, Wang W, Cheng G, Huang L, Chen D, Tao Y, Pan Y, Hao H, Wu Q, Wan D, Liu Z, Wang Y, Yuan Z (2014) High risk of embryo-fetal toxicity: placental transfer of T-2 toxin and its major metabolite HT-2 toxin in BeWo cells. Toxicol Sci 137:168–178
Warth B, Sulyok M, Berthiller F, Schuhmacher R, Fruhmann P, Hametner C, Adam G, Fröhlich J, Krska R (2011) Direct quantification of deoxynivalenol glucuronide in human urine as biomarker of exposure to the Fusarium mycotoxin deoxynivalenol. Anal Bioanal Chem 401:195–200
Warth B, Sulyok M, Fruhmann P, Berthiller F, Schuhmacher R, Hametner C, Adam G, Fröhlich J, Krska R (2012) Assessment of human deoxynivalenol exposure using an LC-MS/MS based biomarker method. Toxicol Lett 211:85–90
Warth B, Sulyok M, Berthiller F, Schuhmacher R, Krska R (2013) New insights into the human metabolism of the Fusarium mycotoxins deoxynivalenol and zearalenone. Toxicol Lett 220:88–94
Weidner M, Welsch T, Hübner F, Schwerdt G, Gekle M, Humpf HU (2012) Identification and apoptotic potential of T-2 toxin metabolites in human cells. J Agric Food Chem 60(22):5676–5684
Weidner M, Hüwel S, Ebert F, Schwerdtle T, Galla HJ, Humpf HU (2013a) Influence of T-2 and HT-2 toxin on the blood-brain barrier in vitro: new experimental hints for neurotoxic effects. PLoS One 8(3):e60484
Weidner M, Lenczyk M, Schwerdt G, Gekle M, Humpf HU (2013b) Neurotoxic potential and cellular uptake of T-2 toxin in human astrocytes in primary culture. Chem Res Toxicol 26(3):347–355
Welsch T, Humpf HU (2012) HT-2 toxin 4-glucuronide as new T-2 toxin metabolite: enzymatic synthesis, analysis, and species specific formation of T-2 and HT-2 toxin glucuronides by rat, mouse, pig, and human liver microsomes. J Agric Food Chem 60:10170–10178
Wu X, Murphy P, Cunnick J, Hendrich S (2007) Synthesis and characterization of deoxynivalenol glucuronide: its comparative immunotoxicity with deoxynivalenol. Food Chem Toxicol 45:1846–1855
Wu Q, Dohnal V, Huang L, Kuča K, Yuan Z (2010) Metabolic pathways of trichothecenes. Drug Metab Rev 42(2):250–267
Wu J, Jing L, Yuan H, Peng S (2011a) T-2 toxin induces apoptosis in ovarian granulose cells of rats through reactive oxygen species-mediated mitochrondrial pathway. Toxicol Lett 202:168–177
Wu Q, Huang L, Liu Z, Yao M, Wang Y, Dai M, Yuan Z (2011b) A comparison of hepatic in vitro metabolism of T-2 toxin in rats, pigs, chickens and carp. Xenobiotica 41(10):863–873
Wu Q, Engemann A, Cramer B, Welsch T, Yuan Z, Humpf HU (2012) Intestinal metabolism of T-2 toxin in the pig cecum model. Mycotoxin Res 28:191–198
Wu Q, Dohnal V, Kuca K, Yuan Z (2013) Trichothecenes: structure-toxic activity relationships. Curr Drug Metab 14:641–660
Yang S, Li Y, Cao X, Hu D, Wang Z, Wang Y, Shen J, Zhang S (2013) Metabolic pathways of T-2 toxin in in vitro and in vitro systems of Wistar rats. J Agric Food Chem 61:9734–9743
Yang W, Yu M, Fu J, Bao W, Wang D, Hao L, Yao P, Nüssler AK, Yan H, Liu L (2014) Deoxynivalenol induced oxidative stress and genotoxicity in human peripheral blood lymphocytes. Food Chem Toxicol 64:383–396
Yoshizawa T, Sakamoto T, Okamoto K (1984) In vitro formation of 3′-hydroxy T-2 and 3′-hydroxy HT-2 toxins from T-2 toxin by liver homogenates from mice and monkeys. Appl Environ Microbiol 47:130–134
Young JC, Trenholm HL, Friend DW, Prelusky DB (1987) Detoxification of deoxynivalenol with sodium bisulfite and evaluation of the effects when pure mycotoxin or contaminated corn was treated and given to pigs. J Agric Food Chem 35:259–261
Yuan Y, Zhou X, Yang J, Li M, Qiu X (2013) T-2 toxin is hydroxylated by chicken CYP3A37. Food Chem Toxicol 62:622–627
Zhang X, Jiang L, Geng C, Cao J, Zhou L (2009) The role of oxidative stress in deoxynivalenol-induced DNA damage in HepG2 cells. Toxicon 54:513–518
Zhou HR, Pestka JJ (2003) Deoxynivalenol-induced apoptosis mediated by p38 MAPK-dependent p53 gene induction in RAW264.7 macrophages. Toxicologist 72:330
Acknowledgments
This work was financially supported by National Basic Research Program of China (973 program; Grant no.2009CB118800) and the project of Excellence FIM UHK as well as the long-term development plan UHHK.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Qing-Hua Wu, Xu Wang, and Wei Yang have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Wu, QH., Wang, X., Yang, W. et al. Oxidative stress-mediated cytotoxicity and metabolism of T-2 toxin and deoxynivalenol in animals and humans: an update. Arch Toxicol 88, 1309–1326 (2014). https://doi.org/10.1007/s00204-014-1280-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-014-1280-0