
Abstract
Significant efforts have been made for the development of new anticancer drugs (protein kinase or proteasome inhibitors, monoclonal humanized antibodies) with presumably low or negligible side effects and high specificity. However, an in-depth analysis of the side effects of several currently used canonical (platin-based drugs, taxanes, anthracyclines, etoposides, antimetabolites) and new generation anticancer drugs as the first line of clinical treatment reveals significant perturbation of glycolysis and oxidative phosphorylation. Canonical and new generation drug side effects include decreased (1) intracellular ATP levels, (2) glycolytic/mitochondrial enzyme/transporter activities and/or (3) mitochondrial electrical membrane potentials. Furthermore, the anti-proliferative effects of these drugs are markedly attenuated in tumor rho (0) cells, in which functional mitochondria are absent; in addition, several anticancer drugs directly interact with isolated mitochondria affecting their functions. Therefore, several anticancer drugs also target the energy metabolism, and hence, the documented inhibitory effect of anticancer drugs on cancer growth should also be linked to the blocking of ATP supply pathways. These often overlooked effects of canonical and new generation anticancer drugs emphasize the role of energy metabolism in maintaining cancer cells viable and its targeting as a complementary and successful strategy for cancer treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite considerable advances in biomedical research and clinical applications in the field, cancer remains a leading cause of mortality worldwide. The severe side effects caused by the frequent use of conventional chemotherapeutics diminish, unfortunately, the patients’ quality of life. Therefore, the development of safe and efficacious alternatives with well-known mechanism of action is mandatory. First-line anticancer drugs such as antibody–drug conjugates and protein kinase (PK) small-molecule inhibitors have become widely used in the clinical treatment of cancer presumably because they are designed to be selective for one protein or cellular process (Ott and Adams 2011; Li et al. 2013; Miller et al. 2013). However, as occurs with other canonical and traditional chemotherapy drugs (platin-based drugs, taxanes, anthracyclines, etoposides, antimetabolites), these new anticancer drugs also have multiple side effects, disturbing important cellular processes present in normal cells, at the doses used during cancer treatment (Hansel et al. 2010; Mellor et al. 2011; Baldo 2013). Thus, for a more comprehensive understanding of the global inhibitory effects of these drugs on cancer and non-cancer cells, an integral functional analysis of the additive or synergistic drug effect on both the protein or pathway for which it was designed and the multiple non-specific inhibition of a priori untargeted pathways or proteins should be undertaken.
In this regard, it has been demonstrated that several cellular processes such as cellular proliferation and survival, apoptosis, endothelial permeability, vascular remodeling and ion channel homeostasis are affected by low doses of monoclonal antibodies, antibody–drug conjugates and PK small-molecule inhibitors (Mellor et al. 2011; Ott and Adams 2011; Baldo 2013). Certainly monoclonal antibodies (mAbs) and PK inhibitors interact directly with their specific receptor- or kinase target. However, structural analysis, performed for instance with the tyrosine kinase (TK) inhibitor imatinib, shows that the drug also interacts directly with other non-kinase proteins in non-tumor cells such as the yeast vacuolar-type H+-ATPase and the human NAD(P)H dehydrogenase quinone oxidoreductase 2 (NQO2) (reviewed in Lee and Wang 2009). Although mAbs bind to their specific extracellular receptors, it has been demonstrated that some mAbs may be transported across the plasma membrane and affect intracellular targets (Dao et al. 2013). In addition, TK inhibitors and mAbs may block non-specifically other transduction pathways such as those mediated by AMPK, platelet-derived growth factor receptor (PDGFR), PI3K-mTOR and MAPK (Mellor et al. 2011; Ott and Adams 2011).
In particular, energy metabolism appears to be highly sensitive to clinical doses of several antineoplastic drugs (reviewed in Rodríguez-Enríquez et al. 2009). This observation becomes relevant by taking into consideration several cellular processes occurring in tumor cells (proliferation, metastasis, macromolecules synthesis, stress-defense, ATPases activation) and non-tumor cells that depend on the intracellular ATP concentration which in turn depend on the ATP production rate by both glycolysis and oxidative phosphorylation (OxPhos) (Müller et al. 1986; Schmidt et al. 1991; Rolfe and Brown 1997). Thus, despite the well-documented genetic and biochemical heterogeneity of cancer cells, it has to be recalled that they all depend on glycolysis and OxPhos for the ATP supply. The downside of targeting ATP supply in cancer cells is that this same function is also essential for non-tumor cells, and hence, this approach becomes extremely toxic. However, this strategy may still be particularly relevant for cancer cells as they demand higher ATP provisions for their accelerated cell proliferation and associated processes (Schmidt et al. 1991), i.e., cancer cells are more susceptible to variations in their ATP supply than non-tumor cells. This central and essential role of the ATP supply has no parallel in other metabolic and signal transduction pathways. In those, the perturbations trigger compensatory mechanisms by turning on and off other interlinked pathways. In contrast, for cellular ATP supply, there are no alternative compensatory ATP-producing pathways. The use of multi-action drugs (acting for instance on their specific targets and on energy metabolism) or a combination of drugs might become an asset in cancer chemotherapy once their modes of action are understood.
Energy metabolism alterations in solid tumors
Oxidative phosphorylation flux and metabolite levels
Oxidative phosphorylation (OxPhos) is the principal ATP producer (>90 %) for several malignant tumor cell types under normoxic conditions (i.e., 21 % atmospheric O2) (Zu and Guppy 2004; Rodríguez-Enríquez et al. 2010). Therefore, drug therapy targeting OxPhos has emerged as an important alternative for growth arrest of oxidative type tumors (reviewed in Moreno-Sánchez et al. 2007; Rodríguez-Enríquez et al. 2011). However, in solid tumors (Fig. 1), the development of unorganized, fragile and leaky vessels promotes intermittent, rather than constant, blood flow and the onset of episodes of hypoxia (from 31 to 86 μM dissolved O2 or 24–66 mmHg under normoxia to 3–13 μM dissolved O2 or 0.1–1 % atmospheric O2 under hypoxia; Vaupel et al. 1989; Höckel and Vaupel 2001) and low-carbon source supply (for instance, extracellular glucose decreases from 5–10 to 0.5–2.5 mM; Tamulevicius and Streffer 1995; Hirayama et al. 2009) in regions distant from the blood vessels. The transient or permanent shortage in O2 and carbon sources may, directly and indirectly, affect OxPhos machinery and induce an energy metabolic shift toward a greater dependence on glycolysis (Sonveaux et al. 2008; Hirschhaeuser et al. 2011; Mathews et al. 2011).

Gradients of glucose and O2 in solid tumors
Long-term (0.1–2 % atmospheric O2, 18–24 h) hypoxia affects OxPhos in HeLa (cervix), MCF-7 (breast), HCT116 and RKO (colon) and RCC4 (renal) cancer cells at different levels of regulation inducing diminution of (1) the contents and activities of the 2OGDH and GA (Rodríguez-Enríquez et al. 2010) and (2) the assembly of the respiratory chain complexes I and IV which in turn influences their enzymatic activities (Chen et al. 2010; Tello et al. 2011). Although OxPhos flux (Rodríguez-Enríquez et al. 2011; reviewed in Moreno-Sánchez et al. 2014) and total O2 consumption (Cairns et al. 2007; Chen et al. 2010; Tello et al. 2011) severely diminish (80–90 %) in cancer cells under long-term hypoxia, cellular viability remains high at 70–80 %. This phenomenon occurs perhaps because rescue pathways are activated (i.e., increased glycolysis, decreased ATP-dependent processes) and cellular proliferation decreases, thus maintaining the intracellular ATP pool constant and high. It has been documented that hypoxia (0.05 % atmospheric O2) and low glucose (1 mM) up-regulate the expression of the mitochondrial proline oxidase (POX) for glutamate and 2OG production. However, it has not been determined whether under hypoxia and low glucose, proline oxidation may support OxPhos flux and in turn help to preserve the high cell viability observed under this stress condition (Liu and Phang 2012; Liu et al. 2012).
HIF-1α stabilization is reached under normoxia in paragangliomas, pheochromocytomas and renal cell cancer in a process mediated by mutations in the Krebs cycle fumarate hydratase or succinate dehydrogenase. The mutant enzymes become inactive promoting the accumulation of succinate and fumarate (Isaacs et al. 2005; Pollard et al. 2005; Selak et al. 2005). These two metabolites competitively inhibit prolyl-hydroxylase (PHD) activity preventing HIF-1α hydroxylation and its proteasomal degradation (Isaacs et al. 2005; Selak et al. 2005).
OxPhos also plays an important role in 3D physiological models such as multi-cellular tumor spheroids (MCTSs, Kunz-Schughart 1999; Rodríguez-Enríquez et al. 2008) and tumor xenotransplants (Menon et al. 2003; Yaromina et al. 2012). The MCTSs resemble the physiologically heterogeneous architecture (hypoxic and normoxic micro-areas) found in the initial avascular stages of solid tumors (Sutherland 1988; Kunz-Schughart et al. 2004). These micro-areas generate O2 (1–120 mmHg) and glucose (3 to >21 μmol/g of cellular protein) gradients between the inner and outer cell layers, which cannot occur in the extensively used monolayer or bi-dimensional culture model (Hirschhaeuser et al. 2010). Thus, the 3D tumor spheroid architecture induces transcriptional and biochemical changes in the energy metabolism and in consequence changes in the sensitivity to metabolic inhibitors (Rodríguez-Enríquez et al. 2008; Mandujano-Tinoco et al. 2013), metastasis onset (Gallardo-Pérez et al. 2014) and the development of anti-apoptotic pathways (Barbone et al. 2012).
It has been reported that HeLa, Hek293 and MCF-7 MCTSs strongly depend (>80 %) on mitochondrial ATP for their growth. Therefore, proliferation is highly sensitive to OxPhos inhibitors (Rodríguez-Enríquez et al. 2008; Mandujano-Tinoco et al. 2013). In contrast, mature MCTSs are destabilized and disaggregated by OxPhos plus glycolytic inhibitors, indicating that both ATP sources are essential for tumor maintenance and survival (Rodríguez-Enríquez et al. 2008). Ductal infiltrating breast (DIB) cancer human biopsies, hormone receptor-positive (HR+) subtype show similar values for mitochondrial and glycolytic protein contents to those of human breast MCF-7 MCTSs (which are also HR+), in which 2OGDH and respiratory complex IV are over-expressed compared to normal tissue (Kim et al. 2014; Pacheco-Velázquez et al. manuscript in preparation). In triple-negative DIB samples, 2OGDH was particularly (2–3 times) over-expressed versus other cancer subtypes, although the glycolytic GLUT-1, HKI and LDH protein contents were similar (Pacheco-Velázquez et al. manuscript in preparation). These data further support a significant role for mitochondria in cancer cell physiology. Consequently, anti-mitochondrial therapy could be an alternative against triple-negative breast cancer subtype. In renal biopsies of oncocytoma and stage-3 urothelial cancer, the mRNA contents of the adenine nucleotide translocator ANT2 and ATP synthase subunit B are up-regulated (Faure-Vigny et al. 1996), which may also indicate an active OxPhos in these cancers.
Glycolytic flux and metabolite levels
Increased glycolysis in tumor cells is associated with the activation of the transcriptional factors HIF-1α, c-MYC and probably H-RAS, all of which promote increased transcription of the genes and increased contents of all glycolytic enzymes/transporters (Gillies et al. 2008; Dang et al. 2009; Marín-Hernández et al. 2009; Chesney and Telang 2013). In fact, HIF-1α induces the over-expression of glycolytic isoforms with high affinity to their substrates and/or activators and low affinity to their physiological inhibitors (Marín-Hernández et al. 2009). This ensures the supply of cytosolic ATP, NADH and glycolytic intermediaries for protein synthesis (alanine, serine, cysteine and glycine), nucleic acid and glycogen synthesis (ribose-5-phosphate, glucose-1-phosphate) and lipid synthesis (glycerol-3-phosphate) even under stress conditions.
Conversely, it has been shown that several glycolytic enzymes have other roles in addition to their canonic metabolic function acting as (1) apoptotic inhibitors (HKI and HKII); (2) transcriptional factors (GAPDH/LDH) regulating DNA replication and repair processes; (3) metastatic and epithelial–mesenchymal transition inducers (HPI); and (4) potential biomarkers for aggressive carcinomas such as lung cancer (ENO) (Marín-Hernández et al. 2009; Funasaka et al. 2009; reviewed in Rodríguez-Enríquez et al. 2009; Gallardo-Pérez et al. 2014). Therefore, the use of anticancer drugs that affect the expression and activity of glycolytic or OxPhos proteins, in addition to their effects on the targeted proteins for which they were designed, may have clinical relevance against glycolytic- or oxidative-dependent tumors, respectively.
Energy metabolism and metastasis
Metastatic tumor cells constitute a subpopulation originated in the primary tumor that displays significant reprogramming in their intermediary and energy metabolism and signaling pathways. These alterations endow the cells with the ability to survive after detachment from the primary tumor, migrate through the circulatory and/or lymphatic systems and eventually colonize distant tissues. Anchorage independence implies the avoidance of anoikis, a form of programmed cell death due to the loss of contact with neighboring cells or the extracellular matrix. Anchorage independence (i.e., anoikis resistance), migration and colonization may be considered separate functional events as they probably result from the subversion of distinct regulatory pathways. Notwithstanding, all of them are subordinated to the energy supply that encompasses the progression from the benign to malignant states. Analysis of energy metabolism in HeLa cells, a metastatic cervix cancer cell line, has shown that OxPhos provides most (>70 %) of the ATP required for cellular work (Rodríguez-Enríquez et al. 2006) such as proliferation and metastasis onset.
As metastasis is a major challenge to cancer therapy, it becomes relevant to dissect those biochemical mechanisms associated with the malignant trait aiming not only at understanding the underlying biochemical mechanisms of metastasis, but also at identifying susceptible targets for drug intervention. Deregulation of many PKs has been shown to affect cell transformation, tumor progression and metastasis. Because PKs constitute regulatory nodes in many signaling pathways, frequently they also affect the reactions that involve directly or indirectly in the energy metabolism. Thus, oncogenic kinases as well as transcription factors have become potential targets for pharmacological intervention.
Src family kinases (SFKs) are non-receptor tyrosine kinases (TKs) that respond to several stimuli all of them bearing on cell transformation, tumor progression and metastasis. Interestingly, SFKs can be activated by reactive oxygen species (ROS), linking up oncogenesis mainly to mitochondrial activity. The p21-activated kinases (PAKs) are serine and threonine PKs also involve in cell transformation, metastasis and angiogenesis (Radu et al. 2014). PAKs modulate the cell cycle through many nuclear signaling pathways and prevent apoptosis by the dual inhibition of caspases and mitochondrial BCL-2.
The transcription factor TP63, a member of the p53 family involved in epithelial morphogenesis and differentiation and a regulator of microRNAs (miRNAs), is generally recognized as a metastasis suppressor factor in epithelial tumors. For instance, knockdown of p63 abrogates the invasion led by specialized breast cancer cells (Cheung et al. 2013) and induces GA 2 expression (Giacobbe et al. 2013). p63 knockout mice exhibit obesity, glucose intolerance and insulin resistance and also display mitochondrial fatty acid oxidation dysfunction (Candi et al. 2014). These data suggest a connection between p63 and the oxidative metabolism, in which p63 may depend on functional mitochondria for promoting metastasis.
Pyruvate kinase isoform M2 (PYKM2) is a glycolytic enzyme over-expressed in highly proliferating cells and most types of cancer cells. It has been shown that the interaction of PYKM2 with phosphotyrosine-containing proteins inhibits its activity and increases upstream glycolytic metabolite levels to support cell proliferation (Mazurek 2011). Exposure to non-physiological PYKM2 activators such as TEPP-46 (a thieno [3,2-b] pyrrole [3,2-d] pyridazinone family member) or DASA-58 (a N,N′-diarylsulfonamide family member) induces inhibition of tumor growth under mild hypoxic conditions (1 % atmospheric O2) and decrease in proliferation-linked metabolites such as ribose-5-phosphate and serine. Binding of these activators to PYKM2 promotes tetramer formation that in turn renders it resistant to inhibition by tyrosine-phosphorylated proteins and suppresses tumor growth (Anastasiou et al. 2012).
However, PYKM2 seems to transcend its metabolic role in tumor glycolysis (Marín-Hernández et al. 2009). (1) Recent results have shown that PYKM2 acquires PK activity, using PEP instead of ATP, upon succinyl-5-aminoimidazole-4-carboxamide-1-ribose-5-phosphate (SAICAR) binding (Keller et al. 2014), an abundant metabolite in proliferating cells. PYKM2 PK activity phosphorylates many proteins such as histones and kinases, particularly Erk1/2, promoting cancer cell proliferation (Keller et al. 2014). (2) MicroRNA-122 (miR-122) is increased in normal liver tissue and decreased in hepatocellular carcinomas. Over-expression of miRNA-122 in hepatocellular cancer cells displays diminished PYKM2 expression (Liu et al. 2014) as well as decreased proliferation and metastasis, and increased sensitivity to anticancer drugs (Bai et al. 2009). Thus, PYKM2 activity in glycolysis (by increasing it) and as PK (by decreasing it) may be attractive novel targets for counteracting tumorigenesis and metastasis.
Another novel anticancer chemotherapeutic approach targets the fine redox balance dealing with oxidative stress. A consensus is emerging in which low ROS levels seem to promote tumor growth and invasiveness, whereas high ROS levels induce tumor cell death. Therefore, pro-oxidant pharmacological interventions based on either increasing ROS production or decreasing ROS scavenging are proving to be effective (Pathania et al. 2014). In this respect, recent results with pancreatic cancer cells have shown that quinazoline derivatives act as redox modulators and substantially increase rates of O2 consumption and ROS production, which in turn induce oxidative stress, apoptosis (Pathania et al. 2014) and tumor vascularity impairment without affecting cellular proliferation. These results suggest that quinazoline derivatives may target specifically those aspects pertaining to anoikis resistance during metastasis (Bilbro et al. 2013), and which seem intimately linked to energy metabolism.
Small-molecule drugs used in oncology that also affect glycolytic and mitochondrial enzymes/transporters
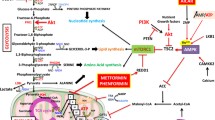
Several commonly used chemotherapeutic drugs such as imatinib, sorafenib, temsirolimus, lapatinib, decitabine, azacytidine, bortezomib, bevacizumab, trastuzumab, cetuximab, rituximab, metformin, phemformin and letrozole (Fig. 2) have a direct effect on several glycolytic and mitochondrial proteins at the transcriptional (PK inhibitors), posttranslational (PK inhibitors, monoclonal humanized antibodies) or kinetic (PK inhibitors, monoclonal humanized antibodies, proteasome inhibitors) levels (Figs. 3, 4).

Chemical structures of some first-line anticancer drugs that block energy metabolism

Glycolytic targets of PK inhibitors and mAbs. GLUT glucose transporter, HK hexokinase, HPI hexosephosphate isomerase, PFK1 phosphofructokinase type 1, ALDO aldolase, TPI triosephosphate isomerase, GAPDH glyceraldehyde-3-phosphate dehydrogenase, PGK phosphoglycerate kinase, PGAM phosphoglycerate mutase, ENO enolase, PYK pyruvate kinase, LDH lactate dehydrogenase, MCT monocarboxylate transporter, Glc glucose, Glc6P glucose 6-phosphate, Fru6P fructose 6-phosphate, Fru16BP fructose 1,6-bisphosphate, DHAP dihydroxyacetone phosphate, G3P glyceraldehyde-3-phosphate, 1,3BPG 1,3-bisphosphoglycerate, 3PG 3-phosphoglycerate, 2PG 2-phosphoglycerate, PEP phosphoenolpyruvate, PYR pyruvate. Red lines indicate drug inhibition; green arrows indicate drug activation

Mitochondrial targets of first-line or experimental drugs in cancer treatment. Cit citrate, Gln glutamine, GA glutaminase, GDH glutamate dehydrogenase, Mal malate, OAA oxaloacetate, 2-OG 2-oxoglutarate, Succ succinate, Fum fumarate
Tyrosine kinase (TK) inhibitors
Imatinib, sunitinib, dasatinib, erlotinib, axitinib, lapatinib, sorafenib, nilotinib, busotinib and temsirolimus (Fig. 2) are currently the most frequently used TK inhibitors in the treatment of cancer (Mellor et al. 2011; Ott and Adams 2011). All of them are signal transduction inhibitors that presumably only affect specific kinases in the tumor cells, but which, unfortunately, also affect other protein functions unrelated to the respective kinase network. For example, imatinib directly interacts with the stem cell factor receptor (c-KIT) and the platelet-derived growth factor receptor (PDGF-R) blocking their respective signal pathways. Also, TK inhibitors directly interact with different oncogenes such as BRC/ABL (imatinib) and B-RAF (sorafenib) affecting their oncogenic function (Arora and Scholar 2005; Fabian et al. 2005; Rix et al. 2007; Kessler et al. 2010; Waller 2010). These observations cast doubt on the cellular specificity of the TK inhibitors.
The intended action of the TK small-molecule inhibitors is the direct interaction with the receptor/enzyme ATP-binding site at relatively low, therapeutically viable, concentrations. For imatinib, computational models with TK crystals revealed a submicromolar inhibition constant (Ki = 0.4 μM, Lin et al. 2013). However, decitanib (IC50 = 9 nM) and nilotinib (IC50 = 45 nM) showed more potency for abolishing erythroleukemia K562 growth than imatinib (IC50 = 400 nM) (Rix et al. 2007). As there is a multitude of ATP-interacting enzymes in cancer metabolism, it can be expected that the TK inhibitors may also interact with the ATP-binding pocket of a variety of nontargeted enzymes such as the glycolytic enzymes HK, PFK-1, PGK and PYK.
Imatinib and lapatinib at therapeutic doses also inhibit several glycolytic steps (Fig. 2; Table 1) including the tumor glycolytic flux-controlling GLUT (Marín-Hernández et al. 2011). Interestingly, in leukemia BCR–ABL-positive (K562) cell line, but not in BCR–ABL-negative (HC-1) cells, imatinib (2.5 μM/96 h) induced a significant decrease in lactate production. The glycolytic flux is 1.3–1.5 times higher in BCR–ABL-positive cells than in their negative counterparts. Moreover, despite significant TK inhibition, imatinib does not affect glycolysis of other BCR–ABL-positive cells (CML-T1) whose glycolysis is significantly lower (1.6 times) than that of BCR–ABL-negative HC-1 cells (Gottschalk et al. 2004). Thus, these data suggest that the imatinib metabolic effect is associated with high glycolytic rates in tumor cells. In other studies, imatinib-positive and imatinib-resistant BCR–ABL cells DA1-3b/M2 cells increase their glycolytic flux correlating with a significant increment in PGK-1 protein and HKII, HPI, PFK1 and ALDO mRNAs contents through a mechanism presumably associated with HIF-1α stabilization (Zhao et al. 2010; Kluza et al. 2011). In other diseases, such as pulmonary arterial hypertension, imatinib and sunitinib also affect 18-fluorodeoxyglucose (FDG) uptake (Zhao et al. 2013) indicating that the imatinib effect on glycolysis is not a typical response of cancer cells.
As a consequence of the glycolysis suppression, imatinib-positive leukemia cells (0.1 μM/week for 5 weeks or 0.1–2.5 μM/96 h) as well as imatinib-negative leukemia cells (same treatment doses) switch their energy metabolism to an oxidative type (Table 2) leading to increased mitochondrial metabolite levels such as succinate, fumarate, malate and glutamate (Gottschalk et al. 2004; Klawitter et al. 2009; Kluza et al. 2011). In these cells, a significant increment in NADH/NAD+, ATP/ADP, UTP/ADP, CTP/CDP and GTP/GDP ratios, energy charge and mRNA contents of several enzymes involved in glutamine oxidation is observed (Table 2); also, perhaps as a consequence of mitochondrial metabolism activation, decreased phosphocreatine and acetate contents are detected (Gottschalk et al. 2004; Klawitter et al. 2009; Kluza et al. 2011) (Table 2). The OxPhos activation induced by imatinib in K562-positive leukemia and HC-1-negative leukemia cells was not observed in imatinib-positive and imatinib-resistant leukemia cells such as DA1-3b/M2 cells, in which imatinib actually induced a significant diminution in the total O2 consumption (Kluza et al. 2011), suggesting decreased mitochondrial energy metabolism. The OxPhos inhibitory effect of imatinib on DA1-3b/M2 cells is supported by the observation that under physiological conditions, the piperazinyl group of imatinib (Fig. 2) is predominantly protonated, thus carrying a net positive charge (Lin et al. 2013). The relevance of the last observation can be fully understood by considering that tumor mitochondria accumulate positive charge lipophilic molecules, to a higher extent than non-tumor mitochondria, due to their greater magnitude of the electrical potential difference across the inner membrane or Δψ (reviewed in Rodríguez-Enríquez et al. 2009). Thus, it can be predicted that imatinib may preferentially accumulate in tumor mitochondria versus normal mitochondria. The molecular mechanisms involved in the apparent imatinib activation or inhibition, at the translational, transcriptional or kinetic levels, of mitochondrial enzymes and metabolism clearly require further investigation.
The OxPhos activation induced by imatinib in K562 and HC-1 leukemia cells has not been observed for other TK small-molecule inhibitors. On the contrary, sorafenib induces a decrease in the ATP intracellular content in hepatoma cells at clinical doses (Table 2; Fiume et al. 2011). Furthermore, in non-tumor cells, imatinib, suratinib, dasatinib, lapatinib and sorafenib cause (1) cardiac failure (Ksienski 2011; Mellor et al. 2011); (2) heart mitochondrial swelling (Chu et al. 2007); (3) inhibition of several heart mitochondrial enzyme activities (COX IC50 = 124 μM; ATP synthase IC50 = 5–96 μM; CI-NADH dehydrogenase IC50 = 31–220 µM; and CII-SDH + CIII IC50 = 3 µM; Will et al. 2008); and (4) lowering in the PYK-M2 protein content (Hegde et al. 2007). Thus, it seems that imatinib may promote differential effects on mitochondrial energy metabolism of cancer cells versus normal cells, activating in the former and depressing in the latter.
Temsirolimus is presumably a selective inhibitor of the mTOR complex, a serine–threonine PK involved in several tumor-promoting signaling pathways and controlling many cellular functions such as proliferation, survival, cell cycle regulation (PI kinase, Akt/PK B) and stabilization of transcription factors such as HIF-1α and VEGF (Koh et al. 2010). In clinical practice, this drug is used in patients with advanced renal cell carcinoma (Ott and Adams 2011; Polivka and Janku 2014). In murine xenograft tumor models, therapeutic doses of temsirolimus increase the content of GLUT1 by 50 % (Ma et al. 2009); other glycolytic proteins were not evaluated.
Epigenetic modifiers
It has been proposed that DNA hypermethylation is associated with tumorigenesis onset (Esteller 2007). As guanine and cytosine are the predominant targets for methylation (Chan et al. 1993; Clark and Melki 2002; Gowher and Jeltsch 2004; Cherepanova et al. 2011), two potent cytosine analogs have been synthesized to block O6-methylguanine-DNA transferase (MGT) or DNA cytosine C5 methyltransferase and tested as anticancer drugs. 5-Azacytidine (AzaC; Fig. 2) and its deoxy derivative 5-aza-2′-deoxycytidine (also called decitabine) are frequently used for treatment of acute myeloid leukemia; other carcinomas (breast, ovary) are also sensitive to these drugs (Xu et al. 2007; Meng et al. 2013).
Adriamycin and trichostatin A potentiate the antitumor effect of decitabine by inducing p53-mediated mitochondrial apoptosis in leukemia cells (Table 2) and pancreas carcinoma without apparent effects on normal cells (Schmelz et al. 2005; Cecconi et al. 2009). In some leukemia cells, decitabine alone increases ROS level and collapses mitochondrial Δψ leading to apoptosis (Table 2; Shin et al. 2012). In contrast, in some OxPhos proteins, such as the mitochondrial citrate carrier in metastatic brain cells and the adenine nucleotide translocator subtype 4 (ANT4) in murine fibroblast, specific proximal promoter regions (CpG) highly susceptible to be methylated have been described (Brower et al. 2009). Accordingly, AzaC or decitabine increases the mitochondrial citrate carrier mRNA and ANT protein contents (Table 2), suggesting that epigenetic abnormalities could negatively affect the OxPhos pathway or that these drugs directly interact with the OxPhos genes/proteins. In other metabolic disorders such as Parkinson’s diseases, decitabine also increases the ROS level (which presumably derives from the mitochondrial metabolism) and the cytochrome c protein expression (Kong et al. 2012).
Proteasome inhibitors
The ubiquitin–26S proteasome pathway is responsible for degradation of the majority of regulatory proteins in mammalian cells including those that control cell cycle progression, apoptosis induction and DNA repair. Proteasomal activity is higher in B-cell chronic lymphocytic leukemia (Masdehors et al. 2000) than in normal lymphocytes promoting an accelerated degradation of key proteins. Therefore, the disruption of proteasomal activity leads to growth arrest and cell death. Bortezomib was the first-generation proteasome inhibitor used in the clinical treatment of multiple myeloma and non-Hodgkin lymphoma (Piperdi et al. 2011). The drug mode of action derives from its reversible binding to the B1 subunit of the catalytic site of 26S proteasome (Shah et al. 2009), leading to inhibition of its proteolytic activity (Blackburn et al. 2010).
Although bortezomib has been considered as a suitable drug for targeted therapy based on its high potency and selectivity for the proteasome, it also has multiple effects in tumor cells. For example, in prostate cancer (LNCaP-Pro5) and malignant B cells, it has been demonstrated that relatively low doses (nanomolar range) of bortezomib stabilizes p53 and Bax (pro-apoptotic proteins), whereas in glioblastoma multiform the drug decreases the mRNA and protein contents of the anti-apoptotic protein Bcl2 (Williams and McConkey 2003; Yin et al. 2005; Liu et al. 2008), leading to tumor arrest. In several carcinomas, bortezomib promotes a severe impairment of OxPhos and autophagy activation prompting a significant mitochondrial and cellular ATP depletion (Table 2). The content of some glycolytic enzymes was unaltered by bortezomib, although determination of the glycolysis flux was not performed (Yin et al. 2005; Selimovic et al. 2013). Bortezomib (0.2 mg/kg weight for 1–3 weeks) induces in rat heart mitochondrial ultrastructural abnormalities and a decrease in cellular respiration (47 %) and, in consequence, a significant decrease in cellular ATP (Nowis et al. 2010).
For other TK small-molecule inhibitors (dasatinib, erlotinib, axitinib, nilotinib, busotinib; see Fig. 2 for chemical structures), information on their effects at the energy metabolism level is not yet available, although significant effects may be expected given their structural analogy with imatinib.
Antibody-based tumor targeting affecting glycolytic and mitochondrial enzymes
The use of monoclonal antibodies (mAb) directed at a particular protein over-expressed in cancer cells has become a common clinical strategy for identification and pharmacological intervention. However, although immunotherapy may result in a more selective strategy than other approaches, its use is associated with undesirable side effects such as cardiac failure (Baldo 2013). In addition, most of the mAbs certainly bind to their extracellular targeted proteins with high affinity, but some of them may cross the plasma membrane and exert their effects on intracellular targets (Dao et al. 2013).
Anti-angiogenic humanized mAbs
Bevacizumab (Avastin®) exerts its effects through different mechanisms including inhibition of the de novo growth of blood vessels along with the regression of newly formed tumor vasculature, both of which altering vascular function. This mAb also has a direct effect on the tumor cells. The molecular mechanism for the direct effect is associated with its extracellular binding (Kd = 1.8 nM) (Papadopoulos et al. 2012) to the vascular endothelium growth factor (VEGF) subtype A, thus blocking the subsequent signal transduction pathways PI3K/MAPK, AKT/PKB, RAS/ERK coupled to TK receptors (VEGFR). This mAb has been employed in the treatment of several cancers including breast, colorectal, lung, brain, kidney and ovarian carcinomas. At the metabolic level, bevacizumab increases the transcription factors associated with glycolysis activation (Table 1) and lowers the mitochondria content in MCTSs derived from human glioblastoma biopsies transplanted in nude rats (Table 2) (Keunen et al. 2011). Also in bevacizumab-resistant tumor cells such as xenograft colorectal cancer, the drug suppresses mitochondrial biogenesis and the content of the OxPhos flux-controlling enzyme, NADH dehydrogenase or respiratory complex I (Xu et al. 2013).
Trastuzumab shows high efficacy in the treatment of breast HER–2neu-positive patients and is potentially effective on other carcinomas (non-small cell lung cancer) (Mazieres et al. 2013). However, many patients eventually develop resistance to the drug after continuous treatment. The TK receptor ERB2B2 (HER–2neu) is 30 % over-expressed in breast cancer (Yu and Hung 2000) and is involved in the activation of several intracellular pathways associated with proliferation (RAS–RAF–MAP kinases), tumor survival (PI3K/AKT kinase) and apoptosis (AKT kinase and NFkB transcription factor activation) (Prenzel et al. 2001; Mendelsohn and Baselga 2000; Papanikolaou et al. 2011). Trastuzumab binds to the extracellular domain of ERB2B2 TK receptor diminishing tumor proliferation, transformation and metastatic potential. At the therapeutic doses used in the clinical treatment (Chan 2007), trastuzumab also affects tumor energy metabolism (Tables 1, 2). Regarding mitochondrial metabolism, it has been reported in ERBB2-positive breast cancer (MCF-7/ERBB2 and MDA-MB-231/ERBB2) that trastuzumab (100 μg/ml) promotes the translocation of the ERBB2 receptor that usually localizes to the plasma membrane and cytosol fractions, to tumor mitochondrial inner membrane (Ding et al. 2012). Although OxPhos performance in the presence of this mAb has not been assessed in tumor cells, in cardiomyocytes exposed to the selective ERBB2 clone 9G6 mAb (5–7.5 μg/mL) for 24 h, OxPhos (measured as Δψ, ATP intracellular content and redox capacity) was partially impaired (Grazette et al. 2004).
A “bifunctional peptide” composed by anti-HER-2 peptide (to neutralize HER-2) fused to a mitochondriotoxic peptide (to impair mitochondrial function) has been engineered against HER-2-overexpressing tumors (Fantin et al. 2005). Presumably, the mitochondriotoxic peptide named BHAP acts at two levels (1) as a lipophilic cation having selectivity for mitochondria with high Δψ (negative inside) and (2) as HER-2 analog blocking the ERBB2 receptor. This peptide (10 μM for 3 h) altered mitochondrial morphology and induced cytochrome c release, the main trigger of apoptosis (Von Ahsen et al. 2000; Ott et al. 2002) in ERBB2 over-expressing BT474 and MDA-MB-453 breast cancer cells (Fantin et al. 2005).
Cetuximab is another mAb that blocks the over-expressed EGF receptor in patients with head and neck as well as colorectal cancers. Cetuximab binding to EGFR blocks the ligand-induced activation of EGFR downstream cell signaling promoting G1-phase arrest and apoptosis. Low doses of cetuximab down regulate HIF-1α, the key transcriptional regulator of tumor glycolysis in epidermoid carcinoma (Luwor et al. 2005). As a consequence, the content of some glycolytic enzymes, glucose consumption and lactate production decreases significantly (Table 1). Although cetuximab induces a significant increase in OxPhos (oligomycin-sensitive respiration), the levels of intracellular ATP diminish by 50 % which probably contribute to the apoptosis induction observed in cetuximab-treated tumor cells.
mAbs directed to tumor over-expressed proteins
Several cancer cells such as type B lymphomas, leukemias and some melanomas maintain highly expressed CD20 in their plasma membrane, which is required for an optimal immune evasive response. CD20 is also involved in the Ca2+ influx across the plasma membrane allowing lymphocyte B-cell activation (Tedder and Engel 1994). Rituximab is a chimeric mAb that recognizes, binds and destroys CD20. However, severe adverse effects such as heart failure, pulmonary toxicity or hepatitis B reactivation leading to fatal outcomes may develop in rituximab-treated patients. Rituximab combined with the bromide analog YM155 (a surviving suppressor) significantly diminishes FDG uptake in WSU-DLC lymphoma (Table 1), which correlates with a marked decrease in the proliferation marker Ki67 (Kita et al. 2012). These results suggest that rituximab interferes with tumor glycolytic metabolism. Furthermore, rituximab alone, or combined with other canonical drugs (cisplatin at 3.3 μM; or cyclophosphamide + bortezomib, 10 nM), induces cytochrome c release and caspase-9 and caspase-3 activation for apoptosis onset, ROS generation and collapse of mitochondrial Δψ (Table 2; Alas et al. 2002; Wang et al. 2008; Eeva et al. 2009).
Phase I, II and III clinical trial new anticancer drugs with severe effects on energy metabolism
The antidiabetes drug metformin (1,1-dimethylbiguanide hydrochloride) has shown potent anti-tumor response in breast, prostate, pancreas and lung carcinomas at similar doses to those required for diabetic patients to reestablish their blood glucose homeostasis (Blandino et al. 2012). Its anti-tumoral effect is related to LKB1/AMPK activation and the subsequent inhibition of mTOR, which in turn suppresses protein synthesis and cell proliferation (Viollet et al. 2012). In several isolated carcinoma cells from breast (MCF-7) and colon (HCT116) and in the human carcinoma-derived cell line (KB cells), metformin at micromolar doses and short-term exposure diminishes total O2 consumption (Table 2; Fig. 4) (Guigas et al. 2004; Buzzai et al. 2007).
Although OxPhos has not been directly evaluated (i.e., measuring the oligomycin-sensitive cellular respiration), it is likely that metformin may affect mitochondrial metabolism as a consequence of its cationic lipophilic chemical nature (Owen et al. 2000). Phenformin (dimethylbiguanide) also affects tumor respiration at micromolar doses. Both phenformin and metformin partially inhibit the activities of the NADH and 2OG dehydrogenases in hepatoma submitochondrial particles without apparent effect on SDH, bc-1, ANT and ATP synthase at similar doses (Gettings et al. 1988; El-Mir et al. 2000; Owen et al. 2000). To explain the selective inhibition of metformin on NAD+-linked dehydrogenases, several reports indicate that the drug (50 μM) diminishes the NADH/NAD+ ratio, compromising the NAD+-dependent enzyme activities. However, the total inhibition of the NADH dehydrogenase activity was observed at 30 mM metformin concentration (Owen et al. 2000; El-Mir et al. 2000; Brunmair et al. 2004), suggesting that the principal target of metformin could occur at the level of the Krebs cycle NADH-producing enzymes. In addition, it has been observed in human fibroblasts and rodent heart myocytes that metformin (0.6–5 mM) increases glycolysis and blocks gluconeogenesis by affecting AMPK and mTOR signaling pathways (Hamann et al. 1993; Fischer et al. 1995).
Mitomycin C is a natural antibiotic used in the treatment of breast and lung cancers (Ardizzoni et al. 1995; Sculier et al. 2001; Mrozek et al. 2008). In established tumor cell lines, mitomycin C diminishes growth of different carcinomas such as lung (SCLC), squamous carcinomas (Ca9-22, HSC-2) and leukemias (HL-60) at relatively low doses (IC50 = 2–37 µM) (Sasaki et al. 2006). Its mechanism of action involves the alkylation of DNA and ROS generation (Paz et al. 2012). Mitomycin C is reduced by the NADPH–cytochrome P-450 reductase in anaerobic conditions (Spanswick et al. 1996); therefore, the drug has been proposed to kill selectively hypoxic tumor cells (Keyes et al. 1985; Cummings et al. 1995). Mitomycin C also collapses mitochondrial Δψ in breast cancer MCF-7 cells, disrupts the mtDNA in rodent carcinoma and inhibits the cellular respiration in Ehrlich ascites cells (Table 2) (Pritsos et al. 1997; Lee et al. 2004). However, in human HL-60 leukemia, similar doses of mitomycin C do not alter mitochondrial morphology (Lee et al. 2004; Sasaki et al. 2006). Also, the drug inhibits tumor glycolysis at micromolar doses (Saito and Kaneko 1968; Table 1).
Letrozole is a third-generation nonsteroidal aromatase inhibitor with, presumably, high specificity for blocking estrogen biosynthesis (den Hollander et al. 2013) and displaying low side effects (Lake and Hudis 2002). This drug is currently used to treat estrogen-receptor-positive breast cancer patients (Haynes et al. 2003; Miller et al. 2012). Letrozole also affects the expression of genes related to the onset of proliferation such as CDC2, CKS2, NUSAP1, PCNA and thymidylate synthetase (Mackay et al. 2007; Miller and Larionov 2011). Chronic treatment (3 months) of letrozole induces severe down-regulation of several OxPhos genes (Table 2). However, there are no data available yet documenting the direct effects of letrozole on protein contents, enzyme activities and flux of OxPhos in cancer cells.
Valproic acid and some of their analogs such as DP-VPA (2-isopropylpentanoic acid–isovaleramide, heptyl-4-yn-valproic acid), inhibit proliferation, increase apoptosis and decrease metastasis and angiogenesis in glioma, breast, colon, prostate and ovarian tumor cells (Takai et al. 2004; Zgouras et al. 2004; Blaheta et al. 2005; Angelucci et al. 2006; Hodges-Gallagher et al. 2007). At the clinical level, valproic acid, in combination with decitabine, cisplatin or topotecan, has demonstrated an effective response in colorectal, melanoma and cervical cancer treatments (Trojnar et al. 2004; Braiteh et al. 2008; Coronel et al. 2011). At higher doses (>200 μM), valproic acid inhibits the ADP-stimulated state three mitochondrial respiration (i.e., OxPhos) in rat liver and behaves as a potent OxPhos uncoupler (Haas et al. 1981). In biopsies from breast cancer patients treated with valproic acid (30 mg/kg daily during 7 days + 21 days more with doxorubicin plus cyclophosphamide, four treatments per day), the drug promoted the up-regulation of at least 89 genes involved in the tumor metabolism; of these, 20 % corresponded to OxPhos genes including flux-controlling OxPhos enzymes such as NADH dehydrogenase (Arce et al. 2006). These observations suggested that epigenetic modification may activate mitochondrial gene expression and OxPhos flux. Alternatively, the drug may directly interact with OxPhos enzymes. Interestingly, opposing effects of valproic acid on energy metabolism in non-tumor cells have been described. In isolated rat hepatocytes, the drug diminished the ATP synthesis (Silva et al. 1997) compromising cellular viability.
Effects of first-line anticancer drugs on cells lacking mtDNA and isolated mitochondria
As discussed throughout this review, it appears that the abrogation of the mitochondrial function is a parallel primary target of several antineoplastic drugs currently used in the clinical practice. In this regard, it has been demonstrated in different tumor (leukemia MOLT-4, fibrosarcoma HT1080 and cervix HeLa) and non-tumor (intestinal epithelium IE6) rho (0) cells that their viability sensitivity toward adaphostin, paclitaxel, cisplatin and adriamycin is lower than in their rho (+) counterparts (Singh et al. 1999; Qian et al. 2005; Le et al. 2007; Patenaude et al. 2007). In MOLT-4 cells, the treatment (10 μM, 30 min) with the preclinical anti-leukemia drug adaphostin, whose action mechanism is associated with ROS production, doubled the mitochondrial ROS production, whereas in rho (0) MOLT-4, adaphostin was unable to increase ROS production (Le et al. 2007). For HT1080 cells treated with paclitaxel (20 nM/16 h), 5-fluorouracil (67 μM/48 h) or vinblastin (20 nM/16 h), 90 % cellular mortality was attained, whereas for rho (0) HT1080 cells the same drug doses induced <30 % cell mortality (Patenaude et al. 2007). In rho (0) HeLa (adriamycin) and in normal rho (0) IE6 cells (cisplatin), the antineoplastic drug was less toxic (IC90 = 1.6 μM and IC50 > 50 μM for HeLa and IE6, respectively) than in their counterparts HeLa (IC90 = 0.75 μM) or IE6 cells (IC50 = 10 μM) (Singh et al. 1999; Qian et al. 2005). In 2008 (ovarian carcinoma) cells rho (0) cytotoxicity to cisplatin was lower (IC50 = 3.5 μM) than in their parental counterparts (IC50 = 0.72 μM) (Montopoli et al. 2011). In contrast, sensitivity to cisplatin (IC50 = 5–6 μM) in tumor cell lines highly dependent on glycolytic metabolism (and presumably with low mitochondrial activity) such as C13 ovarian carcinoma and their rho (0) counterparts was similar (Montopoli et al. 2011). These data strongly suggest that drug cytotoxicity depends on the presence of functional mitochondria.
Furthermore, we have evaluated the effect of the anti-mitochondrial drug Casiopeina II-gly (CasII-gly) (Marín-Hernández et al. 2003, 2012; Hernández-Esquivel et al. 2006; Kachadourian et al. 2010) in combination with first-line canonical antineoplasic drugs in an effort to find chemotherapeutic synergism. Initial results in 24 h drug-treated HeLa cells have revealed that three out of 17 first-line antineoplastic drugs conventionally used at the Instituto Nacional de Cancerología of México induce a synergistic response with CasII-gly, diminishing viability and proliferation rates at nanomolar doses and without apparent effect on normal proliferating cells.
Another set of evidence clearly indicating that OxPhos is a direct target of several antineoplastic drugs is summarized in Table 3. Exposure of isolated mitochondria or permeabilized cells to chemotherapeutic drugs, at similar doses in which their protein- or DNA targets are inhibited, inhibit/activate within seconds or minutes different mitochondrial functions including OxPhos (state 3 respiration), Ca2+ homeostasis, generation and maintenance of mitochondrial ΔΨ, ROS generation, heat generation (i.e., uncoupling) and cytochrome c release (i.e., the initial step for apoptosis). In some of these experiments (Table 3), animals or cells were previously treated with anticancer drug for the indicated times and afterward, mitochondria were isolated or mitochondrial function was determined in permeabilized cells. The results of these last experiments are more difficult to interpret as a direct effect on mitochondrial function, because the long-term interaction between the anticancer drug and its primary target may also induce, indirectly, alterations of the mitochondrial functions. Nevertheless, these observations indicate that the anti-mitochondrial combination therapy (i.e., OxPhos inhibitor plus other canonical or non-canonical anticancer drug) arises as a promising alternative for cancer treatment.
Concluding remarks
Thorough analysis of the information available indicates that the effect of several canonical antineoplastic drugs used as first-line treatment certainly involves inhibition of a particular single specific complex signaling network, but it also involves the strong blocking of OxPhos and, to a lesser extent, glycolysis in tumors (Rodríguez-Enríquez et al. 2000; Marín-Hernández et al. 2006), which in turn could be associated with the severe side effects observed in patients. Therefore, strategies could be redirected to the design of combination therapy including selective OxPhos or/and glycolytic anticancer inhibitors and conventional drug therapy at nanomolar doses to avoid the undesirable side effects. Therefore, the identification of the principal ATP supplier (OxPhos and/or glycolysis) in tumors as the relevant regulator of cancer growth/development emerges as a suitable and strong alternative in the design of new anticancer strategy therapies. This alternative metabolic therapy combined with first-line canonical anticancer drugs may help to improve the traditional therapies.
Abbreviations
- ETC:
-
Electron transport chain
- OxPhos:
-
Oxidative phosphorylation
- 2-OGDH:
-
2-Oxoglutarate dehydrogenase complex
References
Abdel-aleem S, el-Merzabani MM, Sayed-Ahmed M, Taylor DA, Lowe JE (1997) Acute and chronic effects of adriamycin on fatty acid oxidation in isolated cardiac myocytes. J Mol Cell Cardiol 29:789–797. doi:10.1006/jmcc.1996.0323
Aggarwal SK (1993) A histochemical approach to the mechanism of action of cisplatin and its analogues. J Histochem Cytochem 41:1053–1073. doi:10.1177/41.7.8515048
Alas S, Ng CP, Bonavida B (2002) Rituximab modifies the cisplatin-mitochondrial signaling pathway, resulting in apoptosis in cisplatin-resistant non-Hodgkin’s lymphoma. Clin Cancer Res 8:836–845. http://clincancerres.aacrjournals.org/content/8/3/836
Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A et al (2012) Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol 8:839–847. doi:10.1038/nchembio.1060
André N, Braguer D, Brasseur G, Gonçalves A, Lemesle-Meunier D, Guise S, Jordan MA, Briand C (2000) Paclitaxel induces release of cytochrome c from mitochondria isolated from human neuroblastoma cells. Cancer Res 60:5349–5353. http://cancerres.aacrjournals.org/content/60/19/5349
Angelucci A, Valentini A, Millimaggi D, Gravina GL, Miano R, Dolo V, Vicentini C, Bologna M, Federici G, Bernardini S (2006) Valproic acid induces apoptosis in prostate carcinoma cell lines by activation of multiple death pathways. Anticancer Drugs 17:1141–1150. doi:10.1097/01.cad.0000236302.89843.fc
Arce C, Pérez-Plasencia C, González-Fierro A, de la Cruz-Hernández E, Revilla-Vázquez A, Chávez-Blanco A, Trejo-Becerril C, Pérez-Cárdenas E, Taja-Chayeb L, Bargallo E et al (2006) A proof-of-principle study of epigenetic therapy added to neoadjuvant doxorubicin cyclophosphamide for locally advanced breast cancer. PLoS One 1:e98. doi:10.1371/journal.pone.0000098
Ardizzoni A, Addamo GF, Baldini E, Borghini U, Portalone L, De Marinis F, Lionetto R, Conte PF, Bruzzi P, Pennucci MC et al (1995) Mitomycin-ifosfamide-cisplatinum (MIP) vs MIP-interferon vs cisplatinum-carboplatin in metastatic non-small-cell lung cancer: a FONICAP randomised phase II study. Ital Lung Cancer Task Force Br J Cancer 71:115–119. doi:10.1038/bjc.1995.23
Arora A, Scholar EM (2005) Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther 315:971–979. doi:10.1124/jpet.105.084145
Bai S, Nasse MW, Wang SH, Datta J, Kutay H, Yadav A, Nuovo G, Kumar P, Ghoshal K (2009) MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. J Biol Chem 284(46):32015–32027. doi:10.1074/jbc.M109.016774
Baldo BA (2013) Adverse events to monoclonal antibodies used for cancer therapy: focus on hypersensitivity responses. Oncoimmunology 2:e26333. doi:10.4161/onci.26333
Barbone D, Cheung P, Battula S, Busacca S, Gray SG, Longley DB, Bueno R, Sugarbaker DJ, Fennell DA, Broaddus VC (2012) Vorinostat eliminates multicellular resistance of mesothelioma 3D spheroids via restoration of Noxa expression. PLoS One 7:e52753. doi:10.1371/journal.pone.0052753
Barnes K, McIntosh E, Whetton AD, Daley GQ, Bentley J, Baldwin SA (2005) Chronic myeloid leukaemia: an investigation into the role of Bcr-Abl-induced abnormalities in glucose transport regulation. Oncogene 24:3257–3267. doi:10.1038/sj.onc.1208461
Bilbro J, Mart M, Kyprianou M (2013) Therapeutic value of quinazoline-based compounds in prostate cancer. Anticancer Res 33:4695–4700
Blackburn C, Gigstad KM, Hales P, Garcia K, Jones M, Bruzzese FJ, Barrett C, Liu JX, Soucy TA, Sappal DS et al (2010) Characterization of a new series of non-covalent proteasome inhibitors with exquisite potency and selectivity for the 20S beta5-subunit. Biochem J 430:461–476. doi:10.1042/BJ20100383
Blaheta RA, Michaelis M, Driever PH, Cinatl J Jr (2005) Evolving anticancer drug valproic acid: insights into the mechanism and clinical studies. Med Res Rev 25:383–397. doi:10.1002/med.20027
Blandino G, Valerio M, Cioce M, Mori F, Casadei L, Pulito C, Sacconi A, Biagioni F, Cortese G, Galanti S et al (2012) Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat Commun 29:865. doi:10.1038/ncomms1859
Boren J, Cascante M, Marin S, Comín-Anduix B, Centelles JJ, Lim S, Bassilian S, Ahmed S, Lee WN, Boros LG (2001) Gleevec (STI571) influences metabolic enzyme activities and glucose carbon flow toward nucleic acid and fatty acid synthesis in myeloid tumor cells. J Biol Chem 276:37747–37753. doi:10.1074/jbc.M105796200
Boros LG, Lee WN, Cascante M (2002) Imatinib and chronic-phase leukemias. N Engl J Med 347:67–68. doi:10.1056/NEJM200207043470116
Braiteh F, Soriano AO, Garcia-Manero G, Hong D, Johnson MM, Silva LDP, Yang H, Alexander S, Wolff J, Kurzrock R (2008) Phase I study of epigenetic modulation with 5-azacytidine and valproic acid in patients with advanced cancers. Clin Cancer Res 14:6296–62301. doi:10.1158/1078-0432.CCR-08-1247
Brower JV, Lim CH, Han C, Hankowski KE, Hamazaki T, Terada N (2009) Differential CpG island methylation of murine adenine nucleotide translocase genes. Biochim Biophys Acta 1789:198–203. doi:10.1016/j.bbagrm.2008.12.005
Brunmair B, Staniek K, Gras F, Scharf N, Althaym A, Clara R, Roden M, Gnaiger E, Nohl H, Waldhäusl W, Fürnsinn C (2004) Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes 53:1052–1059. doi:10.2337/diabetes.53.4.1052
Bugger H, Guzman C, Zechner C, Palmeri M, Russell KS, Russell RR 3rd (2011) Uncoupling protein downregulation in doxorubicin-induced heart failure improves mitochondrial coupling but increases reactive oxygen species generation. Cancer Chemother Pharmacol 67:1381–1388. doi:10.1007/s00280-010-1441-7
Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB (2007) Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 67:6745–6752. doi:10.1158/0008-5472.CAN-06-4447
Byczkowski JZ, Zychlinski L, Porter CW (1982) Potentiation of the antimitochondrial and antiproliferative effects of bis(guanylhydrazones) by phenethylbiguanide. Cancer Res 42:3592–3595. http://cancerres.aacrjournals.org/content/42/9/3592
Cairns RA, Papandreou I, Sutphin PD, Denko NC (2007) Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc Natl Acad Sci USA 104:9445–9450. doi:10.1073/pnas.0611662104
Candi E, Agostini M, Melino M, Bernassola F (2014) How the TP53 family proteins TP63 and TP73 contribute to tumorigenesis: regulators and effectors. Hum Mutat doi:10.1002/humu.22523
Carvalho C, Correia S, Santos MS, Seiça R, Oliveira CR, Moreira PI (2008) Metformin promotes isolated rat liver mitochondria impairment. Mol Cell Biochem 308:75–83. doi:10.1007/s11010-007-9614-3
Cecconi D, Donadelli M, Dalla Pozza E, Rinalducci S, Zolla L, Scupoli MT, Righetti PG, Scarpa A, Palmieri M (2009) Synergistic effect of trichostatin A and 5-aza-2′-deoxycytidine on growth inhibition of pancreatic endocrine tumour cell lines: a proteomic study. Proteomics 9:1952–1966. doi:10.1002/pmic.200701089
Chan A (2007) A review of the use of trastuzumab (Herceptin) plus vinorelbine in metastatic breast cancer. Ann Oncol 18:1152–1158. doi:10.1093/annonc/mdl476
Chan CL, Wu Z, Ciardelli T, Eastman A, Bresnick E (1993) Kinetic and DNA-binding properties of recombinant human O 6-methylguanine-DNA methyltransferase. Arch Biochem Biophys 300:193–200. doi:10.1006/abbi.1993.1027
Chen Z, Li Y, Zhang H, Huang P, Luthra R (2010) Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 29:4362–4368. doi:10.1038/onc.2010.193
Cherepanova NA, Minero AS, Rakhimova AR, Gromova ES (2011) Mechanism of CpG DNA methyltransferases M. SssI and Dnmt3a studied by DNA containing 2-aminopurine. Nucleosides, Nucleotides Nucleic Acids 30:619–631. doi:10.1080/15257770.2011.583973
Chesney J, Telang S (2013) Regulation of glycolytic and mitochondrial metabolism by ras. Curr Pharm Biotechnol 14:251–260. doi:10.2174/1389201011314030002
Cheung KJ, Gabrielson E, Werb Z, Ewald A (2013) Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 155:1639–1651. doi:10.1016/j.cell.2013.11.029
Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L, Woulfe K, Pravda E, Cassiola F, Desai J et al (2007) Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 370:2011–2019. doi:10.1016/S0140-6736(07)61865-0
Clark SJ, Melki J (2002) DNA methylation and gene silencing in cancer: which is the guilty party? Oncogene 21:5380–5387. doi:10.1038/sj.onc.1205598
Coronel J, Cetina L, Pacheco I, Trejo-Becerril C, González-Fierro A, de la Cruz-Hernandez E, Perez-Cardenas E, Taja-Chayeb L, Arias-Bofill D, Candelaria M et al (2011) A double-blind, placebo-controlled, randomized phase III trial of chemotherapy plus epigenetic therapy with hydralazine valproate for advanced cervical cancer. Preliminary results. Med Oncol 28:S540–S546. doi:10.1007/s12032-010-9700-3
Cuéllar A, Escamilla E, Ramírez J, Chávez E (1984) Adriamycin as an inhibitor of 11 beta-hydroxylase activity in adrenal cortex mitochondria. Arch Biochem Biophys 235:538–543. doi:10.1016/0003-9861(84)90227-3
Cummings J, Spanswick VJ, Smyth JF (1995) Re-evaluation of the molecular pharmacology of mitomycin C. Eur J Cancer 31A:1928–1933. doi:10.1016/0959-8049(95)00364-9
Custódio JB, Cardoso CM, Almeida LM (2002) Thiol protecting agents and antioxidants inhibit the mitochondrial permeability transition promoted by etoposide: implications in the prevention of etoposide-induced apoptosis. Chem Biol Interact 140:169–184. doi:10.1016/S0009-2797(2)00020-0
Dang CV, Le A, Gao P (2009) MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res 15:6479–6483
Dao T, Yan S, Veomett N, Pankov D, Zhou L, Korontsvit T, Scott A, Whitten J, Maslak P, Casey E, et al (2013) Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl Med 5:176ra33. doi:10.1126/scitranslmed.3005661
den Hollander P, Savage MI, Brown PH (2013) Targeted Therapy for Breast Cancer Prevention. Front Oncol 3:250. doi:10.1158/1078-0432.CCR-09-0889
Ding Y, Liu Z, Desai S, Zhao Y, Liu H, Pannell LK, Yi H, Wright ER, Owen LB, Dean-Colomb W et al (2012) Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat Commun 3:1271. doi:10.1038/ncomms2236
Diotte NM, Xiong Y, Gao J, Chua BH, Ho YS (2009) Attenuation of doxorubicin-induced cardiac injury by mitochondrial glutaredoxin 2. Biochim Biophys Acta 1793:427–438. doi:10.1016/j.bbamcr.2008.10.014
Drahota Z, Palenickova E, Endlicher R, Milerova M, Brejchova J, Vosahlikova M, Svoboda P, Kazdova L, Kalous M, Cervinkova Z, Cahova M (2013) Biguanides inhibit complex I, II and IV of rat liver mitochondria and modify their functional properties. Physiol Res 63:1–11
Dykens JA, Jamieson J, Marroquin L, Nadanaciva S, Billis PA, Will Y (2008) Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol Appl Pharmacol 233:203–210. doi:10.1016/j.taap.2008.08.013
Eeva J, Nuutinen U, Ropponen A, Mättö M, Eray M, Pellinen R, Wahlfors J, Pelkonen J (2009) The involvement of mitochondria and the caspase-9 activation pathway in rituximab-induced apoptosis in FL cells. Apoptosis 14:687–698. doi:10.1007/s10495-009-0337-7
El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X (2000) Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 275:223–228. http://www.jbc.org/content/275/1/223
Esteller M (2007) Epigenetics provides a new generation of oncogenes and tumour-suppressor genes. Br J Cancer 96:R26–R30. doi:10.1038/sj.bjc.6602918
Fabian MA, Biggs WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M et al (2005) A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol 23:329–336. doi:10.1038/nbt1068
Fantin VR, Berardi MJ, Babbe H, Michelman MV, Manning CM, Leder P (2005) A bifunctional targeted peptide that blocks HER-2 tyrosine kinase and disables mitochondrial function in HER-2-positive carcinoma cells. Cancer Res 65:6891–6900. http://cancerres.aacrjournals.org/content/65/15/6891
Faure-Vigny H, Heddi A, Giraud S, Chautard D, Stepien G (1996) Expression of oxidative phosphorylation genes in renal tumors and tumoral cell lines. Mol Carcinog 16:165–172. doi:10.1002/(SICI)1098-2744(199607
Ferlini C, Raspaglio G, Mozzetti S, Distefano M, Filippetti F, Martinelli E, Ferrandina G, Gallo D, Ranelletti FO, Scambia G (2003) Bcl-2 down-regulation is a novel mechanism of paclitaxel resistance. Mol Pharmacol 64:51–58. doi:10.1124/mol.64.1.51
Fischer Y, Thomas J, Rösen P, Kammermeier H (1995) Action of metformin on glucose transport and glucose transporter GLUT1 and GLUT4 in heart muscle cells from healthy and diabetic rats. Endocrinology 136:412–420. doi:10.1210/endo.136.2.7835271
Fiume L, Manerba M, Vettraino M, Di Stefano G (2011) Effect of sorafenib on the energy metabolism of hepatocellular carcinoma cells. Eur J Pharmacol 670:39–43. doi:10.1016/j.ejphar.2011.08.038
Floridi A, D’Atri S, Bellocci M, Marcante ML, Paggi MG, Silvestrini B, Caputo A, De Martino C (1984) The effect of gossypol and Lonidamine on electron transport in Ehrlich ascites tumor mitochondria. Exp Mol Pathol 40:246–261. doi:10.1016/0014-4800(84)90081-9
Funasaka T, Hogan V, Raz A (2009) Phosphoglucose isomerase/autocrine motility factor mediates epithelial and mesenchymal phenotype conversions in breast cancer. Cancer Res 69:5349–5356. doi:10.1158/0008-5472.CAN-09-0488
Gallardo-Pérez JC, Rivero-Segura NA, Marín-Hernández A, Moreno-Sánchez R, Rodríguez-Enríques S (2014) GPI/AMF inhibition blocks the metastatic progression of mature multicellular tumor spheroids. Biochim Biophys Acta 1843:1043–1053. doi:10.1016/j.bbamcr.2014.01.013
Gaona-Gaona L, Molina-Jijón E, Tapia E, Zazueta C, Hernández-Pando R, Calderón-Oliver M, Zarco-Márquez G, Pinzón E, Pedraza-Chaverri J (2011) Protective effect of sulforaphane pretreatment against cisplatin-induced liver and mitochondrial oxidant damage in rats. Toxicology 286:20–27. doi:10.1016/j.tox.2011.04.014
Garrido N, Pérez-Martos A, Faro M, Lou-Bonafonte JM, Fernández-Silva P, López-Pérez MJ, Montoya J, Enríquez JA (2008) Cisplatin-mediated impairment of mitochondrial DNA metabolism inversely correlates with glutathione levels. Biochem J 414:93–102. doi:10.1042/BJ20071615
Gettings SD, Reeve JE, King LJ (1988) Possible role of intracellular Ca2+ in the toxicity of phenformin. Biochem Pharmacol 37:281–289. doi:10.1016/0006-2952(88)90730-7
Giacobbe A, Bongiorno-Borbone L, Bernassola F, Terrinoni A, Markert EK, Levine AJ, Feng Z, Agostini M, Zolla L, Agro AF, Notterman DA, Melino G, Peschiaroli A (2013) p63 regulates glutaminase 2 expression. Cell Cycle 12:1395–1405. doi:10.4161/cc.24478
Gillies RJ, Robey I, Gatenby RA (2008) Causes and consequences of increased glucose metabolism of cancers. J Nucl Med 49:24S–42S. doi:10.2967/jnumed.107.047258
Gottschalk S, Anderson N, Hainz C, Eckhardt SG, Serkova NJ (2004) Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clin Cancer Res 10:6661–6668. doi:10.1158/1078-0432.CCR-04-0039
Gowher H, Jeltsch A (2004) Mechanism of inhibition of DNA methyltransferases by cytidine analogs in cancer therapy. Cancer Biol Ther 3:1062–1068. doi:10.4161/cbt.3.11.1308
Grazette LP, Boecker W, Matsui T, Semigran M, Force TL, Hajjar RJ, Rosenzweig A (2004) Inhibition of ErbB2 causes mitochondrial dysfunction in cardiomyocytes: implications for herceptin-induced cardiomyopathy. J Am Coll Cardiol 44:2231–2238. doi:10.1016/j.jacc.2004.08.066
Guerrero-Beltrán CE, Calderón-Oliver M, Martínez-Abundis E, Tapia E, Zarco-Márquez G, Zazueta C, Pedraza-Chaverri J (2010) Protective effect of sulforaphane against cisplatin-induced mitochondrial alterations and impairment in the activity of NAD(P)H: quinone oxidoreductase 1 and γ glutamyl cysteine ligase: studies in mitochondria isolated from rat kidney and in LLC-PK1 cells. Toxicol Lett 199:80–92. doi:10.1016/j.toxlet.2010.08.009
Guigas B, Detaille D, Chauvin C, Batandier C, De Oliveira F, Fontaine E, Leverve X (2004) Metformin inhibits mitochondrial permeability transition and cell death: a pharmacological in vitro study. Biochem J 382:877–884. doi:10.1042/BJ20040885
Haas R, Stumpf DA, Parks JK, Eguren L (1981) Inhibitory effects of sodium valproate on oxidative phosphorylation. Neurology 31:1473–1476. doi:10.1212/WNL.31.11.1473
Hamann A, Benecke H, Greten H, Hatthaei S (1993) Metformin increases glucose transporter protein and gene expression in human fibroblasts. Biochem Biophys Res Commun 196:382–387. doi:10.1006/bbcr.193.2260
Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ (2010) The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov 9:325–338. doi:10.1038/nrd3003
Haynes BP, Dowsett M, Miller WR, Dixon JM, Bhatnagar AS (2003) The pharmacology of letrozole. J Steroid Biochem Mol Biol 87:35–45. doi:10.1016/S0960-0760(03)00384-4
Hegde PS, Rusnak D, Bertiaux M, Alligood K, Strum J, Gagnon R, Gilmer TM (2007) Delineation of molecular mechanisms of sensitivity to lapatinib in breast cancer cell lines using global gene expression profiles. Mol Cancer Ther 6:1629–1640. doi:10.1158/1535-7163.MCT-05-0399
Hernández-Esquivel L, Marín-Hernández A, Pavón N, Carvajal K, Moreno-Sánchez R (2006) Cardiotoxicity of copper-based antineoplastic drugs casiopeinas is related to inhibition of energy metabolism. Toxicol Appl Pharmacol 212:79–88. doi:10.1016/j.taap.2005.06.023
Hickey FB, Cotter TG (2006) Identification of transcriptional targets associated with the expression of p210 Bcr-Abl. Eur J Haematol 76:369–383. doi:10.1111/j.1600-0609.2006.00629.x
Hirayama A, Kami K, Sugimoto M, Sugawara M, Toki N, Onozuka H, Kinoshita T, Saito N, Ochiai A, Tomita M et al (2009) Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res 69:4918–4925. doi:10.1158/0008-5472.CAN-08-4806
Hirschhaeuser F, Menne H, Dittfeld C, West J, Mueller-Klieser W, Kunz-Schughart LA (2010) Multicellular tumor spheroids: an underestimated tool is catching up again. J Biotechnol 148:3–15. doi:10.1016/j.jbiotec.2010.01.012
Hirschhaeuser F, Sattler UG, Mueller-Klieser W (2011) Lactate: a metabolic keyplayer in cancer. Cancer Res 71:6921–6925. doi:10.1158/0008-5472.CAN-11-1457
Höckel M, Vaupel P (2001) Biological consequences of tumor hypoxia. Semin Oncol 28:36–41. http://www.seminoncol.org/article/S0093-7754(01)90211-8/abstract
Hodges-Gallagher L, Valentine CD, Bader SE, Kushner PJ (2007) Inhibition of histone deacetylase enhances the anti-proliferative action of antiestrogens on breast cancer cells and blocks tamoxifen-induced proliferation of uterine cells. Breast Cancer Res Treat 105:297–309. doi:10.1007/s10549-006-9459-6
Iacobazzi V, Infantino V, Palmieri F (2008) Epigenetic mechanisms and Sp1 regulate mitochondrial citrate carrier gene expression. Biochem Biophys Res Commun 376:15–20. doi:10.1016/j.bbrc.2008.08.015
Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, Ratcliffe PJ, Linehan WM, Neckers L (2005) HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8:143–153. doi:10.1016/j.ccr.2005.06.017
Kachadourian R, Brechbuhl HM, Ruiz-Azuara L, Gracia-Mora I, Day BJ (2010) Casiopeína IIgly-induced oxidative stress and mitochondrial dysfunction in human lung cancer A549 and H157 cells. Toxicology 268:176–183. doi:10.1016/j.tox.2009.12.010
Keller KE, Doctor ZM, Dwyer ZW, Lee YS (2014) SAICAR induces protein kinase activity of PKM2 that is necessary for sustained proliferative signaling of cancer cells. Mol Cell 53:700–709. doi:10.1016/j.molcel.2014.02.015
Kessler T, Bayer M, Schwöppe C, Liersch R, Mesters RM, Berdel WE (2010) Compounds in clinical phase III and beyond. In: Liersch R, Berdel WE, Kessler T (eds) Angiogenesis inhibition. Springer, London, pp 137–163
Keunen O, Johansson M, Oudin A, Sanzey M, Rahim SA, Fack F, Thorsen F, Taxt T, Bartos M, Jirik R et al (2011) Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc Natl Acad Sci USA 108:3749–3754. doi:10.1073/pnas.1014480108
Keyes SR, Heimbrook DC, Fracasso PM, Rockwell S, Sligar SG, Sartorelli AC (1985) Chemotherapeutic attack of hypoxic tumor cells by the bioreductive alkylating agent mitomycin C. Adv Enzyme Regul 23:291–307. doi:10.1016/0065-2571(85)90053-6
Kibayashi M, Nagao M, Chiba S (1999) Influence of valproic acid on the expression of various acyl-CoA dehydrogenases in rats. Pediatr Int 41:52–60. doi:10.1046/j.1442-200x.1999.01012.x
Kim MJ, Kim DH, Jung WH, Koo JS (2014) Expression of metabolism-related proteins in triple-negative breast cancer. Int J Clin Exp Pathol 7:301–312. www.ijcep.com. ISSN 1936-2625/IJCEP1311042
Kita A, Mitsuoka K, Kaneko N, Nakata M, Yamanaka K, Jitsuoka M, Miyoshi S, Noda A, Mori M, Nakahara T, Sasamata M (2012) Sepantronium bromide (YM155) enhances response of human B-cell non-Hodgkin lymphoma to rituximab. J Pharmacol Exp Ther 343:178–183. doi:10.1124/jpet.112.195925
Klawitter J, Anderson N, Klawitter J, Christians U, Leibfritz D, Eckhardt SG, Serkova NJ (2009) Time-dependent effects of imatinib in human leukaemia cells: a kinetic NMR-profiling study. Br J Cancer 100:923–931. doi:10.1038/sj.bjc.6604946
Kluza J, Jendoubi M, Ballot C, Dammak A, Jonneaux A, Idziorek T, Joha S, Dauphin V, Malet-Martino M et al (2011) Exploiting mitochondrial dysfunction for effective elimination of imatinib-resistant leukemic cells. PLoS One 6:e21924. doi:10.1371/journal.pone.0021924
Koh MY, Spivak-Kroizman TR, Powis G (2010) HIF-1α and cancer therapy. In: Liersch R, Berdel WE, Kessler T (eds) Angiogenesis inhibition. Springer, London, pp 15–34
Kong M, Ba M, Liang H, Ma L, Yu Q, Yu T, Wang Y (2012) 5′-Aza-dC sensitizes paraquat toxic effects on PC12 cell. Neurosci Lett 524:35–39. doi:10.1016/j.neulet.2012.07.001
Ksienski D (2011) Imatinib mesylate: past successes and future challenges in the treatment of gastrointestinal stromal tumors. Clin Med Insights Oncol 5:365–379. doi:10.4137/CMO.S4259
Kunz-Schughart LA (1999) Multicellular tumor spheroids: intermediates between monolayer culture and in vivo tumor. Cell Biol Int 23:157–161. doi:10.1177/1087057104265040
Kunz-Schughart LA, Freyer JP, Hofstaedter F, Ebner R (2004) The use of 3-D cultures for high-throughput screening: the multicellular spheroid model. J Biomol Screen 9:228–273. doi:10.1177/1087057104265040
Lake DE, Hudis C (2002) Aromatase inhibitors in breast cancer: an update. Cancer Control 9:490-498. http://moffitt.org/File%20Library/Main%20Nav/Research%20and%20Clinical%20Trials/Cancer%20Control%20Journal/v9n6/490.pdf
Le SB, Hailer MK, Buhrow S, Wang Q, Flatten K, Pediaditakis P, Bible KC, Lewis LD, Sausville EA, Pang YP, Ames MM et al (2007) Inhibition of mitochondrial respiration as a source of adaphostin-induced reactive oxygen species and cytotoxicity. J Biol Chem 282:8860–8872. doi:10.1074/jbc.M611777200
Lee SJ, Wang JY (2009) Exploiting the promiscuity of imatinib. J Biol. 8:30. doi:10.1186/jbiol134
Lee CS, Park SY, Ko HH, Han ES (2004) Effect of change in cellular GSH levels on mitochondrial damage and cell viability loss due to mitomycin c in small cell lung cancer cells. Biochem Pharmacol 68:1857–1867. doi:10.1016/j.bcp.2004.06010
Li GN, Wang SP, Xue X, Qu XJ, Liu HP (2013) Monoclonal antibody-related drugs for cancer therapy. Drug Discov Ther 7:178–184. doi:10.5582/ddt.2013.v7.5.178
Lin YL, Meng Y, Jiang W, Roux B (2013) Explaining why Gleevec is a specific and potent inhibitor of Abl kinase. Proc Natl Acad Sci USA 110:1664–1669. doi:10.1073/pnas.1214330110
Liu W, Phang JM (2012) Proline dehydrogenase (oxidase), a mitochondrial tumor suppressor, and autophagy under the hypoxia microenvironment. Autophagy 8:1407–1409
Liu FT, Agrawal SG, Gribben JG, Ye H, Du MQ, Newland AC, Jia L (2008) Bortezomib blocks Bax degradation in malignant B cells during treatment with TRAIL. Blood 111:2797–2805
Liu W, Glunde K, Bhujwalla ZM, Raman V, Sharma A, Phang JM (2012) Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. Cancer Res 72:3677–3686
Liu AM, Xu Z, Shek FH, Wong KF, Lee NP, Poon RT, Chen J, Luk JM (2014) miR-122 targets pyruvate kinase M2 and affects metabolism of hepatocellular carcinoma. PLoS ONE 9:e86872
Lu H, Li X, Luo Z, Liu J, Fan Z (2013) Cetuximab reverses the Warburg effect by inhibiting HIF-1-regulated LDH-A. Mol Cancer Ther 12:2187–2199. doi:10.1158/0008-5472.CAN-12-0080
Luwor RB, Lu Y, Li X, Mendelsohn J, Fan Z (2005) The antiepidermal growth factor receptor monoclonal antibody cetuximab/C225 reduces hypoxia-inducible factor-1 alpha, leading to transcriptional inhibition of vascular endothelial growth factor expression. Oncogene 24:4433–4441. doi:10.1038/sj.onc.1208625
Ma WW, Jacene H, Song D, Vilardell F, Messersmith WA, Laheru D, Wahl R, Endres C, Jimeno A, Pomper MG, Hidalgo M (2009) [18F]fluorodeoxyglucose positron emission tomography correlates with Akt pathway activity but is not predictive of clinical outcome during mTOR inhibitor therapy. J Clin Oncol 27:2697–2704. doi:10.1200/JCO.2008.18.8383
Mackay A, Urruticoechea A, Dixon JM, Dexter T, Fenwick K, Ashworth A, Drury S, Larionov A, Young O, White S et al (2007) Molecular response to aromatase inhibitor treatment in primary breast cancer. Breast Cancer Res 9:R37. doi:10.1186/bcr1732
Mandujano-Tinoco EA, Gallardo-Pérez JC, Marín-Hernández A, Moreno-Sánchez R, Rodríguez-Enríquez S (2013) Anti-mitochondrial therapy in human breast cáncer multi-cellular spheroids. Biochim Biophys Acta 1833:541–551. doi:10.1016/j.bbamcr.2012.11.013
Marín-Hernández A, Gracia-Mora I, Ruiz-Ramírez L, Moreno-Sánchez R (2003) Toxic effects of copper-based antineoplastic drugs (Casiopeinas) on mitochondrial functions. Biochem Pharmacol 65:1979–1989. doi:10.1016/S0006-2952(03)00212-0
Marín-Hernández A, Rodríguez-Enríquez S, Vital-González PA, Flores-Rodríguez FL, Macías-Silva M, Sosa-Garrocho M, Moreno-Sánchez R (2006) Determining and understanding the control of glycolysis in fast-growth tumor cells. Flux control by an over-expressed but strongly product-inhibited hexokinase. FEBS J 273:1975–1988. doi:10.1111/j.1742-4658.2006.05214.x
Marín-Hernández A, Gallardo-Pérez JC, Ralph SJ, Rodríguez-Enríquez S, Moreno-Sánchez R (2009) HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem 9:1084–1101. doi:10.2174/138955709788922610
Marín-Hernández A, Gallardo-Pérez JC, Rodríguez-Enríquez S, Encalada R, Moreno-Sánchez R, Saavedra E (2011) Modeling cancer glycolysis. Biochim Biophys Acta 1807:755–767. doi:10.1016/j.bbabio.2010.11.006
Marín-Hernández A, Gallardo-Pérez JC, López-Ramírez SY, García-García JD, Rodríguez-Zavala JS, Ruiz-Ramírez L, Gracia-Mora I, Zentella-Dehesa A, Sosa-Garrocho M, Macías-Silva M, Moreno-Sánchez R, Rodríguez-Enríquez S (2012) Casiopeina II-gly and bromo-pyruvate inhibition of tumor hexokinase, glycolysis, and oxidative phosphorylation. Arch Toxicol 86:753–766. doi:10.1007/s00204-012-0809-3
Masdehors P, Merle-Béral H, Maloum K, Omura S, Magdelénat H, Delic J (2000) Deregulation of the ubiquitin system and p53 proteolysis modify the apoptotic response in B-CLL lymphocytes. Blood 96:269–274. http://bloodjournal.hematologylibrary.org/content/96/1/269.long
Mathews EH, Liebenberg L, Pelzer R (2011) High-glycolytic cancers and their interplay with the body’s glucose demand and supply cycle. Med Hypotheses 76:157–165. doi:10.1016/j.mehy.2010.09.006
Mazieres J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, Besse B, Blons H, Mansuet-Lupo A, Urban T et al (2013) Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol 31:1997–2003. doi:10.1200/JCO.2012.45.6095
Mazurek S (2011) Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol 43(2011):969–980
Mellor HR, Bell AR, Valentin JP, Roberts RR (2011) Cardiotoxicity associated with targeting kinase pathways in cancer. Toxicol Sci 120:14–32. doi:10.1093/toxsci/kfq378
Mendelsohn J, Baselga J (2000) The EGF receptor family as targets for cancer therapy. Oncogene 19:6550–6565. doi:10.1038/sj.onc.1204082
Meng F, Sun G, Zhong M, Yu Y, Brewer MA (2013) Anticancer efficacy of cisplatin and trichostatin A or 5-aza-2′-deoxycytidine on ovarian cancer. Br J Cancer 108:579–586. doi:10.1038/bjc.2013.10
Menon C, Polin GM, Prabakaran I, Hsi A, Cheung C, Culver JP, Pingpank JF, Sehgal CS, Yodh AG, Buerk DG et al (2003) An integrated approach to measuring tumor oxygen status using human melanoma xenografts as a model. Cancer Res 63:7232–7240. http://cancerres.aacrjournals.org/content/63/21/7232
Miller WR, Larionov A (2011) Molecular effects of oestrogen deprivation in breast cancer. Mol Cell Endocrinol 340:127–136. doi:10.1097/FPC.0b013e32820b853a
Miller WR, Larionov AA, Renshaw L, Anderson TJ, White S, Murray J, Murray E, Hampton G, Walker JR, Ho S et al (2007) Changes in breast cancer transcriptional profiles after treatment with the aromatase inhibitor, letrozole. Pharmacogenet Genomics 17:813–826
Miller WR, Larionov A, Anderson TJ, Evans DB, Dixon JM (2012) Sequential changes in gene expression profiles in breast cancers during treatment with the aromatase inhibitor, letrozole. Pharmacogenomics J 12:10–21. doi:10.1038/tpj.2010.67
Miller MJ, Foy KC, Kaumaya PT (2013) Cancer immunotherapy: present status, future perspective, and a new paradigm of peptide immunotherapeutics. Discov Med 15:166–176. http://www.discoverymedicine.com/Megan-Jo-Miller/2013/03/28/cancer-immunotherapy-present-status-future-perspective-and-a-new-paradigm-of-peptide-immunotherapeutics/
Mironov SL, Ivannikov MV, Johansson M (2005) [Ca2+]i signaling between mitochondria and endoplasmic reticulum in neurons is regulated by microtubules. From mitochondrial permeability transition pore to Ca2+-induced Ca2+ release. J Biol Chem 280:715–721. doi:10.1074/jbc.M409819200
Montopoli M, Bellanda M, Lonardoni F, Ragazzi E, Dorigo P, Froldi G, Mammi S, Caparrotta L (2011) “Metabolic reprogramming” in ovarian cancer cells resistant to cisplatin. Curr Cancer Drug Targets 11:226–235. doi:10.2174/156800911794328501
Moreira PI, Custódio J, Moreno A, Oliveira CR, Santos MS (2006) Tamoxifen and estradiol interact with the flavin mononucleotide site of complex I leading to mitochondrial failure. J Biol Chem 281:10143–10152. doi:10.1074/jbc.M510249200
Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E (2007) Energy metabolism in tumor cells. FEBS J 274:1393–1418. doi:10.1111/j.1742-4658.2007.05686.x
Moreno-Sánchez R, Marín-Hernández A, Saavedra E, Pardo JP, Ralph SJ, Rodríguez-Enríquez S (2014) Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int J Biochem Cell Biol 2725:42–49. doi:10.1016/j.biocel.2014.01.025
Mrozek E, Kolesar J, Young D, Allen J, Villalona-Calero M, Shapiro CL (2008) Phase II study of sequentially administered low-dose mitomycin-C(MMC) and irinotecan (CPT-11) in women with metastatic breast cancer (MBC). Ann Oncol 19:1417–1422. doi:10.1093/annonc/mdn154
Müller M, Siems W, Buttgereit F, Dumdey R, Rapoport SM (1986) Quantification of ATP-producing and consuming processes of Ehrlich ascites tumour cells. Eur J Biochem 161:701–705. doi:10.1111/j.1432-1033.1986.tb10496.x
Nicolay K, Timmers RJ, Spoelstra E, Van der Neut R, Fok JJ, Huigen YM, Verkleij AJ, De Kruijff B (1984) The interaction of adriamycin with cardiolipin in model and rat liver mitochondrial membranes. Biochim Biophys Acta 778:359–371. doi:10.1016/0005-2736(84)90380-8
Nowis D, Maczewski M, Mackiewicz U, Kujawa M, Ratajska A, Wieckowski MR, Wilczyński GM, Malinowska M, Bil J, Salwa P et al (2010) Cardiotoxicity of the anticancer therapeutic agent bortezomib. Am J Pathol 176:2658–2668. doi:10.2353/ajpath.2010.090690
Ott PA, Adams S (2011) Small-molecule protein kinase inhibitors and their effects on the immune system: implications for cancer treatment. Immunotherapy 3:213–227. doi:10.2217/imt.10.99
Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S (2002) Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA 99:1259–1263. doi:10.1073/pnas.241655498
Owen MR, Doran E, Halestrap AP (2000) Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348:607–614. doi:10.1042/0264-6021:3480607
Papadopoulos N, Martin J, Ruan Q, Rafique A, Rosconi MP, Shi E, Pyles EA, Yancopoulos GD, Stahl N, Wiegand SJ (2012) Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis 15:171–185. doi:10.1007/s10456-011-9249-6
Papanikolaou V, Iliopoulos D, Dimou I, Dubos S, Kappas C, Kitsiou-Tzeli S, Tsezou A (2011) Survivin regulation by HER2 through NF-κB and c-myc in irradiated breast cancer cells. J Cell Mol Med 15:1542–1550. doi:10.1111/j.1582-4934.2010.01149.x
Park JH, Kim TH (2005) Release of cytochrome c from isolated mitochondria by etoposide. J Biochem Mol Biol 38:619–623. http://europepmc.org/abstract/MED/16202244
Patenaude A, Deschesnes RG, Rousseau JL, Petitclerc E, Lacroix J, Côté MF, C-Gaudreault R (2007) New soft alkylating agents with enhanced cytotoxicity against cancer cells resistant to chemotherapeutics and hypoxia. Cancer Res 67:2306–2316. doi:10.1158/0008-5472.CAN-06-3824
Pathania D, Sechi M, Palomba M, Sanna V, Berretini F, Sias A, Taheri L, Neamati N (2014) Design and discovery of novel quinazolinedione-based redox modulators as therapies for pancreatic cancer. Biochim Biophys Acta 1840:332–343. doi:10.1016/j.bbagen.2013.08.005
Paz MM, Zhang X, Lu J, Holmgren A (2012) A new mechanism of action for the anticancer drug mitomycin C: mechanism-based inhibition of thioredoxin reductase. Chem Res Toxicol 25:1502–1511. doi:10.1021/tx3002065
Piperdi B, Ling YH, Liebes L, Muggia F, Perez-Soler R (2011) Bortezomib: understanding the mechanism of action. Mol Cancer Ther 10:2029–2030. doi:10.1158/1535-7163.MCT-11-0745
Polivka J Jr, Janku F (2014) Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther 142:164–175. doi:10.1016/j.pharmthera.2013.12.004
Pollard PJ, Brière JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ et al (2005) Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 14:2231–2239. doi:10.1093/hmg/ddi227
Prenzel N, Fischer OM, Streit S, Hart S, Ullrich A (2001) The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr Relat Cancer 8:11-31. doi:10.1677/erc.0.0080011
Pritsos CA, Briggs LA, Gustafson DL (1997) A new cellular target for mitomycin C: a case for mitochondrial DNA. Oncol Res 9:333–337. http://www.biomedsearch.com/nih/new-cellular-target-mitomycin-C/9406239.html
Qian W, Nishikawa M, Haque AM, Hirose M, Mashimo M, Sato E, Inoue M (2005) Mitochondrial density determines the cellular sensitivity to cisplatin-induced cell death. Am J Physiol Cell Physiol 289:C1466–C1475. doi:10.1152/ajpcell.00265.2005
Radu M, Semenova G, Kosoff R, Chernoff J (2014) PAK signaling during the development and progression of cancer. Nat Rev Cancer 14:13–25. doi:10.1038/nrc3645
Rix U, Hantschel O, Dürnberger G, Remsing Rix LL, Planyavsky M, Fernbach NV, Kaupe I, Bennett KL, Valent P, Colinge J, Köcher T, Superti-Furga G (2007) Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 110:4055–4063. doi:10.1182/blood-2007-07-102061
Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S (2000) Distinct pathways for stimulation of cytochrome c release by etoposide. J Biol Chem 275:32438–32443. doi:10.1074/jbc.C000518200
Rodríguez-Enríquez S, Torres-Márquez ME, Moreno-Sánchez R (2000) Substrate oxidation and ATP supply in AS-30D hepatoma cells. Arch Biochem Biophys 375:21–30. doi:10.1006/abbi.1999.1582
Rodríguez-Enríquez S, Vital-González PA, Flores-Rodríguez FL, Marín-Hernández A, Ruiz-Azuara L, Moreno-Sánchez R (2006) Control of celullar proliferation by modulation of oxidative phosphorylation in human and rodent fast-growing tumor cells. Toxicol Appl Pharmacol 215:208–217. doi:10.1016/j.taap.2006.02.005
Rodríguez-Enríquez S, Gallardo-Pérez JC, Avilés-Salas A, Marín-Hernández A, Carreño-Fuentes L, Maldonado-Lagunas V, Moreno-Sánchez R (2008) Energy metabolism transition in multi-cellular human tumor spheroids. J Cell Physiol 216:189–197. doi:10.1002/jcp.21392
Rodríguez-Enríquez S, Marín-Hernández A, Gallardo-Pérez JC, Carreño-Fuentes L, Moreno-Sánchez R (2009) Targeting of cancer energy metabolism. Mol Nutr Food Res 53:29–48. doi:10.1002/mnfr.200700470
Rodríguez-Enríquez S, Carreño-Fuentes L, Gallardo-Pérez JC, Saavedra E, Quezada H, Vega A, Marín-Hernández A, Olín-Sandoval V, Torres-Márquez ME, Moreno-Sánchez R (2010) Oxidative phosphorylation is impaired by prolonged hypoxia in breast and possibly in cervix carcinoma. Int J Biochem Cell Biol 42:1744–1751. doi:10.1016/j.biocel.2010.07.010
Rodríguez-Enríquez S, Gallardo-Pérez JC, Marín-Hernández A, Aguilar-Ponce JL, Mandujano-Tinoco EA, Meneses A, Moreno-Sánchez R (2011) Oxidative phosphorylation as a target to arrest malignant neoplasias. Curr Med Chem 18:3156–3167. doi:10.2174/092986711796391561
Rolfe DF, Brown GC (1997) Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev 77:731–758. http://physrev.physiology.org/content/77/3/731.long
Rumbach L, Warter JM, Rendon A, Marescaux C, Micheletti G, Waksman A (1983) Inhibition of oxidative phosphorylation in hepatic and cerebral mitochondria of sodium valproate-treated rats. J Neurol Sci 61:417–423. doi:10.1016/0022-510X(83)90174-0
Saito T, Kaneko T (1968) Effects of simultaneous application of mitomycin C and prednisolone on respiration and glycolysis of Ehrlich ascites tumor cells in vitro. Tohoku J Exp Med 95:87–106. doi:10.1620/tjem.95.87
Sasaki M, Okamura M, Ideo A, Shimada J, Suzuki F, Ishihara M, Kikuchi H, Kanda Y, Kunii S, Sakagami H (2006) Re-evaluation of tumor-specific cytotoxicity of mitomycin C, bleomycin and peplomycin. Anticancer Res 26:3373–3380. doi:10.1126/scitranslmed.300765
Schmelz K, Wagner M, Dörken B, Tamm I (2005) 5-Aza-2′-deoxycytidine induces p21WAF expression by demethylation of p73 leading to p53-independent apoptosis in myeloid leukemia. Int J Cancer 114:683–695. doi:10.1002/ijc.20797
Schmidt H, Siems W, Müller M, Dumdey R, Rapoport SM (1991) ATP-producing and consuming processes of Ehrlich mouse ascites tumor cells in proliferating and resting phases. Exp Cell Res 94:122–127
Schnekenburger M, Grandjenette C, Ghelfi J, Karius T, Foliguet B, Dicato M, Diederich M (2011) Sustained exposure to the DNA demethylating agent, 2′-deoxy-5-azacytidine, leads to apoptotic cell death in chronic myeloid leukemia by promoting differentiation, senescence, and autophagy. Biochem Pharmacol 81:364–378. doi:10.1016/0014-4827(91)90140-P
Sculier JP, Ghisdal L, Berghmans T, Branle F, Lafitte JJ, Vallot F, Meert AP, Lemaitre F, Steels E, Burniat A et al (2001) The role of mitomycin in the treatment of non-small cell lung cancer: a systematic review with meta-analysis of the literature. Br J Cancer 84:1150–1155. doi:10.1054/bjoc.2001.1742
Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E (2005) Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7:77–85. doi:10.1016/j.ccr.2004.11.022
Selimovic D, Porzig BB, El-Khattouti A, Badura HE, Ahmad M, Ghanjati F, Santourlidis S, Haikel Y, Hassan M (2013) Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell Signal 25:308–318. doi:10.1016/j.cellsig.2012.10.004
Shah JJ, Kuhn DJ, Orlowski RZ (2009) Bortezomib and EGCG: no green tea for you? Blood 113:5695–5696. doi:10.1182/blood-2009-03-204776
Shin DY, Park YS, Yang K, Kim GY, Kim WJ, Han MH, Kang HS, Choi YH (2012) Decitabine, a DNA methyltransferase inhibitor, induces apoptosis in human leukemia cells through intracellular reactive oxygen species generation. Int J Oncol 41:910–918. doi:10.3892/ijo.2012.1546
Silva MF, Ruiter JP, Illst L, Jakobs C, Duran M, de Almeida IT, Wanders RJ (1997) Valproate inhibits the mitochondrial pyruvate-driven oxidative phosphorylation in vitro. J Inherit Metab Dis 20:397–400. doi:10.1023/A:1005398516208
Singh KK, Russell J, Sigala B, Zhang Y, Williams J, Keshav KF (1999) Mitochondrial DNA determines the cellular response to cancer therapeutic agents. Oncogene 18:6641–6646. doi:10.1038/sj.onc.1203056
Song IS, Kim HK, Lee SR, Jeong SH, Kim N, Ko KS, Rhee BD, Han J (2013) Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. Int J Cancer 133:1357–1367. doi:10.1002/ijc.28149
Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF et al (2008) Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 118:3930–3942. doi:10.1172/JCI36843
Spanswick VJ, Cummings J, Smyth JF (1996) Enzymology of mitomycin C metabolic activation in tumour tissue. Characterization of a novel mitochondrial reductase. Biochem Pharmacol 51:1623–1630. doi:10.1016/0006-2952(96)00104-9
Surendran S, Krishnamurthy V (2012) Effect of tanmoxifen on mitochondria—an in vitro study. Am J Pharmatech Res 2:471–482. http://www.ajptr.com/archive/volume-2/april-2012-issue-2/article-161.html
Sutherland RM (1988) Cell and environment interactions in tumor microregions: the multicell spheroid model. Science 240:177–184. doi:10.1126/science.2451290
Sviriaeva IV, Ruuge EK, Shumaev KB (2007) Formation of superoxide radicals in isolated cardiac mitochondria: effect of adriamycin. Biofizika 52:1054–1059. doi:10.1134/S0006350910020119
Takai N, Kawamata N, Gui D, Said JW, Miyakawa I, Koeffler HP (2004) Human ovarian carcinoma cells: histone deacetylase inhibitors exhibit antiproliferative activity and potently induce apoptosis. Cancer 101:2760–2770. doi:10.1002/cncr.20709
Tamulevicius P, Streffer C (1995) Metabolic imaging in tumours by means of bioluminescence. Br J Cancer 72:1102–1112. doi:10.1038/bjc.1995.472
Tedder TF, Engel P (1994) CD20: a regulator of cell-cycle progression of B lymphocytes. Immunol Today 15:450–454. doi:10.1016/0167-5699(94)90276-3
Tello D, Balsa E, Acosta-Iborra B, Fuertes-Yebra E, Elorza A, Ordóñez Á, Corral-Escariz M, Soro I, López-Bernardo E, Perales-Clemente E et al (2011) Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting Complex I activity. Cell Metab 14:768–779. doi:10.1016/j.cmet.2011.10.008
Trojnar MK, Wierzchowska-Cioch E, Krzyzanowski M, Jargiełło M, Czuczwar SJ (2004) New generation of valproic acid. Pol J Pharmacol 56:283–288. http://www.if-pan.krakow.pl/pjp/pdf/2004/3_283.pdf
Tuquet C, Dupont J, Mesneau A, Roussaux J (2000) Effects of tamoxifen on the electron transport chain of isolated rat liver mitochondria. Cell Biol Toxicol 16:207–219. doi:10.1023/A:1007695308257
Vaupel P, Kallinowski F, Okunieff P (1989) Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res 49:6449–6465. http://cancerres.aacrjournals.org/content/49/23/6449
Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F (2012) Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 122:253–270. doi:10.1042/CS20110386
Von Ahsen O, Waterhouse NJ, Kuwana T, Newmeyer DD, Green DR (2000) The ‘harmless’ release of cytochrome c. Cell Death Differ 7:1192–1199. http://www.nature.com/cdd/journal/v7/n12/pdf/4400782a.pdf
Waller CF (2010) Imatinib mesylate. Recent Results Cancer Res 184:3–20. doi:10.1007/978-3-642-01222-8_1
Wang M, Han XH, Zhang L, Yang J, Qian JF, Shi YK, Kwak LW, Romaguera J, Yi Q (2008) Bortezomib is synergistic with rituximab and cyclophosphamide in inducing apoptosis of mantle cell lymphoma cells in vitro and in vivo. Leukemia 22:179–185. doi:10.1038/sj.leu.2404959
Will Y, Dykens JA, Nadanaciva S, Hirakawa B, Jamieson J, Marroquin LD, Hynes J, Patyna S, Jessen BA (2008) Effect of the multitargeted tyrosine kinase inhibitors imatinib, dasatinib, sunitinib, and sorafenib on mitochondrial function in isolated rat heart mitochondria and H9c2 cells. Toxicol Sci 106:153–161. doi:10.1093/toxsci/kfn157
Williams SA, McConkey DJ (2003) The proteasome inhibitor bortezomib stabilizes a novel active form of p53 in human LNCaP-Pro5 prostate cancer cells. Cancer Res 63:7338–7344. http://cancerres.aacrjournals.org/content/63/21/7338
Xu J, Zhou JY, Tainsky MA, Wu GS (2007) Evidence that tumor necrosis factor-related apoptosis-inducing ligand induction by 5-Aza-2′-deoxycytidine sensitizes human breast cancer cells to adriamycin. Cancer Res 67:1203–1211. doi:10.1158/0008-5472.CAN-06-2310
Xu J, Wang J, Xu B, Ge H, Zhou X, Fang JY (2013) Colorectal cancer cells refractory to anti-VEGF treatment are vulnerable to glycolytic blockade due to persistent impairment of mitochondria. Mol Cancer Ther 12:717–724. doi:10.1158/1535-7163.MCT-12-1016-T
Yang Z, Schumaker LM, Egorin MJ, Zuhowski EG, Guo Z, Cullen KJ (2006) Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: possible role in apoptosis. Clin Cancer Res 12:5817–5825. doi:10.1158/1078-0432.CCR-06-1037
Yaromina A, Meyer S, Fabian C, Zaleska K, Sattler UG, Kunz-Schughart LA, Mueller-Klieser W, Zips D, Baumann M (2012) Effects of three modifiers of glycolysis on ATP, lactate, hypoxia, and growth in human tumor cell lines in vivo. Strahlenther Onkol 188:431–437. doi:10.1007/s00066-011-0054-3
Yin D, Zhou H, Kumagai T, Liu G, Ong JM, Black KL, Koeffler HP (2005) Proteasome inhibitor PS-341 causes cell growth arrest and apoptosis in human glioblastoma multiforme (GBM). Oncogene 24:344–354. doi:10.1038/sj.onc.1208225
Yu D, Hung MC (2000) Overexpression of ErbB2 in cancer and ErbB2-targeting strategies. Oncogene 19:6115–6121. doi:10.1038/sj.onc.1203972
Zgouras D, Becker U, Loitsch S, Stein J (2004) Modulation of angiogenesis-related protein synthesis by valproic acid. Biochem Biophys Res Commun 316:693–697. doi:10.1016/j.bbrc.2004.02.105
Zhang D, Tai LK, Wong LL, Chiu LL, Sethi SK, Koay ES (2005) Proteomic study reveals that proteins involved in metabolic and detoxification pathways are highly expressed in HER-2/neu-positive breast cancer. Mol Cell Proteomics 4:1686–1696. doi:10.1074/mcp.M400221-MCP200
Zhao F, Mancuso A, Bui TV, Tong X, Gruber JJ, Swider CR, Sanchez PV, Lum JJ, Sayed N, Melo JV et al (2010) Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1alpha-induced metabolic reprograming. Oncogene 29:2962–2972. doi:10.1038/onc.2010.67
Zhao Y, Liu H, Liu Z, Ding Y, Ledoux SP, Wilson GL, Voellmy R, Lin Y, Lin W, Nahta R, Liu B, Fodstad O, Chen J, Wu Y, Price JE, Tan M (2011) Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res 71:4585–4597. doi:10.1158/0008-5472.CAN-11-0127
Zhao L, Ashek A, Wang L, Fang W, Dabral S, Dubois O, Cupitt J, Pullamsetti SS, Cotroneo E, Jones H et al (2013) Heterogeneity in lung (18)FDG uptake in pulmonary arterial hypertension: potential of dynamic (18)FDG positron emission tomography with kinetic analysis as a bridging biomarker for pulmonary vascular remodeling targeted treatments. Circulation 128:1214–1224. doi:10.1161/CIRCULATIONAHA.113.004136
Zsengellér ZK, Ellezian L, Brown D, Horváth B, Mukhopadhyay P, Kalyanaraman B, Parikh SM, Karumanchi SA, Stillman IE, Pacher P (2012) Cisplatin nephrotoxicity involves mitochondrial injury with impaired tubular mitochondrial enzyme activity. J Histochem Cytochem 60:521–529. doi:10.1369/0022155412446227
Zu XL, Guppy M (2004) Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun 313:459–465. doi:10.1016/j.bbrc.2003.11.136
Acknowledgments
The present work was partially supported by CONACyT-México Grant Nos. 107183 to SRE, 180322 to AMH and 80534 and 123636 to RMS; and Instituto de Ciencia y Tecnología del Distrito Federal Grant No. PICS08.
Conflict of interest
The authors declare there is no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Rodríguez-Enríquez, S., Gallardo-Pérez, J.C., Hernández-Reséndiz, I. et al. Canonical and new generation anticancer drugs also target energy metabolism. Arch Toxicol 88, 1327–1350 (2014). https://doi.org/10.1007/s00204-014-1246-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-014-1246-2