
Abstract
The comet assay (single cell gel electrophoresis) is the most common method for measuring DNA damage in eukaryotic cells or disaggregated tissues. The assay depends on the relaxation of supercoiled DNA in agarose-embedded nucleoids (the residual bodies remaining after lysis of cells with detergent and high salt), which allows the DNA to be drawn out towards the anode under electrophoresis, forming comet-like images as seen under fluorescence microscopy. The relative amount of DNA in the comet tail indicates DNA break frequency. The assay has been modified to detect various base alterations, by including digestion of nucleoids with a lesion-specific endonuclease. We describe here recent technical developments, theoretical aspects, limitations as well as advantages of the assay, and modifications to measure cellular antioxidant status and different types of DNA repair. We briefly describe the applications of this method in genotoxicity testing, human biomonitoring, and ecogenotoxicology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The comet assay for DNA damage appeals to researchers because of its simplicity, sensitivity, versatility, and charm; the images it produces resemble so closely the celestial bodies, that looking down the microscope at disrupted DNA molecules can seem as inspirational as stargazing (Fig. 1). The more formal name for the assay is single cell gel electrophoresis, and while it is true that each individual comet is scored to give a measure of DNA damage, it is often necessary to pool the results of, say, 100 comets to obtain the overall damage level of a population of cells. In brief, a suspension of cells is mixed with agarose to form a thin gel on a microscope slide. The cells are lysed with a detergent (Triton X-100, often with sodium sarcosinate, though this addition is not necessary) to remove cell and nuclear membranes, releasing all soluble cell components. NaCl at >2 M is also present in the lysis solution, to remove histones from the DNA. The gel-embedded bodies resemble nuclei in size and shape and are known as nucleoids. The nucleosomal organisation of the DNA is disrupted by the extraction of histones, but the negative supercoiling of the DNA (imposed by the superhelical turns of the DNA around the histone core) survives as long as the DNA is intact. The model of nucleoid structure developed by Cook and colleagues in the 1970s (Cook et al. 1976) supposes that each DNA molecule is attached at intervals to a nuclear matrix or scaffold, so that it can be seen as a series of loops, each of which is a structural unit. If supercoiling is relaxed in one loop, by a DNA strand break (SB), then—when an electrophoretic field is imposed—that loop is able to extend towards the anode. The more breaks are present, the more DNA loops are relaxed, and the more DNA appears in the comet tail. After staining with a suitable dye, the relative intensity of tail DNA fluorescence is measured as an index of DNA break frequency.

Typical comet images from H2O2-treated lymphocytes, stained with 4′,6-diamidino-2-phenylindole
The first account of a comet assay (though not referred to by that term) was by two Swedish researchers, Östling and Johanson, in 1984 (Ostling and Johanson 1984) and employed lysis and electrophoresis at pH 9.5. They were well aware of the nucleoid story and described the migration of DNA towards the anode as resulting from the relaxation of supercoils when cells were γ-irradiated. A few years later, the method was modified by increasing the pH of electrophoresis to >13 (with 0.3 M NaOH), and this essentially is the comet assay most widely used today (Singh et al. 1988). There have, however, been significant modifications, and the reader is referred to the detailed protocol (currently in use in our laboratory) that was recently published (Collins and Azqueta 2012a).
The versatility and scope of the comet assay
The comet assay has been applied to cells in culture, blood cells taken from animals or humans, haemolymph cells from molluscs and insects, sperm, disaggregated animal tissues, yeast, nuclei released from plant tissue; in fact, any eukaryotic cell type that can be obtained as a single cell or nuclear suspension seems to be amenable to comet assay analysis. It is even possible to produce comets from isolated chromosomes (Cortés-Gutiérrez et al. 2011). As well as measuring DNA SBs, it has been modified to detect damaged bases, by digesting the DNA after lysis with a lesion-specific enzyme that produces a DNA break at the damage site (see section on “Measuring different kinds of DNA damage”). Oxidised and alkylated bases have been studied in this way, as well as the cyclobutane pyrimidine dimers induced by UV irradiation.
It was very quickly realised that the comet assay could be employed to monitor DNA repair as well as DNA damage; in fact, both Ostling and Johanson (1984) and Singh et al. (1988) followed the rejoining of DNA SBs induced by ionising radiation. A further common application is to assess the antioxidant status of cells, by their resistance to damage by reactive oxygen (e.g. H2O2).
There are four main areas of research in which the comet assay has been adopted. It is used in genotoxicity testing, to screen novel drugs, cosmetics, or other chemicals for potential carcinogenic properties; tests can be carried out in vivo (with analysis of various tissues from the experimental animal) or in vitro using suitable cultured cell lines. Equally popular is the application to human biomonitoring: to examine effects at the DNA level of occupational or environmental exposure to hazardous agents, to study effects of, or factors contributing to disease, to investigate individual variations in, for example, DNA repair capacity or antioxidant resistance, or to monitor changes brought about by dietary intervention with, for example, antioxidant micronutrients. Ecogenotoxicology is the third area of application—using a variety of organisms (mussels, earthworms, snails, plants, etc.) as indicators of genetic damage by pollutants, either in the wild or in the laboratory. Finally, the comet assay is a valuable tool in basic research into mechanisms of DNA damage and DNA repair.
Limitations of the comet assay, and misunderstandings
It is wise to point out the limitations of the comet assay at this stage, if only to emphasise that many of these limitations are perceived but not real.
The assay is ideally suited for use with cells such as peripheral blood mononuclear (PBMN) cells which are already in suspension or cultured cells which are either grown in or easily obtained as a suspension. Tissues represent more of a challenge, but methods have been devised, using enzymes and/or physical maceration, to release cells or nuclei of high quality from many animal tissues (Brendler-Schwaab et al. 1994; Sasaki et al. 1997). Methods have been developed for specific human tissues, such as buccal epithelial cells (Szeto et al. 2005), tear duct epithelial cells (Rojas et al. 2000), lens epithelial cells (Osnes-Ringen et al. 2012), and sperm (Hughes et al. 1996). Frozen cells or tissues are generally not suitable, since physical shearing of the DNA occurs as ice crystals form, but DNA can be preserved intact if cells are suspended in freezing medium (culture medium with 10 % foetal bovine serum and 10 % dimethylsulphoxide, DMSO) and frozen slowly to −80 °C. However, a recent report (Al-Salmani et al. 2011) showed that whole blood could be simply frozen, in small aliquots, without DMSO and without prior separation of PBMN cells, and would give reliable DNA strand breakage data on thawing. This is a useful finding, though it is not known whether such samples can be stored indefinitely.
Performing the comet assay on plants requires special care. Nuclei are released by chopping the plant tissue with a sharp blade while on ice (Gichner et al. 2009). It is recommended to perform the nuclear isolation in darkness or under non-actinic red light to avoid white light-induced damage (Pourrut et al. 2011).
Yeast cells have a cellulose cell wall that needs to be disrupted to release the nuclei, but the comet assay has been successfully performed (Oliveira and Johansson 2012).
Mitochondrial DNA is too small to be detected—in fact, it quickly disappears from the gel once the cells have been lysed (Shaposhnikov et al. 2006). In principle, mitochondria and bacteria should not be amenable to comet assay analysis, since they do not have the typical organisation of eukaryotic DNA on a nuclear matrix; comet tails can arise only because DNA loops are anchored in the comet head, which is, we presume, the remnant of the matrix.
The dynamic range of the comet assay is quite limited; in terms of breaks produced by ionising radiation, between a fraction of a Gy and 10 Gy. This represents a range from a few hundred to several thousand breaks in a normal human cell, and it is worth pointing out that the distance between breaks after 10 Gy is about 1 megabase; it is misleading to think in terms of DNA fragmentation. The dynamic range is conveniently physiological; DNA breaks within this range are generally reparable, and cytotoxicity does not need to be considered as a possible cause of DNA damage.
The comet assay has been prone to various misunderstandings. Chief among these is the often expressed belief that by performing the assay at a neutral pH, only double-strand breaks (DSBs) are detected—or, in other words, that an alkaline pH is necessary to reveal single-strand breaks (SSBs). There is a mistaken analogy with methods such as alkaline elution or alkaline unwinding, where a high pH is necessary to allow strand separation; for instance, in alkaline unwinding, each SB acts as a focus for denaturation, and when the alkali is neutralised after a certain period of incubation, the extent of unwinding depends on the number of breaks. The DNA is sheared to small fragments which are either single- or double-stranded, and the proportion of single-stranded DNA is a measure of SB frequency. The comet assay, in contrast, depends on relaxation of supercoils, and so there should be no influence of pH, except for the likely conversion of some alkali-labile sites to breaks at alkaline pH. How strong is the evidence for this interpretation? Ionising radiation induces more SSBs than DSBs—by a factor of 20 times (Bradley and Kohn 1979)—and H2O2 is even more biased towards SSBs (Bradley and Kohn 1979). Both Ostling and Johanson (1984) and Singh et al. (1988), in the original comet assay papers, described experiments with ionising radiation and reported very similar dose responses and limits of detection, even though one method was at effectively neutral pH and the other in strong alkali. We have shown similar sensitivity of neutral and alkaline assays in detecting H2O2- and methylmethanesulphonate (MMS)-induced breaks (Collins et al. 1997a). (It is, however, curious that Singh et al. in their original report (1988) saw no migration under neutral conditions after irradiation with up to 1 Gy of X-rays—enough to produce comet tails at high pH.)
Another common misunderstanding is that comets with small heads and large tails indicate apoptosis; we deal with this in depth later in the article (“Hedgehogs and viability”).
A practical limitation of the comet assay is the number of samples that can be handled in one experiment. Typically, with a standard large electrophoresis tank and 2 gels per slide, 40 gels are the maximum, although higher throughput techniques have been introduced (see subsequent section, “High throughput comet assay methods”).
Scoring comets is tedious, and the charm of the comet assay can be short-lived, especially when throughput is increased, since this entails even more time spent at the microscope. To date, fully automated scoring has not been entirely successful.
The comet assay in its original form detects strand breaks (and alkali-labile sites). SSBs are rapidly repaired and are regarded as not very threatening to the cell in terms of either viability or genetic stability. DNA damage takes various other, arguably more interesting and important forms such as oxidised and alkylated bases, adducts, and cross-links, many of which are potentially mutagenic, and early on the assay was modified to detect some of these.
Measuring different kinds of DNA damage
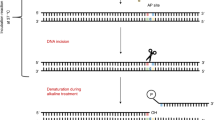
Most simple alterations to DNA bases can be detected by digesting the nucleoid DNA, just after lysis, with a lesion-specific enzyme that removes the base, leaving an apurinic/apyrimidinic (AP)-site; this is then converted to a break by an associated AP lyase activity (or, alternatively, since the AP lyase can be slow-acting, by the high pH to which the DNA is subsequently subjected) (Fig. 2).

The comet assay, modified for the detection of altered bases, by inclusion of a specific nuclease digestion step following lysis
Endonuclease III (EndoIII; thymine glycol DNA glycosylase, EC 4.2.99.18), an Escherichia coli DNA repair enzyme, was the first enzyme to be used (Collins et al. 1993). It was applied in an early human nutritional intervention trial, showing for the first time an effect of supplementation with antioxidants on endogenous DNA damage levels (Duthie et al. 1996). Formamidopyrimidine DNA glycosylase, or FPG (EC 3.2.2.23), was also put to use (Dusinska and Collins 1996); it removes oxidised purines—in particular, 8-oxo-7,8-dihydroguanine (8-oxoGua), and formamidopyrimidines, that is, ring-opened adenine or guanine—but also attacks ring-opened N7 guanine adducts produced by alkylating agents (Li et al. 1997; Speit et al. 2004). The mammalian counterpart of FPG is 8-oxoGua DNA glycosylase or OGG1 (EC 4.2.99.18). OGG1 is more specific than FPG since it is free of any activity against alkylated purines (Smith et al. 2006). The main substrate of 3-methyladenine DNA glycosylase II (AlkA) (EC 3.2.2.21) is 3-methyladenine, although the enzyme can act as a non-specific nuclease (with low efficiency) (Berdal et al. 1998). It was applied with the comet assay to detect damage induced by MMS and revealed a significant level of lesions in human PBMN cells (Collins et al. 2001a). Misincorporated uracil in DNA is detected with uracil DNA glycosylase (UDG: EC 3.2.2.3) (Duthie and McMillan 1997). UV light, especially at lower wavelengths (UVC), induces dimerisation of adjacent pyrimidines in DNA, and these are converted to breaks by T4 endonuclease V (pyrimidine dimer DNA glycosylase, EC 3.1.25.1); this enzyme was used to characterise UV-sensitive mutant cell lines (Collins et al. 1997b).
The use of lesion-specific enzymes has added greatly to the value of the comet assay, especially in human biomonitoring, where FPG in particular has been used in many investigations of oxidative damage to DNA. There are, however, some important considerations when using the enzyme approach:
-
Purified enzymes can be obtained, but they tend to be less stable than crude extracts. Since the enzymes are generally prepared from over-producing bacterial strains in which the enzyme is a substantial fraction of total protein, non-specific nucleases in a crude extract are not a serious problem.
-
The concentration at which the enzyme is used is critically important. It should obviously be high enough to detect all the lesions present, but not so high that non-specific nucleases interfere. A titration experiment is necessary, using cells that are known to contain the lesion of interest—ideally, cells treated with an appropriate agent, such as UVC (for T4 endonuclease V), MMS (for AlkA), or photosensitiser Ro 19-8022 plus light (for FPG and OGG1). In the case of endonuclease III (or FPG if the photosensitiser is not available), cells can be treated with H2O2 and incubated to allow repair of SBs, leaving oxidised bases as a substrate for testing the enzyme. A plateau should be reached at a certain enzyme concentration, reflecting optimal detection of lesions.
-
It is usual to have sample gels incubated with enzyme buffer, alongside the gels incubated with the enzyme. The comet score from the buffer incubation is subtracted from the score with the enzyme, to give ‘net enzyme-sensitive sites’ (expressed as % DNA in tail). This is valid, so long as the scores are on the linear part of the dose–effect curve (% DNA in tail plotted against DNA break frequency or X-ray dose). But if cells are treated with an agent that induces a substantial number of SBs as well as altered bases, so that the total score with enzyme is beyond the linear part of the curve, simply subtracting the +buffer score from the +enzyme score will result in an underestimation of enzyme-sensitive sites.
-
It is advisable to include in experiments cells that have a known amount of the appropriate damage, as a reference standard (see section “Reference standards”).
-
As has been pointed out already, the enzymes are not entirely specific, and this should be borne in mind when interpreting results. The fact that apurinic/apyrimidinic (AP) lyase activity is generally present with the glycosylase should not decrease specificity, since AP-sites that are already present in the DNA should, in theory at least, be converted to SBs by the alkaline conditions and so be included in the buffer incubation score.
Cross-links between DNA molecules, or between DNA and protein, have the opposite effect from SBs, since they inhibit the extension of broken DNA loops into a comet tail. They can therefore be studied through their ability to block the normal migration of DNA following ionising irradiation or H2O2 treatment.
High throughput comet assay methods
Although the comet assay is simple and economical in terms of materials and equipment, it is labour-intensive, since there is a limit to the number of samples that can be processed in one experiment, imposed by the number of slides that can be placed in the electrophoresis tank (and simply adding another electrophoresis tank leads to an unmanageable number of slides). Various attempts have been made to improve throughput, by combining multiple gels on one substrate, and/or by reducing the volume of agarose gel per sample. McNamee et al. (2000) placed 12 gels, 80 μl in volume, on a 10 × 6.5 cm piece of GelBond plastic film, and fitted four of these in one tank, giving a capacity per experiment of 48 samples. A logical extension of this was to decrease the volume of agarose per sample to a few μl, so that 48 or even 96 gels could be placed on one GelBond film (and up to almost 400 samples electrophoresed in a single run) (Gutzkow et al. 2013).
Stang and Witte (2009) developed special 96-well multi-chamber plates with an agarose-containing bottom plate to which cells attach. The bottom plate is subsequently detached from the chamber structure and undergoes lysis, electrophoresis, etc., in the comet assay. Again, almost 400 samples can be run simultaneously.
Often the need is not to maximise the number of gels per experiment but to increase throughput while maintaining flexibility, that is, allowing different gels to have individual treatment. For this, the medium throughput method with 12 mini-gels set on a microscope slide is appropriate (Shaposhnikov et al. 2010). A device that clamps a gasket and incubation wells onto the slide makes it possible to incubate gels with different concentrations of reagent, different reagents, different lesion-specific endonucleases, or different DNA probes (in the case of the fluorescent in situ hybridisation (FISH)-comet assay).
Validation of the new high throughput methods against the standard comet assay is important. Stang and Witte (2009) obtained similar results with cells treated with MMS or H2O2 using their multi-chamber plates or standard slides. We recently ran parallel assays of cells treated with MMS or X-rays, using the standard assay, slides with 12 mini-gels, or GelBond films with mini-gels, and found no significant effect of format on results (Azqueta et al. 2013a).
Mercey et al. (2010) prepared agarose-coated substrate plates with a micropattern (created by UV etching) that creates sites for attachment and growth of one or a few cells in a regular array. Treatment with test-chemicals, and post-treatment culture, are carried out on the plate, which is then subjected to normal comet assay procedures. In a rather similar approach, Wood et al. (2010) created arrays of microwells of a few tens of microns, using a stamp placed in the setting agarose. Cells then settle passively into the micro-wells. By making the micro-wells in agarose on a GelBond film and superimposing a bottomless multiwell plate, chambers are created for individual incubation of cell arrays on the same substrate for subsequent lysis and electrophoresis.
Making it easier to run many samples simultaneously is of course advantageous—but it does increase the burden of scoring, which can then become the ‘rate-limiting step’. Commercial automated comet assay scoring systems exist, and claims are made for some of the high throughput modifications that automated scoring is facilitated, but in our experience, little time is saved because of the need for manual checking of the results of the automated scoring.
Critical factors influencing comet assay performance
With the aim of encouraging standardisation of comet assay protocols, two research groups (one of them ours) independently set out to define the factors that are most important in determining comet assay performance and influencing results; their conclusions were very similar (Ersson and Möller 2011; Azqueta et al. 2011a).
Agarose concentration has a marked effect. The lowest concentration tested was 0.4 %, which gives a relatively high % tail DNA (with cells containing DNA breaks) but is too fragile to be recommended. As agarose concentration is increased to 1.3 %, the % tail DNA steadily decreases.
The period of lysis in the Triton and high salt solution appears not to be important. Generally, a minimum period of 1 h is employed, but overnight lysis is common, and in some cases, gels are stored in lysis solution for weeks with no apparent effect on results.
Alkaline incubation time is, however, critical. When varied over the range 10–60 min, there was a steady increase in % tail DNA with time (Ersson and Möller 2011) or an approach to a plateau after 40 min (Azqueta et al. 2011a). Untreated cells also showed a response to incubation time.
The greatest effects (on comets from both treated and untreated cells) were seen with variations in electrophoresis voltage and duration (Ersson and Möller 2011; Azqueta et al. 2011a). The relative tail intensity rises steeply with voltage and with time (Fig. 3). Voltage gradient is the important parameter, and specifically the voltage gradient over the platform on which the slides are placed. There is very little change in voltage between the electrode and the edge of the platform, as there is a considerable depth of electrophoresis solution and therefore low resistance compared with the depth of a few mm of solution over the slides. Within limits, increasing voltage gradient will compensate for a shorter electrophoresis time, and vice versa.

Effect of varying electrophoresis voltage (a) or time (b). TK-6 lymphoblastoid cells were untreated (dark shading) or treated with 70 μM H2O2 for 5 min on ice (light shading) (Redrawn from Azqueta et al. 2011a, with permission from Elsevier.)
Many researchers aim at a current of 300 mA and adjust the current by varying the volume of electrophoresis solution. However, changes in current have very little effect on comet appearance—and the effect they do have is explained by the change in voltage gradient when the depth of solution is altered. A very high current could significantly increase the temperature. It is generally advised that the temperature should be 15 °C or below, and it is common to run the electrophoresis in a cold room. Tanks with built-in temperature control are becoming available and should make for more consistent results within a laboratory and between laboratories using the same equipment.
If the enzyme digestion step is included, the concentration and time of incubation are important. As mentioned above, at too high a concentration, non-specific breakage may occur (especially when a crude enzyme extract is used, since other nucleases are likely to be present at low concentrations). Assuming that the supplier has indicated an appropriate concentration, or it has been ascertained by titration, the enzyme should be tested with different incubation times. Ideally, a plateau should be reached at a certain time (typically, about 30 min), beyond which there is no increase in breaks (Ersson and Möller 2011). This test can be done with cells treated with a chemical, or radiation, to induce the required damage; if there is a significant background level of such damage in normal cells, as with base oxidation, then PBMN cells can be used for the test.
Scoring of comets is a potential cause of variation; different stains are in use, varying in fluorescence intensity, and comet scores can be affected by microscope quality and adjustments, ageing of UV lamp, and settings (such as threshold for fluorescence detection) within the image analysis software. It is thought that such factors account for much of the variation recorded in the European Comet Assay Validation Group (ECVAG) trials (Ersson et al. 2013). We systematically tested three scoring methods (Azqueta et al. 2011b)—conventional computerised image analysis (in which comets are selected by the operator), automated image analysis (comets selected and analysed without operator involvement), and visual scoring, in which the operator classifies each (randomly selected) comet into one of 5 categories (0; no discernible tail, to 4; small head, most DNA in tail) and the category values of 100 comets are summed, giving an overall damage score of between 0 and 400. We found that, while visual scoring systematically overestimates low levels of damage, heavily damaged comets tend to be missed when automated image analysis is used. However, when we compared the ability of the different scoring methods to give quantitative estimates of damage caused by MMS or H2O2, we concluded that there was essentially no difference in sensitivity, and—judging by a Bland–Altman analysis—results from all three are equally valid and interchangeable (Azqueta et al. 2011b).
Cell/comet density needs to be taken very seriously. Too few cells in the gel are obviously bad news; but too many can cause problems, too. Overlapping comets are very difficult to score and are systematically excluded by good image analysis systems. Getting the right density is a matter of experience, but we give some advice below.
Guidelines
(A non-exhaustive list, based on the preceding section)
-
Aim at about 104 cells per gel (conventional method) or about 250 when using minigels. Note that some cells are lost during processing (especially washing and centrifugation) (Ho et al. 2011).
-
Dilute cells in agarose to a final concentration of between 0.6 and 0.8 %. Do not reuse the stock agarose solution more than 3 or 4 times, as it tends to become more concentrated through evaporation; store in small aliquots.
-
Lysis should be at 4 °C and can be for 1 h, overnight, or longer (but beware of gels becoming detached after prolonged lysis).
-
Enzyme incubation conditions should be optimised and then carefully adhered to. If enzyme is added to gels on ice, allow 10 min for the incubation to reach 37 °C in the incubator.
-
Alkaline incubation at 4 °C for 40 min is recommended (though 20 min is acceptable).
-
Electrophoresis should be carried out in a cold room, for 20 min at 1.15 V/cm (across the platform) or 30 min at 0.8 V/cm.
-
After neutralisation and washing, allow the gels to dry at room temperature. When the gel is dry, it is much thinner, and focusing on comets is simpler.
-
Avoid scoring comets near the edge of the gel or near bubbles in the gel, as they tend to be anomalous. (In the case of minigels, the edge represents a substantial proportion of the whole gel, but ‘edge effects’ can be minimised by not allowing gels to dry during processing and by dehydrating them in ethanol immediately after neutralisation and washing following electrophoresis.)
Expressing results
The comet assay provides not just the overall level of damage in a sample of cells, but data on individual cells. Sometimes it is important to know how individual cells respond to a damaging agent. For instance, human PBMN cells treated with H2O2 do not behave homogeneously; a sub-population are relatively resistant, remaining undamaged at H2O2 concentrations that produce a high % of tail DNA in the rest of the cell population (Azqueta et al. 2011b; Fig. 4). The phenomenon has not been properly investigated, but it might reflect varying levels of antioxidants between different PBMN cell types.

Heterogeneous behaviour of PBMN cells treated with H2O2. The panel on the left shows the distribution of comets from PBMN cells treated with increasing concentrations of H2O2 (5 min on ice). Comets were scored visually into 5 classes, from undamaged (0) to highly damaged (4). Even at higher H2O2 concentrations, some lymphocytes are still undamaged. In contrast, the lymphoblastoid TK-6 cells treated with MMS in the right-hand panel progress as a homogeneous group from a low level of damage (class 0/1) through intermediate classes to almost all in category 4 at the highest MMS concentration (Redrawn from Azqueta et al. 2011b, with permission from Oxford University Press)
Generally, though, we can ignore individual comet data and simply take the mean or median comet score as a measure of damage in the sample. This then becomes the ‘experimental unit’ for statistical analysis. Whether to use mean or median depends on circumstances. Median can be useful if there are just a few comets with high levels of damage and there is reason to believe that they are anomalous. On the other hand, if the distribution is fairly homogeneous, the mean is appropriate. (With visual scoring, this choice does not arise; the overall score is simply the sum of individual comet scores.) The selection of appropriate statistical tests to analyse comet assay results is a complex issue, dealt with comprehensively by Lovell and Omori (2008) or Lovell (2009), and rather than attempting to summarise this topic here, we refer the reader to these excellent papers.
When measuring base damage with lesion-specific enzymes, it is customary to subtract buffer incubation score from the enzyme incubation score, as described above, to obtain the measure of the net enzyme-sensitive sites. (Normally, there is only a slight increase in breaks above the 'lysis only' score when nucleoids are incubated with buffer; if the increase is substantial, the buffer should be discarded.) If the scores are on the nonlinear portion of the dose–effect curve, a calibration curve (see below) should be used to convert all comet scores to DNA break frequencies before performing the subtraction; otherwise enzyme-sensitive sites will be underestimated.
Various parameters to choose from
When using computerised image analysis, the output will include a variety of different comet parameters, including tail length, percentage of total fluorescence in head and tail, and various versions of tail moment. Tail length can be useful at low levels of damage, but it quickly reaches a maximum; once the tail is established, its length tends not to change, since it is defined by the DNA loop length. The percentage of DNA in the tail is the most useful parameter, since it is linearly related to DNA break frequency over a wide range of damage (see “Calibration”, below), and it is immediately apparent, given a particular % tail DNA, what sort of comet is being described. Tail moment is at first sight attractive; it is essentially tail intensity x length summed over the whole extent of the tail, and so combines two measures in a single value. However, in practice, it does not differ from % tail DNA in sensitivity, and it suffers from the serious disadvantage that there are no standard units for tail moment. A particular tail moment value tells us nothing about the kind of comet or therefore the level of damage being presented.
Variability, accuracy, and precision
Researchers of a chemical bent tend to be suspicious of the comet assay as a ‘soft’, biological, subjective assay. Surely high-performance liquid chromatography (HPLC) or gas chromatography–mass spectrometry (GC–MS) is more accurate. The issue came to a head in the mid-1990s when measurements of the common oxidation product 8-oxoGua in human cells using the comet assay, alkaline unwinding, or alkaline elution (with FPG) were compared with determinations using HPLC or GC–MS. It appeared that chromatographic methods were often reporting orders of magnitude more 8-oxoGua than were the enzymic methods (Collins et al. 1997c). This was recognised as a serious problem, and the European Standards Committee on Oxidative DNA Damage (ESCODD) was set up to try to resolve it. Over a period of about 6 years, with the help of an EC ‘Concerted Action’ contract, we distributed a series of samples of oligonucleotides, DNA, DNA with experimentally induced 8-oxoGua, liver tissue, cultured cells, and cells with added 8-oxoGua, for partners to analyse (ESCODD 2002a, b, 2003, ESCODD et al. 2005). To summarise, chromatographic methods (at least HPLC) were good at measuring relatively high levels of 8-oxoGua induced experimentally; but when measuring the very low levels found in cells, these methods had to contend with a serious and variable artefact of 8-oxoGua introduced into the DNA during sample preparation. However hard we tried, with various antioxidant regimes, we could not eliminate the artefact, and it is probably fair to conclude that, while the comet assay may be less precise than chromatographic methods, it is more accurate at measuring background levels of 8-oxoGua, which are likely to be less than 1 per 106 Gua.
(A common illustration of accuracy and precision uses the analogy of a dart board; the darts thrown by the comet assay may not all hit the same spot, but they are centred on the bull’s eye and in that sense are accurate. In contrast, as was shown in the ESCODD project, HPLC can be very precise, but is a long way off target when measuring background levels of damage because of the spurious oxidation occurring during sample preparation (ESCODD 2003).)
Variability is studied at different levels:
-
Intra-experimental variation should be minimal, if a good protocol is followed, and equipment is suitable and in good condition. It is assessed by comparing results from replicate gels prepared with cells from a single sample.
-
Inter-experimental variation can be monitored and corrected for by the use of reference standards (see below, and Fig. 5).
Fig. 5 
Inter-experimental variation; net FPG-sensitive sites, measured in reference samples of PBMN cells, either untreated (circles) or treated with photosensitiser plus light to induce 8-oxoGua (squares), in a series of experiments (unpublished data)
-
Inter-laboratory variation has been addressed in various trials, under the auspices of ESCODD and, latterly, ECVAG, with generally disappointing results (ESCODD et al. 2005; Forchhammer et al. 2010, 2012, Johansson et al. 2010, Ersson et al. 2013). Adopting a standard protocol should minimise differences between laboratories, although scoring remains a likely source of variation, since quantitation of comets depends on staining intensity, microscope quality, illumination source, and image analysis software.
-
Intra-individual variation, for instance in samples of PBMN cells taken from human subjects, on different occasions. If such samples are run in a single experiment, inter-experimental variation is eliminated; variation between replicates of the same sample will indicate intra-experimental variation; what remains is the true variation.
-
Inter-individual variation, within a population. Normally, it is not possible to analyse more than a few samples in one experiment, and so it is important to include reference standards in each experiment, and if necessary to carry out a normalisation procedure.

Figure 6 illustrates both intra- and inter-individual variation, with data from individual subjects sampled on two occasions; in two separate trials, we measured DNA damage and DNA repair capacity.
In a series of experiments conducted in two laboratories, with cells treated with MMS or X-rays, and different formats (2 gels per slide, or 12 or 24 mini-gels), CVs were calculated for each set of experiments in each laboratory; they varied between 4.8 and 16 %, with an average of 10 % (Azqueta et al. 2013a). We suggest that, in experiments with a moderate level of damage, a CV of 10 % represents acceptable variability. (CVs are likely to be higher when levels of damage are low, i.e. comets from undamaged cells, and lower when the damage is high and the comet assay is near saturation.)

Inter- and intra-individual variation in DNA damage (a; FPG-sensitive sites) and DNA base excision repair (b). BER was measured using the in vitro assay described in the section “Measuring DNA repair”. PBMN cells were isolated from subjects on two occasions several weeks apart (sample 1 and sample 2). Subjects came from two different studies: a the control group in a coffee intervention study (ms in preparation), and b pre-intervention samples in a kiwifruit crossover intervention study (Gaivão et al. 2009, from which the figure is redrawn, with permission from Springer)
Calibration
In most cases, the % tail DNA is an adequate measure of DNA damage. But sometimes it is informative to express damage in terms of actual DNA break frequency. This can be done by means of a calibration curve. The yield of DNA breaks induced by ionising radiation in most types of cell is well established, on the basis of alkaline sucrose sedimentation studies, at 0.3 breaks per 109 Dalton per Gy (Ahnström and Erixon 1981). Treating cells with different doses of X- or γ-irradiation up to about 10 Gy reveals a more or less linear relationship between % tail DNA and radiation dose, up to about 75 % tail DNA (Collins et al. 2008). It is therefore possible to convert values of % tail DNA to ‘breaks per 109 Dalton’ or, using simple conversion factors, to ‘breaks per 106 base pairs,’ or ‘breaks per cell’. When carrying out a calibration experiment, it is important to irradiate cells on ice to prevent any rejoining of breaks by cellular repair; they can in fact be irradiated after embedding in agarose (before lysis). Calibration curves produced in different laboratories are similar but not identical (Collins et al. 2008). Some of the variation arises from errors in calibration of the radiation source, as is demonstrated in a recent comparative study coordinated under the EC-funded COMICS project (Brunborg et al. in preparation).
Most laboratories do not have ready access to a source of ionising radiation. Unfortunately, chemical DNA-damaging agents are not recommended for calibration purposes. H2O2 produces strand breaks, but the yield varies widely from cell type to cell type and even from cell to cell, presumably because of differing levels of antioxidants. Alkylating agents induce low levels of frank breaks with higher levels of base damage, and consequently, breaks arise as repair intermediates—adding to the potential variability and making them unsuitable for calibration.
Reference standards
When carrying out a study of multiple samples analysed on different dates (normal practice in human biomonitoring studies), it is advisable to include reference standards to check for inter-experimental variation. The standards—positive and negative—should be prepared from a single large batch of cells (cultured cells or PBMN cells are suitable), treated for example with radiation or H2O2 (if SBs are the lesion of interest in the samples), or with Ro 19-8022 (Hoffmann-La Roche) plus light (for oxidised purines), as well as untreated cells. The cells are suspended in freezing medium, split into small aliquots, and frozen slowly to −80 °C for storage. Sufficient aliquots should be prepared for the complete series of experiments. The reference standards make it possible to identify experiments with anomalously high or low levels of damage; repeat assays can then be carried out on affected samples (see Fig. 5). It is also possible to normalise the % tail DNA results from all samples against the standard, as follows:
-
Find the median value for the positive reference standard % tail DNA across the whole series of experiments (m).
-
Divide the reference standard value for each experiment by m; this is then the correction factor, p, for that experiment.
-
Divide the % tail DNA for each sample by p.
-
Whether this normalisation process is useful can be judged by whether the statistical significance of the experimental result is improved.
Measuring antioxidant resistance
One of the earliest (and simplest) applications of the comet assay in human biomonitoring was the assessment of antioxidant status, by challenging cells ex vivo with an inducer of oxidative damage and measuring the yield of DNA breaks. In the first such study, volunteers took a single dose of 500 mg of vitamin C at breakfast; PBMN cells were isolated from blood taken before and after this dose and irradiated with 2 Gy from a 60Co source: the comet assay then gave the yield of SBs, which was lower following vitamin C supplementation (Green et al. 1994). Ex vivo resistance to H2O2, also, can be affected by a single dose of antioxidant or antioxidant-rich food, such as vitamin C, vitamin E, or β-carotene (Panayiotidis and Collins 1997), a flavonoid-rich meal (Boyle et al. 2000), or dealcoholized red wine (Arendt et al. 2005), whereas to demonstrate an effect of an antioxidant on endogenous DNA oxidation typically takes weeks of daily supplementation (compare the effects of a single kiwifruit dose (Collins et al. 2001b) with a 3-week daily intervention with kiwifruit (Collins et al. 2003)). This test of antioxidant efficacy is also useful in cell culture experiments, demonstrating for instance DNA-protective effects of carotenoids (Lorenzo et al. 2009).
Measuring DNA repair
As we stated earlier, the comet assay has been used since the very earliest days to follow the repair of DNA damage. Rejoining of single-strand breaks induced by ionising radiation or by H2O2 is a simple cellular process; thousands of breaks per cell can be repaired in a matter of half an hour in typical cultured mammalian cells. This means that if precise monitoring of repair kinetics is required, for example, when comparing two cell types, care must be taken to ensure that the initial repair events are not missed. Even a short period of incubation at 37 °C before the first sample is taken will lead to a significant decrease from the initial level of damage. A solution is to prepare a set of slides with cells embedded in agarose, before treating the cells with radiation or H2O2—and to treat the cells on ice. One slide is then placed in lysis solution while the others are quickly transferred to warm medium in a 37 °C incubator, to be placed in lysis solution at the required intervals in the time course. Delay in the commencement of repair resulting from cold shock is not noticeable; and cells are metabolically competent at repair at least for a few hours after embedding (Alapetite et al. 1999; Collins and Horvathova 2001).
Repair of base damage is accomplished by base excision repair (BER), starting with recognition and removal of the damaged base by a glycosylase, followed by cleavage at the baseless sugar (AP-site), removal of the residue of the nucleoside, insertion of one or a few nucleotides, and ligation (Lindahl and Wood 1999). It typically takes a few hours to replace altered bases induced by oxidising or alkylating agents. To follow this repair with the comet assay requires the use of FPG or other lesion-specific enzymes. The damage should be as ‘clean’ as possible, that is, limited to the specific lesion under consideration. A suitable agent for creating base oxidation is visible light in the presence of the photosensitiser Ro 19-8023, which induces predominantly 8-oxoGua. MMS is a useful inducing agent for studying repair of alkylation damage.
Nucleotide excision repair (NER) (Lindahl and Wood 1999) is the process that deals with lesions that distort the double helix—notably bulky adducts and dimerised pyrimidines (induced by UV(C)). Repair of the latter can be followed using T4 endonuclease V in combination with the comet assay. (There is unfortunately no simple enzymic method to detect bulky adducts.) The time course for completion of NER is typically a few hours.
Studying repair of DNA cross-links is more complicated, as the movement of DNA into a comet tail is blocked. However, if cells treated with cross-linker are incubated and, at intervals, X- or γ-irradiated or H2O2-treated prior to performing the comet assay, the % tail DNA (induced by the X-rays) increases with incubation time, indicating the removal of the inhibitory cross-links (Spanswick et al. 2010).
So far, we have discussed cellular repair, often referred to as a ‘challenge assay’. It is relatively straightforward to perform, though if a large number of samples are to be analysed (as in biomonitoring studies), setting up the necessary incubations and preparing gels at frequent intervals are laborious and may be simply unfeasible. There are theoretical problems with the challenge assay, too. Assume we are comparing repair rates in two groups of individuals (for instance, cancer cases and controls, or subjects taking either antioxidant supplements or placebo). The diagram (Fig. 7) shows mean damage levels measured in PBMN cells from the two groups, A and B, after treating them with H2O2. The initial level of damage varied between the two groups, perhaps because group B has higher antioxidant status than group A. After 60 min, both A and B have removed three-quarters of the lesions initially present. Are the two groups equally good at repair because they have removed the same fraction of damage? Or is group B better because the level of damage at the end is lower? Or is group A better because a greater number of lesions were removed? Measuring only a late repair time point is not optimal (though it is often done). The initial rate of lesion removal is biochemically sound, but difficult to measure accurately and really requires the starting level of damage to be the same. The half-time for damage removal might be a more reliable index.

Kinetics of removal of DNA damage by two subjects, a, b, with different susceptibility to damage (shown by different levels of incident damage after the same dose of damaging agent). Both have repaired the same fraction of the incident damage after 60 min. (From Collins and Azqueta 2012b, with permission from Elsevier)
In fact there is an alternative, more biochemical approach—the in vitro DNA repair assay, in which a cell-free extract is prepared and incubated with a DNA substrate containing specific lesions (Fig. 8). It is the nature of the substrate that defines the particular repair pathway being followed. Damaged DNA substrates can be oligonucleotides, plasmids, or—in the case of the comet assay—nucleoids. BER of oxidative base damage employs nucleoids with 8-oxoGua (Collins et al. 2001c). To measure NER, either nucleoids from cells treated with the diolepoxide of benzo(a)pyrene (BPDE) (Langie et al. 2006) or nucleoids with UV damage (Gaivão et al. 2009) can be used. We consider UV to be preferable, as doses can be accurately measured, and the yield of dimers per Jm−2 is known, while on the other hand, BPDE induces a variety of kinds of damage and the yield of damage depends on the cell type.

The comet in vitro DNA repair assay; scheme shown is for assessing capacity for BER of oxidised guanine
In any comparative study with the in vitro repair assay, it is important to start with the same concentration of cells in each extract. The measured repair activity depends on the protein concentration in the extract but is not directly proportional, and adjusting measured rates against protein concentration is of dubious accuracy.
Studying damage and repair at the level of genes
It is natural to wonder about the location of specific genes or regions of DNA within the comet tail, and the first such experiments, with human lymphocytes, were reported by Santos et al. (1997), using FISH with probes specific for centromeric and telomeric DNA as well as three segments of the gene MGMT (coding for the repair enzyme O6-methylguanine methyltransferase). Signals from the DNA probes for telomeric DNA were seen over the comet head region, while in the case of centromeric DNA (several megabases long), FISH signals formed long strings of dots extending from head into tail. The MGMT signals typically formed a linear array. We also employed probes to the MGMT gene (Horvathova et al. 2004)—labelling the two ends with different colours—and found that one end was almost always located close to the head, while the other end was found to a certain extent in the tail when breaks were present. Individual chromosomes were probed by Rapp et al. (2000); those with the highest density of active genes were concentrated in the comet head, after UV(A) irradiation of lymphocytes—perhaps because sites of transcription are located at the nuclear matrix (Cook 1999). An account of the different kinds of probes that can be used, as well a review of findings, has been published (Shaposhnikov et al. 2009).
In addition to elucidating the structural organisation of nuclear DNA, FISH-comets have been examined in connection with human disease. For instance, several cancer-related genes tend to show preferential location in the comet tail after genotoxic treatment of cells, indicating an elevated sensitivity to damage (Glei et al. 2009).
FISH has allowed investigation of the kinetics of repair of individual genes after very low doses of damage—focusing on the notion that transcribed genes are preferentially repaired. (This important concept was established many years ago (Bohr et al. 1985) but in experiments done with supralethal doses of damage.) McKenna et al. (2003) first showed that the rejoining of radiation-induced breaks in the tumour protein p53 (TP53, tumour suppressor) gene was faster than total genomic repair. Preferential repair of this gene has been shown by others, after H2O2 treatment or irradiation, in various cell types (Horvathova et al. 2004; Kumaravel and Jha 2006; Fig. 9). A more extensive study, involving a range of transcribed and non-expressed genes, has yet to be carried out.

Time-course of repair of total and specific DNA studied with the comet assay in combination with FISH. Human PBMN cells were treated with H2O2 and incubated for 20 min. Rejoining of breaks in the genome overall is indicated by the decrease in % tail DNA (broken line, squares). Comets were hybridised with labelled probes for the two ends of the TP53 gene (triangles and inverted triangles); signals from these probes were scored as either in the tail (i.e. in DNA loops containing breaks) or in the head (intact). From Horvathova et al. (2004), with permission from Oxford University Press
Measuring methylation status
Thirty years ago, the link between DNA methylation and cancer was highlighted by Feinberg and Vogelstein (1983), who found hypomethylation of specific genes to be a feature of cells from tumour tissues compared with analogous normal tissue from the same patients. Methylation of cytosines in CpG dinucleotides in particular regions (CpG islands, with high CpG density) is important in regulating gene expression, and changes in gene expression are a feature of transformed phenotypes. Two groups have applied the comet assay to the study of DNA methylation. There are two restriction endonucleases, HpaII and MspI, that recognise the sequence 5′-CCGG-3′ but are inhibited by the presence of methylated cytosine. Wentzel et al. (2010) showed that comets are formed from HepG2 cells when treated at the nucleoid stage (after lysis) with either enzyme and that incubation of cells with 5-azacytidine, a demethylating agent, significantly changed the comet tail intensity. Wasson et al. (2006) used the same enzymes but with a different approach. Demethylation in cultured colon carcinoma cells was achieved by the more natural method of folate deprivation for 14 days, folate being a cofactor in the synthesis of the methyl donor S-adenosylmethionine. Global hypomethylation was demonstrated by a great increase in % tail DNA (after digestion with HpaII/MspI) compared with folate-replete cultures, which was reversed when folate was restored for 7 days. Wasson et al. took the investigation further, by preparing comets and then using FISH to identify the TP53 gene (or rather the DNA region containing that gene) within the comet; it appeared that this region was fully demethylated in only 7 days, whereas globally demethylation was only partial at this time. Applying the comet assay in this way could provide an additional, epigenetic endpoint to human biomonitoring studies.
Viability
Guidelines for in vivo and in vitro genotoxicity testing with the comet assay were published by Tice et al. (2000). They recommended that, in in vitro testing, doses of a test chemical that decrease viability of cells by more than 30 % should be avoided; and similarly, in in vivo experiments, ‘cell viability in the target tissue that is below 70–80 % of that in the control animals may be considered excessive.’ These guidelines are sound and have generally been accepted, though some would allow up to 50 % loss of viability (Burlinson 2012). The criterion has been extended to the study of white blood cells in human biomonitoring; cells are expected to show a viability of 80 % or more to be accepted for analysis. However, the term ‘viability’ tends to be loosely used. It means, literally, ‘capacity for living’. The most commonly used ‘viability test’ is the trypan blue exclusion assay; normal healthy cells are impermeable to this dye, and it is assumed that cells that take up the dye and appear darkly stained under the microscope must be dead. But cells can very easily acquire damage to the membrane which is then repaired, without loss of viability. For example, we found (ESCODD 2003) that when HeLa cells were gently scraped from the culture dish, over 80 % were trypan blue positive, compared with fewer than 20 % if the cells were trypsinised. Yet there was no sign of excessive DNA damage in the scraped compared with the trypsinised cells, and in a separate experiment scraped or trypsinised cells, when replated and incubated, attached and survived equally well.
It is necessary, though, to assess viability, or cytotoxicity, when testing for DNA-damaging effects, to avoid false classification of a chemical as a genotoxin, when in fact the DNA damage caused is secondary to cytotoxicity. It is generally accepted that the best measures of cytotoxicity are based on assays of cell proliferation (Kirkland 2011).
Hedgehogs and viability
The more highly damaged comets have most of the DNA in the tail, with a small head, and are often referred to as ‘hedgehog’ comets. Because some image analysis systems apparently have difficulty in registering these comets (because the head is ill-defined or seems to have separated from the tail), it is not unusual in papers to read that ‘hedgehog comets are excluded from the comet analysis’. Other papers report hedgehog comets as representing apoptotic cells or use their presence as an indication of cytotoxicity. There are several arguments against these interpretations.
-
Apoptosis involves fragmentation of the DNA to the size of oligonucleotides. This is, of course, far beyond the range of detection of the comet assay. ‘Hedgehog’ comets appear when the break frequency is around 3 breaks per 109 daltons (e.g. after X- or γ-irradiation with 10 Gy).
-
Mitochondrial DNA, which is much larger than apoptotic fragments, disperses very quickly during lysis (Shaposhnikov et al. 2006), and so apoptotic fragments would not be detectable after electrophoresis.
-
Apoptosis is irreversible. However, ‘hedgehog’ comets produced by treating cells with H2O2 have disappeared if the cells are incubated for an hour. We have shown that this is not because the DNA of heavily damaged cells has disintegrated and dispersed, leaving a few relatively undamaged cells to recover; there are as many comets after the incubation as before (Lorenzo et al. 2013).
Hedgehog comets can represent an early stage of apoptosis, before massive breakdown has begun (an idea first mooted by Olive et al. 1993). But it is a serious mistake to regard them as indicative of apoptosis, or, by extension, of cytotoxicity.
Applications
Genotoxicity testing
Various organisations and regulatory bodies have an interest in monitoring chemicals for genotoxicity. The European Food Safety Authority (EFSA) has recently published guidelines for testing chemicals found in foods or food-related products (EFSA Scientific Committee 2011), and the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) has recommended procedures for testing the safety of pharmaceuticals (ICH 2011). Of broader relevance are the REACH (Registration, Evaluation, Authorisation and Restriction of Chemical substances) regulations, which require logging of safety information on chemicals in use above a certain level at the European Chemicals Agency (ECHA 2013, http://echa.europa.eu/web/guest/regulations/reach/): many chemicals in everyday use have not so far been properly tested. There is broad agreement over testing strategies (Fig. 10); the initial testing should be in vitro, using the Ames test for reverse mutations in bacteria, and a test for chromosome damage or mutation in mammalian cells. If one or both of these tests is positive, in vivo testing (with rodents) is required, and the in vivo comet assay is acceptable as one of a battery of tests—even though it is not yet covered by OECD guidelines. (JACVAM, the Japanese Centre for the Validation of Alternative Methods, is currently running an international validation trial in preparation for OECD acceptance.) The comet assay is rapid and relatively simple and applicable to any tissues that can be prepared as single cell suspensions. It is regarded as an ‘indicator test,’ particularly useful as a follow-up test to provide information on mechanisms of genotoxicity.

Genotoxicity testing strategy, showing proposed role for comet assay (adapted from EFSA Scientific Committee 2011, with permission)
While the ‘official’ role of the comet assay is limited, it is widely used in the initial screening of pharmaceuticals, cosmetics (for which in vivo testing is banned) and other chemicals for potentially mutagenic effects. In a test of a range of chemicals with HepG2 cells (which retain some biotransformation enzyme activities), the comet assay was as sensitive (in the sense of being able to detect known mutagens/carcinogens, avoiding ‘false negatives’) as assays for micronuclei, chromosome aberrations, and sister chromatid exchanges (Uhl et al. 1999). The comet assay and the chromosome aberration test gave similar verdicts in a study of 13 candidate drugs (Hartmann et al. 2003).
An interesting exercise was carried out by Kirkland and Speit (2008). They found in the literature reports of comet assay tests on 67 chemicals that are known carcinogens but give equivocal or negative results in the micronucleus assay: 90 % were positive in the comet assay. Also, 78 % of non-carcinogens appeared as negative—a relatively high specificity (defined as ability to identify correctly non-genotoxic compounds) giving a reasonably low risk of ‘false positives’.
‘Sensitivity’ has another meaning, namely the ability to detect effects at low concentrations. The criterion for a good genotoxicity test is that it should be able to detect positive compounds at concentrations that are relatively non-cytotoxic: Cell survival can be assessed as the number of live cells present at a certain time (e.g. 24 or 48 h) after treatment, compared with untreated cultures.
It is clear that the sensitivity of the comet assay (in both senses of the word) is limited by the fact that it detects only SBs (and alkali-labile sites), while many genotoxic chemicals cause other kinds of damage, notably base modifications or adducts, which do not appear as breaks until chemical degradation or cellular processing (DNA repair) takes place. Therefore, it should be possible to enhance sensitivity by including digestion with FPG, which converts not just oxidised purines but other kinds of base damage to strand breaks.
In a recent paper (Azqueta et al. 2013b), we describe experiments to test this idea, using six known genotoxic chemicals, two cytotoxic but non-genotoxic chemicals, and three non-cytotoxic, non-genotoxic chemicals. MMS, methylnitrosoruea (MNU), benzo(a)pyrene (B(a)P), and 4-nitroquinoline-1-oxide (4NQO) are known to damage DNA, but give very low levels of SBs at non-cytotoxic concentrations; the yield of breaks after FPG incubation was greatly enhanced—in the case of MMS by a factor of about 25 at a concentration (<10 μM) that has virtually no effect on cell number (Fig. 11). Etoposide, another known genotoxin, showed a dose-dependent increase in SBs with dose, but no increase in FPG-sensitive sites—consistent with its mode of action as a topoisomerase inhibitor rather than a direct DNA-damaging agent. Cyclophosphamide is a bifunctional alkylating agent, cross-linking DNA strands, and so disrupting DNA synthesis. Cross-linking prevents DNA loops from extending under electrophoresis; FPG does nothing to increase the low level of SBs. Triton X-100 is cytotoxic and genotoxic (the latter being secondary to cell killing); SBs showed an increase in cytotoxic concentrations, with no effect of FPG. The other negative compounds, fluometuron, D-mannitol, Tris, and EDTA behaved as expected, with no SBs or FPG-sensitive sites at the concentrations tested (mM or higher) (Fig. 11).

Enhanced detection of DNA damage with FPG. TK-6 cells were treated with different concentrations of MMS (top), Triton X-100 (middle), or EDTA (bottom) for 3 h. Cytotoxicity (broken line) was assessed as relative suspension growth (cell number) at 48 h. DNA breaks (circles) and FPG-sensitive sites (triangles) were measured with the comet assay. From Azqueta et al. 2013b, with permission from Oxford University Press
This modified comet assay, if implemented in genotoxicity testing regimes, would significantly reduce the possibility of falsely classifying genotoxic compounds as negative. It would certainly add to the value of the comet assay as an indicator test, since the presence of FPG-sensitive sites is clearly indicative of base damage.
Human biomonitoring
Most of the published reports using the comet assay in human biomonitoring are based on white blood cells, usually isolated PBMN cells (more often than not referred to as lymphocytes). As mentioned earlier, other tissues (buccal epithelial cells, tear duct epithelial cells, lens epithelial cells, sperm, etc.) require specialised techniques, or deviations from the standard comet assay protocol. When studies are focused on disease, tissue samples may be available (though the lack of corresponding samples from healthy controls is a serious constraint).
There are several recent reviews which illustrate the scope of comet assay applications in molecular epidemiology and summarise the main findings (Dusinska and Collins 2008; Hoelzl et al. 2009; Valverde and Rojas 2009). Here, we briefly describe the main approaches.
Descriptive, or cross-sectional studies: The aim is to take a group of subjects (random, or fitting to some preordained criterion) and to measure biomarkers (including, in this context, DNA damage and/or repair) in order to test a hypothesis. For instance, such a study could seek correlations between oxidised DNA bases or DNA repair on the one hand, and smoking status (smokers versus non-smokers, or number of cigarettes smoked), or sex, or age, or body mass index, or country of residence, or nutritional factors … the possibilities are endless. It can be tempting to claim causal connections where only an association is justified by the data. We found a significant negative correlation between serum concentration of carotenoids (a dietary antioxidant) and DNA-based oxidation (EndoIII-sensitive sites) (Collins et al. 1998)—but concluded that a reasonable interpretation would be that carotenoids are a marker of overall consumption of fruits and vegetables, and this could be the real determinant of DNA damage levels. Correlations between different biomarkers can be informative, especially when genetic polymorphism data are available. It is possible to relate levels of DNA damage or repair to variations in genes coding for phase I/II metabolism enzymes, or enzymes involved in antioxidant protection, or DNA repair pathways (Dusinska et al. 2006; Slyskova et al. 2007; Ryk et al. 2008; Langie et al. 2010).
Intervention studies: A claim that a particular factor—such as a dietary antioxidant, or an environmental pollutant—is responsible for maintaining a certain level of DNA damage can be tested in an intervention study, in which subjects are treated with the specific agent and the effect on DNA damage or repair is monitored. Simply measuring the biomarkers before and after the treatment is not enough; any changes observed could have happened anyway, for various reasons. A better design is the placebo-controlled trial, in which parallel groups are treated with the agent under investigation or with a placebo (or simply with no treatment). It is important that the two groups should not show differences in the measured biomarker at the start of the trial, and however carefully the groups are matched, this sometimes occurs. The best design is therefore a crossover trial. Here, parallel, randomised groups take treatment or placebo (or no treatment) for a set period, and after a washout phase, the groups are reversed so that the subjects originally given the treatment receive the placebo, and vice versa. Thus, all subjects will have been given both placebo and treatment, and so can act as their own controls, which makes for stronger statistics. Giving a nutritional supplement is easy to justify, on the basis of what is already known, or suspected, of the beneficial (or at least harmless) effect of the supplement. Treating subjects with environmental pollutants is obviously ethically problematic, though it has been done with wood smoke (Danielsen et al. 2008) and with air polluted with traffic fumes (Bräuner et al. 2007).
Cohort (prospective) studies: These longitudinal studies are designed to follow a specific section of the population (for example, people born in a certain year—a birth cohort, or particular professions, such as nurses or doctors) for long enough to accumulate significant instances of disease. The relevant biomarkers are measured at recruitment to the study (and possibly at intervals thereafter), or, alternatively, samples taken at recruitment are stored for later analysis. Ultimately, those subjects suffering from the disease of interest can be investigated to identify biomarkers in which they differ from subjects free of the disease. The approach was notably successful in identifying the level of chromosome aberrations or of micronuclei in white blood cells as predictive markers of the risk of cancer (Hagmar et al. 1998; Bonassi et al. 2007). So far, there are few reports of this approach involving the comet assay, in part because the assay has only quite recently been used in this way; but there are certainly some large ongoing cohort studies in which the intention is to follow the participants for many years.
Case–control studies: Patients with a certain disease are compared with disease-free subjects who otherwise match the patients in basic parameters such as age and sex. (Other matching criteria will depend on the nature of the investigation; a lung cancer study in which patients and controls are matched for smoking status would obviously be aimed at identifying relevant factors other than smoking.) There is always a danger of confusing cause and effect; for instance, a low level of DNA repair in cancer patients compared with controls might suggest that poor repair capability leads to a higher steady state level of DNA damage and consequently a greater chance of mutation and transformation; but equally, DNA repair activity might be depressed as a result of the disease. To avoid this difficulty, so-called nested case–control studies can be carried out within a large prospective study. A number of subjects suffering from a disease are carefully matched (for age, sex, smoking status, etc.) with subjects free of the disease within the same cohort. Contemporaneous as well as historic biomarker measurements are compared between the two groups. If a difference is seen between biomarkers measured early in the study when all subjects were disease-free, it is a reasonable assumption that the difference is significant in the aetiology of the disease.
The ComNet project (http://www.comnetproject.org) (Collins et al. 2012) was set up to coordinate the activities of researchers using the comet assay in biomonitoring. Too often, reported studies are based on rather small numbers of subjects, and disagreement between different studies is common and inevitable. The aim is to collect data from as many studies as possible, to carry out a pooled analysis to establish baseline levels of DNA damage of different sorts (which might vary from country to country), and to answer questions such as
-
Are there differences in DNA damage and repair between men and women?
-
Does DNA damage (measured in white blood cells) increase with age?
-
Does DNA repair capacity decline with age?
-
Are higher levels of damage seen in white blood cells of smokers?
Ecogenotoxicology
The vulnerability of the natural environment to human interference—deliberate or unforeseen—is of course a matter of grave concern. Pollution can affect the ecosystem in various ways, one of which is via genotoxic effects, the study of which is known as ecogenotoxicology. The emphasis of environmental toxicology differs from that of epidemiology; instead of focusing on health and disease in individuals, the emphasis is at the species or population level, and factors that affect population size, reproduction, or the gene pool are important. In addition, selected organisms can act as indicators of the presence of toxins—for instance, radioactive compounds, or heavy metals, or pesticide residues—that could have consequences for human populations. These organisms are referred to as sentinel organisms and include snails, mussels, fish, mice, earthworms, insects, and certain plants, covering a wide range of aquatic and terrestrial habitats.
The comet assay is increasingly being applied—often alongside other biomarker assays—in monitoring for toxic effects in these sentinel species (Jha 2008). There are different kinds of investigation. Generally, the first stage is to carry out laboratory experiments, exposing the selected organisms to known DNA-damaging agents, at different concentrations or doses, to determine the sensitivity of the organism (and the appropriateness of the comet assay). Specific procedures may need to be developed to isolate suitable cell types (or nuclei) from the chosen organism. Subsequently, ‘clean’ specimens can be placed in specific locations (those suspected of being contaminated, as well as control, uncontaminated environments) for an appropriate period before being analysed for DNA damage. The ultimate stage in this approach is to collect wild specimens from the test areas; this will give information on the effects of long-term natural exposure.
Conclusions
The comet assay is deceptively simple. It can be performed proficiently after a few days’ tuition or can easily be self-taught. But it is easy unknowingly to adopt bad habits—some of which we have highlighted in this article—and interpretation of results can be invalid if the theoretical principles underlying the assay are misunderstood.
The comet assay is still developing and diversifying. Recent innovations include the high throughput versions of the method, the study of epigenetics in terms of methylation, comets from isolated chromosomes, and the identification of individual genes within the comet.
Probably genotoxicity testing is still the most common application of the assay, and it is gradually gaining acceptance with regulatory bodies. We believe that its usefulness as an ‘indicator assay’, giving information about mechanisms of action of potential carcinogens, is enormously enhanced when it is combined with lesion-specific enzymes. There are many such enzymes, including some artificially modified, still to be tested, and some will surely be added to the repertoire in the future. An enzymic method for detecting bulky adducts would be particularly useful.
Human biomonitoring has been a very productive area of research based on the comet assay. Comparability between laboratories is a besetting problem, though adoption of a standard protocol and a common scoring method should make for more homogeneity. The ComNet project will undertake a pooled analysis of biomonitoring results, attempting to accommodate or allow for the disparities.
The comet assay is underexploited in the area of ecogenotoxicology—in part, surely, because of the difficulty of collecting and transporting samples from sites far from the lab. A ‘field comet assay’, supporting all stages from sample collection to image analysis in the back of a van, would be a real boon.
The outstanding problem is, as it has been for many years, the tedious business of comet scoring. The comet community is waiting for a reliable, affordable, fully automated image analysis system.
Meanwhile, the latest developments and research findings are communicated at the International Comet Assay Workshop every second year, where expert and novice researchers gather to enthuse about their favourite assay.
References
Ahnström G, Erixon K (1981) Measurement of strand breaks by alkaline denaturation and hydroxyapatite chromatography. In: Friedberg EC, Hanawalt PC (eds) DNA Repair. A Laboratory Manual of Research Procedures. Marcel Dekker, New York, pp 403–418
Alapetite C, Thirion P, de la Rochefordière A, Cosset J-M, Moustacchi E (1999) Analysis by alkaline comet assay of cancer patients with severe reactions to radiotherapy: defective rejoining of radioinduced DNA strand breaks in lymphocytes of breast cancer patients. Int J Cancer 83:83–90
Al-Salmani K, Abbas HHK, Schulpen S, Karbaschi M, Abdalla I, Bowman KJ, So KK, Evans MD, Jones GDD, Godschalk RW, Cooke MS (2011) Simplified method for the collection, storage, and comet assay analysis of DNA damage in whole blood. Free Rad Biol Med 51:719–725
Arendt B, Ellinger S, Kekic K, Geus L, Fimmers R, Spengler U, Muller WU, Goerlich R (2005) Single and repeated moderate consumption of native or dealcoholized red wine show different effects on antioxidant parameters in blood and DNA strand breaks in peripheral leukocytes in healthy volunteers: a randomized controlled trial [ISRCTN68505294]. Nutr J 4:33
Azqueta A, Gutzkow KB, Brunborg G, Collins AR (2011a) Towards a more reliable comet assay; Optimising agarose concentration, unwinding time and electrophoresis conditions. Mutat Res 724:41–45
Azqueta A, Meier S, Priestley C, Gutzkow KB, Brunborg G, Sallette J, Soussaline F, Collins A (2011b) The influence of scoring method on variability in results obtained with the comet assay. Mutagenesis 26:393–399
Azqueta A, Gutzkow KB, Priestley CC, Meier S, Walker JS, Brunborg G, Collins A (2013a) A comparative performance test of standard, medium- and high-throughput comet assays. Toxicol in Vitro 27:768-773
Azqueta A, Arbillaga L, Lopez de Cerain A, Collins A (2013b) Enhancing the sensitivity of the comet assay as a genotoxicity test, by combining it with bacterial repair enzyme FPG. Mutagenesis 28:271-277
Berdal KG, Johansen RF, Seeberg E (1998) Release of normal bases from intact DNA by a native DNA repair enzyme. EMBO J 17:363–367
Bohr VA, Smith CA, Okumoto DS, Hanawalt PC (1985) DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell 40:359–369
Bonassi S, Znaor A, Ceppi M, Lando C, Chang WP, Holland N, Kirsch-Volders M, Zeiger E, Ban S, Barale R, Bigatti MP, Bolognesi C, Cebulska-Wasilewska A, Fabianova E, Fucic A, Hagmar L, Joksic G, Martelli A, Migliore L, Mirkova E, Scarfi MR, Zijno A, Norppa H, Fenech M (2007) An increased micronucleus frequency in peripheral blood lymphocytes predicts the risk of cancer in humans. Carcinogenesis 28:625–631
Boyle SP, Dobson VL, Duthie SJ, Kyle JAM, Collins AR (2000) Absorption and DNA protective effects of flavonoid glycosides from an onion meal. Eur J Nutr 39:213–223
Bradley MO, Kohn KW (1979) X-ray induced DNA double strand break production and repair in mammalian cells as measured by neutral filter elution. Nucl Acids Res 7:793–804
Bräuner EV, Forchhammer L, Moller P, Simonsen J, Glasius M, Wahlin P, Raaschou-Nielsen O, Loft S (2007) Exposure to ultrafine particles from ambient air and oxidative stress-induced DNA damage. Env Health Persp 115:1177–1182
Brendler-Schwaab SY, Schmezer P, Liegibel U, Weber S, Michalek K, Tompa A, Pool-Zobel BL (1994) Cells of different tissues for in vitro and in vivo studies in toxicology: compilation of isolation methods. Toxicol In Vitro 8:1285–1302
Burlinson B (2012) The in vitro and in vivo comet assays. In: Parry JM, Parry EM (eds) Genetic toxicology: principles and methods, pp 143–163
Collins AR, Azqueta A (2012a) Single-cell gel electrophoresis combined with lesion-specific enzymes to measure oxidative damage to DNA. Meth Cell Biol 112:69–92
Collins AR, Azqueta A (2012b) DNA repair as a biomarker in human biomonitoring studies; further applications of the comet assay. Mutat Res 736:122–129
Collins AR, Horvathova E (2001) Oxidative DNA damage, antioxidants and DNA repair; applications of the comet assay. Biochem Soc Trans 29:337–341
Collins AR, Duthie SJ, Dobson VL (1993) Direct enzymic detection of endogenous oxidative base damage in human lymphocyte DNA. Carcinogenesis 14:1733–1735
Collins A, Cadet J, Epe B, Gedik C (1997a) Problems in the measurement of 8-oxoguanine in human DNA. Report of a workshop, DNA Oxidation, held in Aberdeen, UK, 19–21 January, 1997. Carcinogenesis 18:1833–1836
Collins AR, Mitchell DL, Zunino A, de Wit J, Busch D (1997b) UV-sensitive rodent mutant cell lines of complementation groups 6 and 8 differ phenotypically from their human counterparts. Environ Mol Mutagen 29:152–160
Collins AR, Dobson VL, Dusinská M, Kennedy G, Stetina R (1997c) The comet assay: what can it really tell us? Mutat Res 375:183–193
Collins AR, Olmedilla B, Southon S, Granado F, Duthie SJ (1998) Serum carotenoids and oxidative DNA damage in human lymphocytes. Carcinogenesis 19:2159–2162
Collins AR, Dusinska M, Horska A (2001a) Detection of alkylation damage in human lymphocyte DNA with the comet assay. Acta Biochim Polon 48:611–614
Collins AR, Dusinska M, Horvathova E, Munro E, Savio M, Stetina R (2001b) Inter-individual differences in DNA base excision repair activity measured in vitro with the comet assay. Mutagenesis 16:297–301
Collins BH, Horska A, Hotten PM, Riddoch C, Collins AR (2001c) Kiwifruit protects against oxidative DNA damage in human cells and in vitro. Nutr Cancer 39:148–153
Collins AR, Harrington V, Drew J, Melvin R (2003) Nutritional modulation of DNA repair in a human intervention study. Carcinogenesis 24:511–515
Collins AR, Azqueta Oscoz A, Brunborg G, Gaivão I, Giovannelli L, Kruszewski M, Smith CC, Stetina R (2008) The comet assay: topical issues. Mutagenesis 23:143–151
Collins A, Anderson D, Coskun E, Dhawan A, Dusinska M, Koppen G, Kruszewski M, Moretti M, Rojas E, Speit G, Valverde M, Bonassi S (2012) Launch of the ComNet (comet network) project on the comet assay in human population studies during the International Comet Assay Workshop meeting in Kusadasi, Turkey (September 13–16, 2011). Mutagenesis 27:385–386
Cook PR (1999) The organization of replication and transcription. Science 284:1790–1795
Cook PR, Brazell IA, Jost E (1976) Characterization of nuclear structures containing superhelical DNA. J Cell Sci 22:303–324
Cortés-Gutiérrez EI, Dávila-Rodríguez MI, Fernández JL, López-Fernández C, Gosálbez A, Gosálvez J (2011) New application of the comet assay: chromosomeGÇôComet assay. J Histochem Cytochem 59:655–660
Danielsen PH, Bräuner EV, Barregard L, Sällsten G, Wallin M, Olinski R, Rozalski R, Møller P, Loft S (2008) Oxidatively damaged DNA and its repair after experimental exposure to wood smoke in healthy humans. Mutat Res 642:37–42
Dusinska M, Collins A (1996) Detection of oxidised purines and UV-induced photoproducts in DNA of single cells, by inclusion of lesion-specific enzymes in the comet assay. Alt Lab Animals 24:405–411
Dusinska M, Collins AR (2008) The comet assay in human biomonitoring: gene environment interactions. Mutagenesis 23:191–205
Dusinska M, Dzupinkova Z, Wsolova L, Harrington V, Collins AR (2006) Possible involvement of XPA in repair of oxidative DNA damage deduced from analysis of damage, repair and genotype in a human population study. Mutagenesis 21:205–211
Duthie SJ, McMillan P (1997) Uracil misincorporation in human DNA detected using single cell gel electrophoresis. Carcinogenesis 18:1709–1714
Duthie SJ, Ma A, Ross MA, Collins AR (1996) Antioxidant supplementation decreases oxidative DNA damage in human lymphocytes. Cancer Res 56:1291–1295
EFSA Scientific Committee (2011) Scientific opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA J 9:2379. doi:10.2903/j.efsa.2011.2379
Ersson C, Möller L (2011) The effects on DNA migration of altering parameters in the comet assay protocol such as agarose density, electrophoresis conditions and durations of the enzyme or the alkaline treatments. Mutagenesis 26:689–695
Ersson C, Møller P, Forchhammer L, Loft S, Azqueta A, Godschalk RWL, van Schooten F-J, Jones GDD, Higgins JA, Cooke MS, Mistry V, Karbaschi M, Phillips DH, Sozeri O, Routledge MN, Nelson-Smith K, Riso P, Porrini M, Matullo G, Allione A, Stępnik M, Ferlińska M, Teixeira JP, Costa S, Corcuera L-A, López de Cerain A, Laffon B, Valdiglesias V, Collins AR, Möller L (2013) An ECVAG inter-laboratory validation study of the comet assay: inter- and intra-laboratory variation of DNA strand breaks and FPG-sensitive sites in human mononuclear cells. Mutagenesis 28:279-286
ESCODD (2002a) Comparative analysis of baseline 8-oxo-7,8-dihydroguanine in mammalian cell DNA, by different methods in different laboratories: an approach to consensus. Carcinogenesis 23:2129–2133
ESCODD (2002b) Inter-laboratory validation of procedures for measuring 8-oxo-7,8-dihydroguanine/8-oxo-7,8-dihydro-2′-deoxyguanosine in DNA. Free Rad Res 36:239–245
ESCODD (2003) Measurement of DNA oxidation in human cells by chromatographic and enzymic methods. Free Rad Biol Med 34:1089–1099
ESCODD, Gedik CM, Collins A (2005) Establishing the background level of base oxidation in human lymphocyte DNA: results of an interlaboratory validation study. FASEB J 19:82–84
Feinberg AP, Vogelstein B (1983) Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301:89–92
Forchhammer L, Johansson C, Loft S, Möller L, Godschalk RWL, Langie S, Jones GDD, Kwok RWL, Collins AR, Azqueta A, Phillips DH, Sozeri O, Stepnik M, Palus J, Vogel U, Wallin H, Routledge MN, Handforth C, Allione A, Matullo G, Teixeira JP, Costa S, Riso P, Porrini M, Møller P (2010) Variation in the measurement of DNA damage by comet assay measured by the ECVAG inter-laboratory validation trial. Mutagenesis 25:113–123
Forchhammer L, Ersson C, Loft S, Möller L, Godschalk RWL, van Schooten FJ, Jones GDD, Higgins JA, Cooke M, Mistry V, Karbaschi M, Collins AR, Azqueta A, Phillips DH, Sozeri O, Routledge MN, Nelson-Smith K, Riso P, Porrini M, Matullo G, Allione A, Stepnik M, Komorowska M, Teixeira JP, Costa S, Corcuera LA, Lopez de Cerain A, Laffon B, Valdiglesias V, Møller P (2012) Inter-laboratory variation in DNA damage using a standard comet assay protocol. Mutagenesis 27:665–672
Gaivão I, Piasek A, Brevik A, Shaposhnikov S, Collins AR (2009) Comet assay-based methods for measuring DNA repair in vitro; estimates of inter- and intra-individual variation. Cell Biol Toxicol 25:45–52
Gichner T, Znidar I, Wagner ED, Plewa MJ (2009) The use of higher plants in the comet assay. In: Anderson D, Dhawan A (eds) The comet assay in toxicology. Royal Society of Chemistry, London, pp 98–119
Glei M, Hovhannisyan G, Pool-Zobel BL (2009) Use of comet-FISH in the study of DNA damage and repair: review. Mutat Res 681:33–43
Green MHL, Lowe JE, Waugh APW, Aldridge KE (1994) Effect of diet and vitamin C on DNA strand breakage in freshly-isolated human white blood cells. Mutat Res 316:91–102
Gutzkow KB, Langleite TM, Meier S, Graupner A, Collins A, Brunborg G (2013) High throughput comet assay using 96 minigels. Mutagenesis 28:333-340
Hagmar L, Bonassi S, Stromberg U, Brogger A, Knudsen LE, Norppa H, Reuterwall C (1998) Chromosomal aberrations in lymphocytes predict human cancer: a report from the European Study Group on Cytogenetic Biomarkers and Health (ESCH). Cancer Res 58:4117–4121
Hartmann A, Agurell E, Beevers C, Brendler-Schwaab S, Burlinson B, Clay P, Collins A, Smith A, Speit G, Thybaud V, Tice RR (2003) Recommendations for conducting the in vivo alkaline Comet assay. Mutagenesis 18:45–51
Ho CK, Choi SW, Siu PM, Benzie IFF (2011) Cryopreservation and storage effects on cell numbers and DNA damage in human lymphocytes. Biopres Biobank 9:343–347
Hoelzl C, Knasmüller S, Misik M, Collins A, Dusinska M, Nersesyan A (2009) Use of single cell gel electrophoresis assays for the detection of DNA-protective effects of dietary factors in humans: recent results and trends. Mutat Res 681:68–79
Horvathova E, Dusinska M, Shaposhnikov S, Collins AR (2004) DNA damage and repair measured in different genomic regions using the comet assay with fluorescent in situ hybridization. Mutagenesis 19:269–276
Hughes CM, Lewis SEM, McKelvey-Martin VJ, Thompson W (1996) A comparison of baseline and induced DNA damage in human spermatozoa from fertile and infertile men, using a modified comet assay. Molec Human Repro 2:613–619
ICH harmonised tripartite guideline S2 (R1). Guidance on genotoxicity testing and data interpretation for pharmaceuticals intended for human use. http://www.ich.org/products/guidelines/safety/article/safety-guidelines.html. 2011. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use
Jha AN (2008) Ecotoxicological applications and significance of the comet assay. Mutagenesis 23:207–221
Johansson C, Møller P, Forchhammer L, Loft S, Godschalk RWL, Langie SAS, Lumeij S, Jones GDD, Kwok RWL, Azqueta A, Phillips DH, Sozeri O, Routledge MN, Charlton AJ, Riso P, Porrini M, Allione A, Matullo G, Palus J, Stepnik M, Collins AR, Möller L (2010) An ECVAG trial on assessment of oxidative damage to DNA measured by the comet assay. Mutagenesis 25:125–132
Kirkland D (2011) Improvements in the reliability of in vitro genotoxicity testing. Expert Opin Drug Metab Toxicol 7:1513–1520
Kirkland D, Speit G (2008) Evaluation of the ability of a battery of three in vitro genotoxicity tests to discriminate rodent carcinogens and non-carcinogens: III. Appropriate follow-up testing in vivo. Mutat Res 654:114–132
Kumaravel TS, Jha AN (2006) Reliable comet assay measurements for detecting DNA damage induced by ionising radiation and chemicals. Mutat Res 605:7–16
Langie SAS, Knaapen AM, Brauers KJJ, van Berlo D, van Schooten F-J, Godschalk RWL (2006) Development and validation of a modified comet assay to phenotypically assess nucleotide excision repair. Mutagenesis 21:153–158
Langie SAS, Wilms LC, Hämäläinen S, Kleinjans JCS, Godschalk RWL, van Schooten FJ (2010) Modulation of nucleotide excision repair in human lymphocytes by genetic and dietary factors. Br J Nutr 103:490–501
Li Q, Laval J, Ludlum DB (1997) FPG protein releases a ring-opened N-7 guanine adduct from DNA that has been modified by sulfur mustard. Carcinogenesis 18:1035–1038
Lindahl T, Wood RD (1999) Quality control by DNA repair. Science 286:1897–1905
Lorenzo Y, Azqueta A, Luna L, Bonilla F, Dominguez G, Collins AR (2009) The carotenoid b-cryptoxanthin stimulates the repair of DNA oxidation damage in addition to acting as an antioxidant in human cells. Carcinogenesis 30:308–314
Lorenzo Y, Costa S, Collins AR, Azqueta A (2013) The comet assay, DNA damage, DNA repair and cytotoxicity: hedgehogs are not always dead. Mutagenesis. doi: 10.1093/mutage/get018
Lovell DP (2009) Statistical analysis of comet assay data. In: Anderson D, Dhawan A (eds) The comet assay in toxicology. Royal Society of Chemistry, London, pp 424–450
Lovell DP, Omori T (2008) Statistical issues in the use of the comet assay. Mutagenesis 23:171–182
McKenna DJ, Rajab NF, McKeown SR, McKerr G, McKelvey-Martin VJ (2003) Use of the comet-FISH assay to demonstrate repair of the TP53 gene region in two human bladder carcinoma cell lines. Radiat Res 159:49–56
McNamee JP, McLean JRN, Ferrarotto CL, Bellier PV (2000) Comet assay: rapid processing of multiple samples. Mutat Res 466:63–69
Mercey E, Obeïd P, Glaise D, Calvo-Muñoz ML, Guguen-Guillouzo C, Fouqué B (2010) The application of 3D micropatterning of agarose substrate for cell culture and in situ comet assays. Biomaterials 31:3156–3165
Olive PL, Frazer G, Banath JP (1993) Radiation-induced apoptosis measured in TK6 human B lymphoblast cells using the comet assay. Rad Res 136:130–136
Oliveira R, Johansson B (2012) Quantitative DNA damage and repair measurement with the yeast comet assay. In: Bjergbæk L (ed) DNA repair protocols, Meth Mol Biol 920. Humana Press, New York, pp 101–109
Osnes-Ringen Ø, Azqueta AO, Moe MC, Zetterström C, Röger M, Nicolaissen B, Collins AR (2012) DNA damage in lens epithelium of cataract patients in vivo and ex vivo. Acta Ophthalm. doi:10.1111/j.1755-3768.2012.02500.x [Epub ahead of print]
Ostling O, Johanson KJ (1984) Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. Biochem Biophys Res Commun 123:291–298
Panayiotidis M, Collins AR (1997) Ex vivo assessment of lymphocyte antioxidant status using the comet assay. Free Rad Res 27:533–537
Pourrut B, Séverine J, Silvestre J, Pinelli E (2011) Lead-induced DNA damage in Vicia faba root cells: potential involvement of oxidative stress. Mut Res Genet Toxicol Environ Mutagen 726:123–128
Rapp A, Bock C, Dittmar H, Greulich K-O (2000) UV-A breakage sensitivity of human chromosomes as measured by COMET-FISH depends on gene density and not on the chromosome size. J Photochem Photobiol B Biol 56:109–117
Rojas E, Valverde M, Lopez MC, Naufal I, Sanchez I, Bizarro P, Lopez I, Fortoul TI, Ostrosky-Wegman P (2000) Evaluation of DNA damage in exfoliated tear duct epithelial cells from individuals exposed to air pollution assessed by single cell gel electrophoresis assay. Mutat Res 468:11–17
Ryk C, Routledge MN, Allan JM, Wild CP, Kumar R, Lambert B, Hou S (2008) Influence of DNA repair gene polymorphisms on the initial repair of MMS-induced DNA damage in human lymphocytes as measured by the alkaline comet assay. Environ Mol Mutagen 49:669–675
Santos SJ, Singh NP, Natarajan AT (1997) Fluorescence in situ hybridization with comets. Exp Cell Res 232:407–411
Sasaki YF, Izumiyama F, Nishidate E, Ohta T, Ono T, Matsusaka N, Tsuda S (1997) Simple detection of in vivo genotoxicity of pyrimethamine in rodents by the modified alkaline single-cell gel electrophoresis assay. Mutat Res 392:251–259
Shaposhnikov S, Larsson C, Henriksson S, Collins A, Nilsson M (2006) Detection of Alu sequences and mtDNA in comets using padlock probes. Mutagenesis 21:243–247
Shaposhnikov S, Frengen E, Collins AR (2009) Increasing the resolution of the comet assay using fluorescent in situ hybridization: a review. Mutagenesis 24:383–389
Shaposhnikov S, Azqueta A, Henriksson S, Meier S, Gaivão I, Huskisson NH, Smart A, Brunborg G, Nilsson M, Collins AR (2010) Twelve-gel slide format optimised for comet assay and fluorescent in situ hybridisation. Toxicol Lett 195:31–34
Singh NP, McCoy MT, Tice RR, Schneider EL (1988) A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 175:184–191
Slyskova J, Dusinska M, Kuricova M, Soucek P, Vodickova L, Susova S, Naccarati A, Tulupova E, Vodicka P (2007) Relationship between the capacity to repair 8-oxoguanine, biomarkers of genotoxicity and individual susceptibility in styrene-exposed workers. Mut Res Genet Toxicol Environ Mutagen 634:101–111
Smith CC, O’Donovan MR, Martin EA (2006) hOGG1 recognizes oxidative damage using the comet assay with greater specificity than FPG or ENDOIII. Mutagenesis 21:185–190
Spanswick V, Hartley J, Hartley J (2010) Measurement of DNA interstrand crosslinking in individual cells using the single cell gel electrophoresis (Comet) assay. In: Fox Keith R (ed) Drug-DNA interaction protocols, Meth Mol Biol 613. Humana Press, London, pp 267–282
Speit G, Schütz P, Bonzheim I, Trenz K, Hoffmann H (2004) Sensitivity of the FPG protein towards alkylation damage in the comet assay. Toxicol Lett 146:151–158
Stang A, Witte I (2009) Performance of the comet assay in a high-throughput version. Mutat Res 675:5–10
Szeto YT, Benzie IFF, Collins AR, Choi SW, Cheng CY, Yow CMN, Tse MMY (2005) A buccal cell model comet assay: development and evaluation for human biomonitoring and nutritional studies. Mutat Res 578:371–381
Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu JC, Sasaki YF (2000) Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen 35:206–221
Uhl M, Helma C, Knasmüller S (1999) Single-cell gel electrophoresis assays with human-derived hepatoma (Hep G2) cells. Mutat Res 441:215–224
Valverde M, Rojas E (2009) Environmental and occupational biomonitoring using the comet assay. Mutat Res 681:93–109
Wasson GR, McGlynn AP, McNulty H, O’Reilly SL, McKelvey-Martin VJ, McKerr G, Strain JJ, Scott J, Downes CS (2006) Global DNA and p53 region-specific hypomethylation in human colonic cells is induced by folate depletion and reversed by folate supplementation. J Nutr 136:2748–2753
Wentzel JF, Gouws C, Huysamen C, Ev Dyk, Koekemoer G, Pretorius PJ (2010) Assessing the DNA methylation status of single cells with the comet assay. Anal Biochem 400:190–194
Wood DK, Weingeist DM, Bhatia SN, Engelward BP (2010) Single cell trapping and DNA damage analysis using microwell arrays. Proc Natl Acad Sci 107:10008–10013
Acknowledgments
The data shown in Fig. 6a were from reference control samples included with samples analysed for Kraft Foods as part of an intervention trial. We thank Naouale el-Yamani and Yolanda Lorenzo for access to unpublished data.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Azqueta, A., Collins, A.R. The essential comet assay: a comprehensive guide to measuring DNA damage and repair. Arch Toxicol 87, 949–968 (2013). https://doi.org/10.1007/s00204-013-1070-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-013-1070-0