
Abstract
Citrinin (CIT), produced by several Penicillium, Aspergillus, and Monascus species, has been detected as contaminant in feeds, grains, and other food commodities. CIT can co-occur with ochratoxin A (OTA), a mycotoxin also known for its nephrotoxicity, and this raises concern regarding possible combined effects. But, in contrast to OTA, data on CIT contamination in foods for human consumption are scarce, and CIT biomonitoring has not been conducted so far due a lack of suitable methods for human specimen. Thus, it was the aim of the present study to develop sensitive methods for the analysis of CIT in human blood and urine to investigate human exposure. To this end, we assessed different methods of sample preparation and instrumental analysis for these matrices. Clean-up of blood plasma by protein precipitation followed by LC–MS/MS-based analysis allowed robust detection of CIT (LOD 0.07 ng/mL, LOQ 0.15 ng/mL). For urine, sample clean-up by an immunoaffinity column (CitriTest®) proved to be clearly superior to SPE with RP18 material for subsequent analysis by LC–MS/MS. For CIT and its metabolite dihydrocitrinone (HO-CIT), the LOD and LOQ determined by external calibration curves in matrix were 0.02 and 0.05 ng/mL for CIT, and those for HO-CIT were 0.05 and 0.1 ng/mL urine. The newly developed method was applied in a small pilot study: CIT was present in all plasma samples from 8 German adults, at concentrations ranging from 0.11 to 0.26 ng/mL. The molar (nM) concentrations of CIT are similar to those measured for OTA in these samples as a result of dietary mycotoxin intake. CIT was detected in 8/10 urines (from 4 adults and 6 infants) in a range of 0.16–0.79 ng/mL, and HO-CIT was present in 5/10 samples at similar concentrations. Thus, CIT is excreted in urine as parent compound and also as metabolite. These first results in humans point to the need for further studies on CIT exposure.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Citrinin (CIT) is produced by several fungal species of the genera Penicillium, Aspergillus, and Monascus, and this mycotoxin has been detected as a natural contaminant in grains, foods, feeds and other commodities (Frank 1992; Xu et al. 2006). CIT is known to exert nephrotoxic effects in several animal species, and it has been implicated as a potential causative agent in human endemic Balkan nephropathy together with ochratoxin A (OTA), another nephrotoxic mycotoxin (IARC 1986; Petkova-Bocharova et al. 1991). This view is supported by the co-occurence of CIT and OTA in foods from this region (Flajs and Peraica 2009; Vrabcheva et al. 2000). Both mycotoxins have been detected also in grains in Germany (Schmidt et al. 2003) and in breakfast cereals in France (Molinie et al. 2005).
Yet, compared with reports and surveys on OTA contamination of foods and feeds, there are relatively few data on the occurrence of CIT in commodities intended for human consumption (Table 1 in Flajs and Peraica 2009; Xu et al. 2006). This is due to the fact that there are presently no legal requirements to measure CIT in food and feed (EFSA 2012) and because CIT analysis in these matrices is not trivial (Schmidt et al. 2003; Xu et al. 2006). On the other hand, there is a clear need to assess human exposure to CIT, also in the light of possible combined toxic effects of CIT and OTA (EFSA 2012; Flajs and Peraica 2009; Frank 1992).
In principle, human exposure to mycotoxins can be assessed by two approaches: analysis of levels in foods combined with food intake values and/or by measuring levels in blood or urine samples (biomonitoring). For OTA, such data are available for many European countries (EC/SCOOP 2002) and document that dietary exposure to this mycotoxin is widespread in the general population. OTA is frequently detected in blood (plasma or serum samples) from people in many countries and show some variation for different geographical locations and often with higher OTA blood levels in people from endemic (BEN) versus non-endemic regions of the Balkan and in patients diagnosed with kidney disease (data reviewed in Aslam et al. 2012; Degen 2011; Duarte et al. 2011; Märtlbauer et al. 2009). More variable OTA levels were reported in urine samples (Duarte et al. 2011; Gilbert et al. 2001; Muñoz et al. 2010; Scott 2005). As urine sampling is non-invasive, OTA analysis in this matrix is a promising alternative as a biomarker of exposure, especially in infants, although the relationship at the individual level between dietary intake and urinary biomarker needs to be developed further, including temporal variations in the presented levels (Duarte et al. 2011).
In contrast, biomonitoring of CIT has not been conducted so far, probably due to a lack of suitable analytical methods. In his review, H. K. Frank pointed out that “reports on the occurrence of citrinin in the human organism are not available”. This is still true, except for a short report on the (qualitative) detection of CIT in one human urine, yet with no details on the origin of the human sample and the CIT concentration (Orti et al. 1986), and one positively tested urine when a new multi-mycotoxin method was applied to 40 samples (Ediage et al. 2012). Therefore, it was the aim of the present study to develop sensitive methods suitable for the analysis of CIT in human blood and urine.
Materials and methods
Reagents, biochemicals, and standards
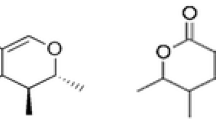
Methanol, acetonitrile (ACN), and acetic acid (96 %) were obtained from Merck (Darmstadt, Germany). Solvents used as mobile phase were HPLC and LC–MS grade, respectively. Citrinin (CIT) (CAS 518-75-2) (5 mg, purity >98 %) was purchased from Sigma-Aldrich (Taufkirchen, Germany), and solutions of CIT prepared in methanol were calibrated spectrophotometrically at 321 nm using the molar extinction coefficient 5,490 M−1 cm−1 (Molinie et al. 2005). Dihydrocitrinone (HO-CIT) (CAS 65718-85-6), a metabolite of citrinin (purity 98.9 %), was from AnalytiCon Discovery GmbH (Potsdam, Germany). Working standard solutions of CIT and HO-CIT (see Fig. 1 for structures) were prepared weekly from a stock solution of 200 μg CIT/mL ACN and 500 μg HO-CIT/mL ACN, by dilutions in methanol in a range of 1–100 ng/mL. CIT in methanol solutions showed some instability after 3-month storage at −20 °C, evident as additional peaks in HPLC. This was much less pronounced, but in line with observations by Clarke et al. (2006) on degradation of CIT in aqueous methanolic solutions. Experience for HO-CIT is lacking.

Citrinin (left) and dihydrocitrinone (right)
Sample collection
In this pilot study, healthy volunteers were asked to provide blood and urine samples, after signing an informed consent sheet. Blood samples were collected in winter 2010 in Dortmund, Germany, from a group of 5 men and 4 women (ages ranged between 16 and 58 years). Plasma was obtained from whole blood by centrifugation at 2,380 relative centrifugal force (rcf) (Labofuge 400R Heraeus, Germany) and kept frozen at −20 °C until analysis. In 2012, urine samples from 10 volunteers were collected: 4 male adults (20–58 years) and 6 infants (age below 2 years). Urine samples (20 mL) were centrifuged at 3,940 rcf and then frozen and stored at −20 °C.
Enzymatic cleavage of conjugates
To account for the possible presence of phase II metabolites (i.e. glucuronide or sulphate conjugates), all urine and plasma samples were enzymatically treated with β-Gluc/ArylSult prior to further sample clean-up (Muñoz et al. 2010).
Sample extraction
Plasma samples
The method for blood plasma consisted of a protein precipitation step as follows: 1 mL plasma was mixed with acetonitrile (1:1, v/v). The sample was then centrifuged at 9,300 rcf (Centrifuge 5415D, Eppendorf, Germany) for 3 min. 1 mL of the upper layer was evaporated to dryness under a gentle N2 stream at a temperature of 40 °C. The sample was then reconstituted in 300 μL methanol for chromatographic analysis.
Urine samples
Due to the complex urine matrix and probably low concentrations of analytes, sample clean-up was considered necessary. Thus, for the analysis of CIT, two clean-up methods were tested: C18 solid-phase extraction (SPE) and immunoaffinity column (IAC) extraction.
Solid-phase extraction C18 (SPE): 5 mL urine was mixed with 5 mL of 1 mM acetic acid in water. The mixture was loaded onto a C18 cartridge (LiChrolut® RP-18, 40–63 μm, 500 mg, 3 mL from Merck), previously conditioned with 2 mL methanol and 2 mL water. The column was rinsed with 5 mL water followed by 5 mL of methanol/water (1:4, v/v) and dried by passing air through it. CIT was eluted with 3 mL methanol. The eluate was evaporated to dryness under a stream of nitrogen at 40 °C, and the residue was dissolved in 500 μL methanol and filtered through a 0.45-μm-pore-size Teflon syringe filter prior to HPLC analysis.
Immunoaffinity column (IAC) extraction: With the aim to compare the SPE with another frequently used clean-up method for biological samples, IAC for CIT was used as clean-up step for urine samples. CitriTest® columns (Vicam®, Watertown, MA, USA) were used for the clean-up. Five mL human urine was diluted with 5 mL of 1 mM acetic acid in water; the sample was mixed in a rotary shaker for 15 min and then quantitatively loaded on the CitriTest® column at a flow rate of 1 drop/s. The column was washed twice with 5 mL distilled water, then CIT was eluted (flow rate 1 drop/s) from the column using 4 mL of methanol. The eluate was evaporated to dryness under a stream of nitrogen at 40 °C, and the residue was dissolved in 500 μL methanol. Thus, the enrichment factor was 10.
HPLC analysis with fluorescence detection
Sample extracts and standards were analysed using an HPLC system (Shimadzu, Germany), consisting of two LC-10AS pumps, RF-10Axl fluorescence detector, SIL-10ADvp auto-injector, CBM-20A controller, and Shimadzu LC solution software. A C18 column Nucleodur® Sphinx EC 125/3 (3-μm material) from Macherey & Nagel (Düren, Germany) was used for the separation of the analytes. The injection volume was 40 μL, and the analysis was performed at a column temperature of 22 °C. The mobile phases (flow rate 0.4 mL/min) were the following: phase A water/methanol/acetic acid (96 %) (69.5:30:0.5, v/v/v) and phase B methanol/acetic acid (96 %) (99.5:0.5, v/v). The stepwise gradient was 0–4 min 100 % A, 4.5–17.5 min 45 % A, 18–20 min 20 % A, re-equilibration 21–30 min 100 % A. The fluorescence detector was set at 330-nm excitation and 500-nm emission wavelengths (Reinhard and Zimmerli 1999). The retention time for CIT was 12.5 min (Fig. 2).

Chromatogram of CIT standard solution (100 ng/mL in methanol) analysed by HPLC with fluorescence detection
LC–MS/MS analysis
Liquid chromatography with tandem mass spectrometry was used to measure CIT and its metabolite HO-CIT in urine and plasma samples. Detection was performed with a 1,200-L Quadrupole MS/MS equipped with an electrospray ionization (ESI) source and a Prostar® Varian HPLC system and Varian MS Workstation version 6.9.1 data system (Agilent Technologies, Germany). The following settings were used: nitrogen as drying gas (19 psi), gas temperature 200 °C, and argon used as collision gas (2.0 mTorr). Chromatographic separation was carried out at 21 °C on a Nucleosil® 100-5 C18 HD 125 × 3 mm column with ammonium formate 1 mM in water (mobile phase A) and ammonium formate 1 mM in methanol (mobile phase B) as eluents in the following gradient: 0–1 min 20 % B, 1–3 min 20–75 % B, 3–11 min 75 % B, 11–12 min 75–100 % B, 12–14 min 100 % B (column wash), 14–14.5 min 100–20 % B, and from 14.5 to 20 min 20 % B (re-equilibration). The flow rate was 0.3 mL/min. ESI–MS/MS was executed by multiple reaction monitoring (MRM) in negative ion mode. The specific transitions of precursor ion and product ion were as follows: 249.0 → 204.7 m/z (loss of the carboxyl group) and 249.0 → 176.7 for CIT. The optimized collision energy (CE) was 15 and 19 eV, respectively. For HO-CIT, the transitions of precursor and product ions were 265.0 → 221.0 and 265.0 → 176.7 m/z with an optimized CE of 16 and 20 eV, in that order. The retention time for CIT and its metabolite HO-CIT was 7.3 and 6.8 min (Fig. 3).

Chromatogram of CIT (0.20 ng/mL, t R = 7.3 min) and HO-CIT (1.12 ng/mL t R = 6.8 min) in a human urine sample. The analysis was performed by using IAC sample preparation and LC–MS/MS
Results and discussion
Validation parameters
Instrumental detection limit
Two methods of analysis were tested: HPLC with fluorescence detection and LC–MS/MS. They were compared with regards to the instrumental limits of detection, in other words, the lowest amounts of CIT that are detectable by two systems available in our laboratory. LC–MS/MS showed better results in terms of sensitivity than HPLC with fluorescence detection: the lowest amount measured with fluorescence by injecting 40 μL was 400 pg, that is, 40× higher than the lowest amount measured with LC–MS/MS (10 pg). Since tandem mass spectrometric detection offered better analytical conditions, further analyses were performed using this system.
Matrix effect
Matrix effects can occur when molecules co-elute with the compound(s) of interest, affecting the ionization efficiency of the electrospray interface. The matrix effect was assessed by post-extraction addition method: this requires sample extracts with the analyte of interest added post-extraction compared with pure solutions prepared in mobile phase containing equivalent amounts of the analyte of interest. The difference in response (AUC) between the post-extraction sample and the pure solution divided by the response of the pure solution times 100 determines the degree of matrix effect in percentage occurring to the analyte in question under chromatographic conditions (Taylor 2005). When using human urine samples with non-detectable CIT, high ion suppression was observed after the C18-SPE. On the other hand, a minimal suppression was registered when urine clean-up was done by immunoaffinity columns (Table 1).
Recovery
The recovery rate was tested for both the C18-SPE and the IAC extraction method. Human urine samples (with non-detectable CIT) were spiked with known amounts of CIT (5, 10, or 200 ng/mL) and processed as described above. At higher concentrations, CIT was almost entirely recovered after C18-SPE clean-up (95 and 97 %). However, at a low concentration (5 ng/mL), the recovery rate was only 10 %. This can be related to the strong matrix effect observed in the C18-SPE post-extracted spiked samples, which makes the detection of CIT in human urine at low concentrations difficult.
A different situation was observed for clean-up of human urine with immunoaffinity columns. At a concentration of 5 ng CIT/mL urine, the recovery rate was higher than 70 %. Similar results were found even at much lower (100×) analyte concentrations. A summary of CIT recovery rates after immunoaffinity clean-up is shown in Table 2.
The outcomes of the recovery tests performed for both clean-up methods demonstrated the efficiency of the IAC over C18-SPE at low analyte levels. An advantage of the IAC used (CitriTest®) is the cross-reactivity with the CIT–metabolite HO-CIT. A test performed at an OH-CIT concentration of 1 ng/mL (n = 3) results in a recovery rate of 106 ± 3 %. Therefore, the immunoaffinity clean-up was selected as the most suitable method for the extraction of CIT and HO-CIT from human urine samples.
Precision
The intra-assay repeatability was determined in urine with no background levels of CIT and HO-CIT. It was spiked with concentrations of 0.5 and 1.0 ng/mL. These spiked samples were analysed by LC–MS with IAC sample preparation ten and five times, respectively. Table 3 shows the values for the intra-assay repeatability.
For the intra-day repeatability, the same urine was applied. It was spiked with concentrations of 0.25 and 1.0 ng/mL and analysed as before. The samples were measured five times for both concentrations. Table 4 shows the data for the intra-day repeatability.
Accuracy
The relative recovery rates (Table 5) were calculated for all concentrations (0.25, 0.5, and 1.0 ng/mL) applied in the repeatability measurements in spiked urine samples with the method of IAC and LC–MS/MS.
Sensitivity of the method
Blood plasma samples: Since only unchanged CIT was found in blood of rats collected after administration of radiolabeled mycotoxin (Dunn et al. 1983), the metabolite HO-CIT was not measured in this matrix. The method for CIT involves a protein precipitation step followed by an enrichment of the analyte. Quantification was done by spiking plasma that shows no detectable CIT background. The LOD and LOQ of CIT were determined based on the lowest quantity of analyte that can be clearly distinguished (LOD) from the background (S/N = 3) or quantified (LOQ; S/N ≥ 6). In plasma, the LOD for CIT was 0.07 ng/mL and LOQ was 0.15 ng/mL.
Urine samples: CIT and its metabolite HO-CIT were quantified by using external calibration curves in matrix. The calibration curves are linear in the range of 0.5 to 20 ng/mL for CIT and 1–20 ng/mL for HO-CIT (quantification range), with coefficients of determination (R 2) of 0.9978 and 0.9999 for CIT and HO-CIT, respectively. Spiked samples were used to define the minimum detectable level: the LOD and LOQ were determined based on the lowest quantity of analyte that can be clearly distinguished (LOD) from the background (S/N = 3) or quantified (LOQ; S/N ≥ 6). For CIT, the LOD in urine was 0.02 ng/mL and the LOQ was 0.05 ng/mL. For HO-CIT, the LOD and LOQ were 0.05 and 0.10 ng/mL, respectively. These detection levels are considerably lower than those reported for CIT in urine (LOD 2.88 ng/mL; LOQ 5.76 ng/mL) by using a different sample clean-up method for subsequent multi-mycotoxin analysis (Ediage et al. 2012).
For urine, sample preparation with IAC (CitriTest®) and analysis of extracts with high-performance liquid chromatography and tandem mass spectrometric detection generated clean chromatograms, and no other interfering peaks were observed close to CIT. In relation to the validation parameters, the clean-up with IAC showed a low variation for CIT (RSD < 10 %) and the recovery levels were sufficiently high for analysis even at low analyte concentrations (Table 2). This is the first description of a method for the sensitive detection of CIT and its metabolite HO-CIT in human biological samples. The method was then applied in a pilot study to obtain first insights into CIT levels and metabolite occurrence in human urine and blood plasma samples.
Pilot study
CIT and its metabolite HO-CIT were determined in urines from a small cohort of 10 persons: 4 German adults and 6 Turkish infants. The age of the adults was between 20 and 60 years; all infants were below 2 years at the time of the sample collection. Urines were processed using the IAC-based method described above, and aliquots of all samples were also hydrolysed before analysis to account for the possible presence of glucuronides and/or sulphates. Quantification of the analytes was done by means of a calibration curve in matrix. Eight of ten urines were tested positive for CIT, and the metabolite was detected in 50 % of the samples. The results obtained in the human urines are summarized in Table 6.
These results show that CIT is mainly excreted with urine as parent compound and in some cases also as metabolite (HO-CIT). Dihydrocitrinone has been identified previously as a urinary metabolite of CIT in rats (Dunn et al. 1983). The analysis of human urine samples with and without enzymatic hydrolysis provided no evidence for the presence of conjugates in this matrix. This is in line with studies performed in rats, where the concentration of CIT in urine did not change (increase) after an enzymatic hydrolysis (Dunn et al. 1983).
CIT was also analysed in blood samples of eight adult German volunteers (4 males and 4 females). These samples have been previously analysed by a validated method for the presence of OTA (Muñoz et al. 2010), and it was detected in all 8 samples. Now, with the newly developed method, CIT could be detected in all plasma samples from males and females, with values ranging from 0.10 to 0.25 ng/mL. The metabolite OH-CIT was not analysed in plasma, since only unchanged CIT was found in blood of rats in a kinetic study (Dunn et al. 1983). The CIT concentrations measured in human plasma were similar or somewhat lower than the OTA levels when expressed as nanomolar concentrations to account for differences in the molecular weights of CIT and OTA (Fig. 4). The results from this small survey are indicative of a frequent co-occurrence of two nephrotoxic mycotoxins (OTA and CIT) in blood of German adults, albeit at low nM levels.

CIT and OTA concentrations (nM) in blood plasma of German adults (M are males; F are females). 1 nM OTA = 0.40 ng/mL; 1 nM CIT = 0.25 ng/mL
Conclusions
A sensitive method for the analysis of citrinin in human urine and blood samples has been developed and validated. As shown in a small pilot study, the method is suitable for biological monitoring and allows detection of the mycotoxin (and its metabolite) in a high percentage of randomly collected samples. To the best of our knowledge, these are the first data on the frequent presence of citrinin and its main metabolite dihydrocitrinone in humans. It will be interesting to determine the enzymatic route(s) for the formation of this metabolite in mammalian species and its toxicity in relation to citrinin. Such studies are currently underway in our laboratory. Finally, additional studies are needed to identify the major dietary sources of human exposure to citrinin and on the resulting internal levels in different populations, along with studies on the co-occurrence of ochratoxin A.
References
Aslam M, Rivzi SAH, Beg AE, Blaszkewicz M, Golka K, Degen GH (2012) Analysis of ochratoxin A blood levels in bladder cancer cases and healthy persons from Pakistan. J Toxicol Environ Health Part A 75(19–20):1176–1184. doi:10.1080/15287394.2012.707602
Clarke BR, Capon RJ, Lacey E, Tennant S, Gill JH (2006) Citrinin revisited: from monomers to dimers and beyond. Org Biomol Chem 4(1520):1528. doi:10.1039/b600960c
Degen GH (2011) Tools for investigating workplace related risks from mycotoxins. World Mycotoxin Journal 4:315–327
Duarte SC, Pena A, Lino CM (2011) Human ochratoxin A biomarkers—from exposure to effect. Crit Rev Toxicol 41:187–212
Dunn BB, Stack ME, Park DL, Joshi A, Friedman L, King RL (1983) Isolation and identification of dihydrocitrinone, a urinary metabolite of citrinin in rats. J Toxicol Environ Health 12:283–289
EC/SCOOP: European Commission (2002) Assessment of dietary intake of ochratoxin A by the population of EU member states. Report of the scientific cooperation, task 3.2.7. Directorate-general health and consumer protection, european commission. Available at http://ec.europa.eu/food/fs/scoop/index_en.html
Ediage EN, Mavungu JD, Song S, Wu A, Van Peteghem C, De Saeger S (2012) A direct assessment of mycotoxin biomarkers in human urine samples by liquid chromatography tandem mass spectrometry. Anal Chim Acta 741:58–69. doi:10.1016/j.aca.2012.06.038
EFSA (2012) EFSA Panel on Contaminants in the Food Chain (CONTAM); scientific opinion on the risks for public and animal health related to the presence of citrinin in food and feed. EFSA Journal 10(3):2605. [82 pp.] doi:10.2903/j.efsa.2012.2605. Available at: www.efsa.europa.eu/efsajournal
Flajs D, Peraica M (2009) Toxicological properties of citrinin. Arh Hig Rada Toksikol 60:457–464
Frank HK (1992) Citrinin. Zeitschrift für Ernährungswissenschaften 31:164–177
Gilbert J, Brereton P, MacDonald S (2001) Assessment of dietary exposure to ochratoxin A in the UK using a duplicate diet approach and analysis of urine and plasma samples. Food Addit Contam 18:1088–1093
IARC (1986) Citrinin. In: Some naturally occurring and synthetic food components, coumarins, ultraviolet radiation. Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, 40: 67–82; IARC, Lyon, France
Märtlbauer E, Usleber E, Dietrich R, Schneider E (2009) Ochratoxin A in human blood serum—retrospective long-term data. Mycotoxin Res 25:175–186
Molinie A, Faucet V, Castegnaro M, Pfohl-Leszkowicz A (2005) Analysis of some breakfast cereals on the French market for their contents of ochratoxin A, citrinin and fumonisin B1: development of a method for simultaneous extraction of ochratoxin A and citrinin. Food Chem 92:391–400
Muñoz K, Blaszkewicz M, Degen GH (2010) Simultaneous analysis of ochratoxin A and its major metabolite ochratoxin α in plasma and urine for an advanced biomonitoring of the mycotoxin. J Chromatogr B 897:2623–2629
Orti DL, Hill RH, Liddle JA, Needham LL, Vickers L (1986) High performance liquid chromatography of mycotoxin metabolites in human urine. J Anal Toxicol 10:41–45
Petkova-Bocharova T, Castegnaro M, Michelon J, Maru V (1991) Ochratoxin A and other mycotoxins in cereals from an area of Balkan endemic nephropathy and urinary tract tumours in Bulgaria. IARC Sci Publ 115:83–87
Reinhard H, Zimmerli B (1999) Reversed-phase liquid chromatographic behaviour of the mycotoxins citrinin and ochratoxin A. J Chromatogr A 862:147–159
Schmidt S, Brockmeyer A, Knor K, Thielert G (2003) Vorkommen von citrinin in Getreide. Mycotoxin Res 19:129–133
Scott PM (2005) Biomarkers of human exposure to ochratoxin A. Food Addit Contam 22(Suppl 1):99–107
Taylor PJ (2005) Matrix effects: the Achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clin Biochem 38:328–334
Vrabcheva T, Usleber E, Petkova-Bocharova T, Nikolov I, Chernozemsky I, Dietrich R, Märtlbauer E (2000) Citrinin in the diet of young and healthy persons living in Balkan endemic nephropathy areas. Mycotoxin Res 16(Suppl 2):150–153
Xu B, Jia X, Gu L, Sung C (2006) Review on the qualitative and quantitative analysis of the mycotoxin citrinin. Food Control 17:271–285
Acknowledgments
The excellent technical assistance of Iris Glaeser and Shadi Reidt from the Analytical Chemistry Unit of IfADo is gratefully acknowledged.
Conflict of interest
None of the authors have any conflict of interest in relation to publication of this paper. K. Muñoz received a stipend from CONYCIT/DAAD. All other funding for this work was internal.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Blaszkewicz, M., Muñoz, K. & Degen, G.H. Methods for analysis of citrinin in human blood and urine. Arch Toxicol 87, 1087–1094 (2013). https://doi.org/10.1007/s00204-013-1010-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-013-1010-z