
Abstract
Ammonium perfluorooctanoate (APFO), a processing aid used in the production of fluoropolymers, produces hepatomegaly and hepatocellular hypertrophy in rodents. In mice, APFO-induced hepatomegaly is associated with increased activation of the xenosensor nuclear receptors, PPARα and CAR/PXR. Although non-genotoxic, chronic dietary treatment of Sprague–Dawley (S–D) rats with APFO produced an increase in benign tumours of the liver, acinar pancreas, and testicular Leydig cells. Most of the criteria for establishing a PPARα-mediated mode of action for the observed hepatocellular tumours have been previously established with the exception of the demonstration of increased hepatocellular proliferation. The present study evaluates the potential roles for APFO-induced activation of PPARα and CAR/PXR with respect to liver tumour production in the S-D rat and when compared to the specific PPARα agonist, 4-chloro-6-(2,3-xylidino)-2-pyrimidinylthioacetic acid (Wy 14,643). Male S-D rats were fed APFO (300 ppm in diet) or Wy 14,643 (50 ppm in diet) for either 1, 7, or 28 days. Effects of treatment with APFO included: decreased body weight; hepatomegaly, hepatocellular hypertrophy, hepatocellular hyperplasia (microscopically and by BrdU labelling index), and hepatocellular glycogen loss; increased activation of PPARα (peroxisomal β-oxidation and microsomal CYP4A1 protein); decreased plasma triglycerides, cholesterol, and glucose; increased activation of CAR (CYP2B1/2 protein) and CAR/PXR (CYP3A1 protein). Responses to treatment with Wy 14,643 were consistent with increased activation of PPARα, specifically: increased CYP4A1 and peroxisomal β-oxidation; increased hepatocellular hypertrophy and cell proliferation; decreased apoptosis; and hypolipidaemia. With the exception of decreased apoptosis, the effects observed with Wy 14,643 were noted with APFO, and APFO was less potent. These data clearly demonstrate an early hepatocellular proliferative response to APFO treatment and suggest that the hepatomegaly and tumours observed after chronic dietary exposure of S-D rats to APFO likely are due to a proliferative response to combined activation of PPARα and CAR/PXR. This mode of action is unlikely to pose a human hepatocarcinogenic hazard.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Perfluorooctanoate (PFOA, C8F15O2 −) is a processing aid used in the production of fluoropolymers, typically as the ammonium salt (ammonium perfluorooctanoate, APFO). The toxicology of PFOA has been reviewed (Kennedy et al. 2004; Lau et al. 2007). PFOA has been confirmed to activate and to be a ligand for the peroxisome proliferator-activated receptor α (PPARα) (Bjork and Wallace 2009; Nakamura et al. 2009; Maloney and Waxman 1999; Rosen et al. 2008a; Takacs and Abbott 2007; Vanden Heuvel et al. 2006; Wolf et al. 2008). The marked increase in liver mass observed in rodents after treatment with PFOA is believed to be due, in part, to increases in hepatocellular peroxisomes and smooth endoplasmic reticulum as a direct result of activation of xenosensor nuclear receptors such as PPARα, the constitutive androstane receptor (CAR), and the pregnane X receptor (PXR) (Pastoor et al. 1987; Maloney and Waxman 1999; Rosen et al. 2008a, b). PFOA (as the ammonium salt, APFO) also increased the incidence of hepatocellular adenoma in one of the two chronic dietary studies in Sprague–Dawley rats. In a lifetime feeding study in male rats, hepatocellular adenomas were seen in rats fed 300 ppm (equivalent to daily intake of approximately 15 mg/kg) for 2 years (Biegel et al. 2001). In another two-year feeding study in which both male and female rats were fed diets containing either 30 or 300 ppm APFO (daily intake of approximately 1.5 and 15 mg/kg, respectively), no increase in hepatocellular tumours was observed (Sibinski et al. 1983).
It is well documented that sustained activation of xenosensor receptors such as PPARα, CAR and PXR may lead to development of hepatocellular tumours in long-term studies in rodents (Lake 2009). It is hypothesized that the hepatomegaly and liver tumours observed in rats dosed with APFO are the result of activation of PPARα nuclear receptor. Activation of PPARα may increase liver weight through increasing peroxisomal mass as well as expanding the smooth endoplasmic reticulum. Other factors may also contribute to the hepatomegaly observed following treatment of rodents with PFOA. In rodent liver, compounds that activate the nuclear receptors CAR or PXR also may increase liver mass through induction of cytochromes and increases in cytochromal proteins. The majority of genes for which mRNA expression levels are changed in response to dosing with PFOA in mice are regulated by PPARα, and many of those not regulated by PPARα are regulated by CAR (Rosen et al. 2008a, b). In addition, PFOA has been shown to increase mitochondrial mass via mitochondrial proliferation in rats and monkeys, and this also may account partially for liver weight increase (Berthiaume and Wallace 2002; Butenhoff et al. 2002; Walters et al. 2009).
In addition, in rodent liver activation of xenosensor nuclear receptors, PPARα, CAR, and PXR, may increase liver size by increasing replicative DNA synthesis (cell proliferation) and, sometimes in addition, by decreasing apoptosis (Klaunig et al. 2003; Lake 2009). These latter effects of PPARα activation may lead to clonal expansion of preneoplastic foci and, ultimately, liver carcinogenesis in rodents. Interestingly, in studies utilizing mice humanized with respect to the xenosensor nuclear receptors, the activation of the human PPARα, CAR, and PXR does not appear to lead to cell proliferation (Cheung et al. 2004; Gonzalez and Shah 2008; Shah et al. 2007; Ross et al. 2010). The evidence supports PFOA as a ligand capable of activating PPARα and increasing peroxisomal mass and fatty acid β-oxidation, and expansion of the smooth endoplasmic reticulum. Similarly, activation of CAR by PFOA has been demonstrated in mice as evidenced by the induction of CAR-regulated genes. The interactions of chemicals with these receptors are complex, not in the fact that they regulate common genes, but also because a single chemical agent may interact with two or more receptors simultaneously. However, in order for PFOA-activated PPARα/CAR to be supported as a causal influence for the observed hepatic tumours on chronic dosing of rats with APFO, evidence of increased replicative DNA synthesis and decreased apoptosis has been lacking.
Biegel et al. (2001) attempted to observe cell proliferation in male rats fed 300 ppm APFO over the two-year dosing period at intervals beginning after the first 30 days on diet. Hepatic cell proliferation was not increased at the time points for which it was measured. It has been suggested that the earliest time point of 30 days may have been too late to observe a burst of early cell proliferation in response to dietary treatment. Thus, observing for cell proliferation soon after exposures begin has been warranted.
The objective of the work reported herein was to characterize PFOA-induced hepatomegaly in male rats, particularly with respect to cell proliferation and decreased apoptosis. A potent nuclear receptor agonist, 4-chloro-6-(2,3-xylidino)-2-pyrimidinylthioacetic acid (Wy 14,643), was used as a positive control. In an initial study, technical issues were encountered with some preparations for immunohistochemical staining for proliferation and for the TUNEL assay for apoptosis. Therefore, the first study was replicated in most aspects, and results are reported for both the initial (study 1) and the replicate (study 2).
Materials and methods
Two studies were conducted which replicated the dosing scheme and key parameters of major interest. In the first study (study 1), the data from two key endpoints, apoptosis and cell proliferation [bromodeoxyuridine (BrdU) labelling index], were not sufficiently robust due to technical problems in histological preparations. Therefore, a second study (study 2) was initiated that replicated most, but not all, of the endpoints included in the first study. Study 1 included several clinical chemistry and protein expression parameters that were not examined in the second study. The results from both studies will be presented here.
Test materials
The test substance, ammonium perfluorooctanoate (APFO, FC-143, Lot 332, 98% purity), was supplied by 3M Company (St. Paul, MN, USA). The positive control substance for activation of PPARα nuclear receptor, 4-chloro-6-(2,3-xylidino)-2-pyrimidinylthioacetic acid (Wy 14,643, 98% purity) was obtained from Alexis Biochemicals (Enzo Life Sciences, Inc., Exeter, UK). Both materials were chemically stable during the course of the experiments.
Laboratory animals and husbandry
A sufficient number of male 7- to 8-week-old Sprague–Dawley (CD) rats were obtained from Charles River UK Ltd. (Margate, Kent, UK). On arrival at the laboratory, the rats were housed 2/cage on sawdust in solid-bottomed polypropylene cages. The rats were acclimatized in the testing facility for at least 5 days before use. The Sprague–Dawley (CD) rat was the test system of choice for this study because previous work with APFO used this strain. In addition, the testing facility has extensive experience in the use of this strain of rat, and the strain is well accepted by the regulatory authorities.
The room temperature was maintained within a range of 19–23°C with a relative humidity within a range of 40–70%. There were a nominal 14–15 air changes per hour, and the light/dark cycle was 12/12 h. In-life procedures undertaken during the course of the studies were subject to the provisions of the United Kingdom Animals (Scientific Procedures) Act 1986. The studies complied with all applicable sections of the Act and associated Codes of Practice for the Housing and Care of Animals used in Scientific Procedures and the Humane Killing of Animals in the Act.
Diets
RMI powdered diet (Special Diet Services Ltd., Stepfield, Witham, Essex, UK) was used, and the diet specifications were retained at the testing facility. The test diets contained either 300 ppm APFO or 50 ppm Wy 14,643. Diets were prepared fresh weekly, and samples were retained for possible future analysis. Drinking water was taken from the local supply and provided in bottles. Drinking water was provided ad libitum.
Experimental design
The rats were uniquely numbered by ear-punch and randomly assigned to groups up to 1 week after arrival. Each study consisted of 1 control and 2 test groups, each containing 30 male rats. Control rats received powdered RMI diet ad libitum. The test groups of rats were administered either APFO at 300 ppm or Wy 14,643 at 50 ppm in the diet for either 1, 7 or 28 days. Ten rats from each group were killed on days 2, 8, and 29. Rats were implanted with osmotic pumps (Alzet 2ML 1, Alzet Corp., Cupertino, CA, USA) containing BrdU 15 mg/mL in phosphate-buffered saline (PBS, pH 7.4) 5 days before termination while still receiving the treatment regimen. Rats killed after exposure to diet for 1 day were administered BrdU (15 mg/mL in PBS, 5 mL/kg bodyweight) by subcutaneous injection 2 h prior to killing.
Clinical observations
Prior to the start of the study, all rats were observed to ensure that they appeared physically normal and exhibited normal activity. Each rat was observed at least once daily during the study. Clinical abnormalities of individual rats were recorded. The body weight of each rat was recorded at the start of the study. The rats were weighed weekly on the same day of the week. All rats were weighed prior to killing. Food consumption was measured every time the diet jars were changed/refilled and when the rats were killed. Rats on diet for 7 days or greater had their diet jars weighed on the same day each week, regardless of refilling, so that weekly food consumption could be recorded.
Terminal procedures
On the day of killing, the rats were weighed and transferred to the post-mortem room. The rats were euthanized by CO2 asphyxiation. For analysis of PFOA in plasma, blood was taken and plasma was prepared and stored pending analysis as described below. The liver was removed from each rat and weighed. Liver samples (and duodenal samples as a positive for BrdU incorporation) were taken for BrdU immunohistopathology and analysis of the apoptotic index [two 2 mm strips, 1 from the left and 1 from the median lobe, placed in neutral buffered formalin (NBF) for 48 h], for H&E histopathology (two 2 mm strips, 1 from the left and 1 from the median lobe, placed in NBF for 1 week), for DNA determination (1 sample, 0.25 g, flash frozen in liquid nitrogen and stored at −70°C), and for possible future use (1 sample, 1 g flash frozen in liquid nitrogen and stored at −70°C). The remainder was weighed and scissor-minced in ice-cold 1.15% (w/v) KCl prior to subcellular fractionation, and samples of homogenate and heavy pellet were stored at −70°C.
Parameters measured
Clinical chemistries Blood was taken by cardiac puncture in lithium/heparin-coated tubes, mixed for 10 min, then cooled on ice. Plasma was obtained following centrifugation and stored at approximately −70°C until analysis. In study 1 only, plasma samples were assayed for alanine aminotransferase (ALT), aspartate aminotransferase (AST), total cholesterol, glucose, and triglycerides using a Roche Cobas Integra® 400 automated analyzer calibrated according to manufacturer’s instructions.
Histopathology Following fixation, all samples were processed and 5-μm sections were cut and stained with H&E. For apoptosis, the method used was an indirect TUNEL labelling assay (Roche 11 684 817910). Sections were analysed for BrdU incorporation as a measure of cell proliferation using an indirect BrdU labelling assay. For both apoptosis and BrdU incorporation, the results were expressed as the percentage of labelled cells as well as normalized as a percentage of the control value, with control representing 100%.
Biochemical analyses Peroxisome proliferation was evaluated by CN-insensitive acyl CoA oxidation determined spectrophotometrically in liver heavy pellet using palmitoyl CoA as a substrate (Bronfmann et al. 1979). Results were expressed as nmol NAD+ reduced/min/mg protein. The DNA content of the liver was measured spectrophotometrically in whole tissue using the diphenylamine reaction (Burton 1956). Results were expressed as mg DNA/g liver and mg DNA/whole liver.
In study 1 only, determination of the expression of cytochrome P450 isoforms representing activation of the nuclear receptors PPARα (CYP4A1), CAR (CYP2B1/2), and PXR (CYP3A1) was performed using SDS–PAGE and Western blotting using liver microsomes.
The protein concentration of tissue heavy pellets was determined in aqueous solutions using a modification of the method of Lowry et al. (1951) and bovine serum albumin standards.
Plasma perfluorooctanoate (PFOA) concentrations Rat plasma PFOA concentrations were measured by LC–MS/MS utilizing reverse-phase liquid chromatography on a Waters Alliance 2795 HPLC system interfaced with a Waters Quattro micro mass spectrometer in negative electrospray ionization mode. The negative ion collision-induced transition monitored (MRM) for PFOA was mass 412.88 > 168.93.
A gradient elution was carried out on a Waters Xterra C18, 2.5 μm, 2.1 × 30 mm column at a flow rate of 0.25 mL/min. Mobile phase A was 10 mM ammonium acetate and mobile phase B was 100% HPLC-grade acetonitrile. Initial conditions were held for 30 s at 70% mobile phase A, followed by a linear gradient to 80% mobile phase B out to 3 min. Conditions were switched back to 70% mobile phase A at 3.1 min, and the column reconditioned out to 6 min. PFOA had a retention time of approximately 3.4 min.
Rat plasma standards were prepared by spiking known amounts of PFOA in acetonitrile into control rat plasma (volume of spiking solution used was no more than 5% total volume). The standard concentration range used for the rat plasma samples was linear from 5 to 500 μg/mL with an r 2 value >0.99. Ten microlitres of each standard and sample were diluted (600×) into 100% HPLC-grade acetonitrile, vortexed, and centrifuged (1,500×g) to precipitate the protein. Twenty microlitres of the supernatant from each of the standards and the samples were injected onto the HPLC system. Quantification was carried out using Waters QuanLynx and MassLynx software versions 4.0. The limit of quantitation was 5 μg/mL.
Statistical analysis: Data were analysed using the two-sided Student’s t test with significance at P < 0.05.
Results
Body weights
In both study 1 and study 2, body weights among rats fed 300 ppm APFO were decreased on days 8 and 29 (Table 1). Decreases were 12% and 11% on day 8 for studies 1 and 2, respectively, and 23 and 13% on day 29 for studies 1 and 2, respectively (Table 1). Rats fed 50 ppm Wy 14,643 showed body weights that were not statistically significantly different than controls in both study 1 and study 2; although mean body weights were 2–6% lower than control means.
Food consumption and test compound intakes
In study 1, food consumption for rats fed APFO was decreased on a g per rat basis with statistical significance throughout the feeding period (Table 2). On a g of food per kilogram body weight basis, in study 2, APFO-treated rats tended to eat more food than did the controls from weeks 2 through 4 (81, 80, and 72 g/kg/days compared to 69, 64, and 59 g/kg/days in the controls, weeks 2, 3, and 4, respectively).
Feeding of Wy 14,643 did not appear to significantly alter the food consumption of rats. In study 1, food consumption by rats fed Wy 14,643 was somewhat lower than controls during weeks 1 and 2. However, in study 2, the Wy 14,643 rats tended to eat more food per body weight than the controls during weeks 3 and 4.
Approximate daily test material intake during the study for APFO in study 1 was 17–22 mg/kg (mean 19 mg/kg) and for study 2 was 22–24 mg/kg (mean 23 mg/kg). For Wy 14,643, intakes were 3.1–3.4 mg/kg for study 1 (mean 3.3 mg/kg) and 3.3–4.1 mg/kg for study 2 (mean 3.7 mg/kg).
Clinical observations
No adverse clinical observations were seen in any of the rats in either study 1 or study 2.
Liver weights
Increased liver weights, both absolute and relative to body weight, were seen in rats fed APFO and Wy 14,643 for either 7 or 28 days in both studies (Table 3). Relative to controls, with both chemical treatments, liver weight increases at day 29 were generally greater than those seen at day 8; however, the differences were not striking. For studies 1 and 2, respectively, on day 29, the absolute liver weights reached 114 and 143% of the controls with APFO-fed rats, and 167 and 170% with Wy 14,643. The corresponding respective increases in liver-to-body-weight ratios were 147 and 166% for APFO-fed rats, and 179 and 180% for Wy 14,643-fed rats.
Cell proliferation
Technical problems with fixation and immunostaining for BrdU led to the loss of a number of critical samples from study 1. Only 3 control livers from day 8 were successfully stained, and no control data were available for day 29. For liver sample preparations from APFO- and Wy 14,643-treated rats, all day 2 and 8 samples were adequately stained for evaluation, and 5 and 9 samples from day 29 were adequately stained, respectively. Although control samples were decreased for day 8 and unavailable for day 29, a control labelling index value of approximately 1.5–2.0 is typical for male rats of this age (laboratory historical data, CXR Biosciences, unpublished). With the inclusion of data from study 2, it is possible to draw conclusions from the data obtained. Both Wy 14,643 and APFO increased the hepatocellular labelling index (S-phase) by approximately 9- and 3-fold, respectively, 1 day after commencement of treatment (Table 4). At day 8, these levels of S-phase activity were maintained in the treated groups. Assuming that control liver S-phase activity at 29 days would be similar to that at 8 days, the hepatocellular labelling indices of livers from APFO-treated animals appeared to return to control levels; while the labelling indices of livers from Wy 14,643-exposed animals appeared to be maintained at elevated levels.
Data from study 2 were adequate for evaluation. On day 2, APFO and Wy 14,643 increased the hepatocellular labelling index (S-phase) by approximately 2- and 4-fold, respectively (Table 4). On day 8, the levels of S-phase activity were increased to 5- and 9-fold, respectively. The hepatocellular labelling indices of livers from APFO-treated animals returned to twice the control values after 28 days of treatment, while the labelling indices of livers from Wy 14,643-exposed rats appeared to be maintained at elevated levels (8-fold control levels).
Taken together, the results from studies 1 and 2 showed that hepatic BrdU labelling index was clearly increased over control values to the greatest extent on day 8 for both APFO and Wy 14,643 treatments. APFO treatment increased the labelling index by approximately 5-fold compared to controls on day 8, while Wy 14,643 increased the labelling index by approximately 10-fold. For APFO, labelling indices are near background rates for male rats on days 2 and 29, even though they were statistically significantly higher than time-respective controls. Labelling indices for livers from Wy 14,643 rats were higher than for the time-respective APFO-treated rat livers.
Apoptosis
Technical problems due to poor fixation of tissues precluded evaluation of hepatocellular apoptosis in study 1. Table 5 presents the effect of APFO and Wy 14,643 on hepatic apoptotic indices from study 2. Wy 14,643 decreased the apoptotic indices at all time points. The decrease was only statistically significant after 28 days of treatment. There was no statistically significant decrease in apoptosis with APFO at any of the time points studied.
DNA content of the liver
Data for liver DNA concentration (mg/g liver) and total DNA content per liver are provided in Table 6. In studies 1 and 2, both chemicals produced a modest decrease in DNA concentration (mg/g liver), with the possible exception of day 2 in study 2. However, APFO administration did not affect the total liver content of DNA in both studies. The administration of Wy 14,643 led to increases of 18–29% and 48–63% of control values on days 8 and 29, respectively.
Clinical chemistry
Clinical chemistry values were obtained only in study 1 and are presented in Table 7. Plasma concentrations of ALT and AST for rats treated with APFO and Wy 14,643 were not increased when compared to controls and, in some cases, were slightly decreased with statistical significance. Plasma total cholesterol was statistically significantly decreased with both APFO and Wy 14,643 at all time points compared to control values with the exception of the day 2 value for APFO-treated rats, which was lower than the control value but lacked statistical significance. Both APFO and Wy 14,643 treatments showed decreased triglycerides to a similar extent when compared to controls on days 8 and 29. Triglyceride concentrations following 2 days of treatment with either compound appeared normal; although the value for APFO-treated rats was increased with statistical significance. Both APFO and Wy 14,643 showed similar statistically significant decreases in plasma glucose concentrations following 8 and 29, but not 2, days of treatment.
Peroxisome proliferation
Administration of either APFO or Wy 14,643 to rats resulted in marked increases in hepatic peroxisomal β-oxidation at all time points and in both studies (Table 8). The activities on days 8 and 29 were several fold higher than those on day 2. At the doses employed, both compounds maximally increased peroxisomal β-oxidation by approximately 8- to 10-fold.
Induction of cytochrome P450s
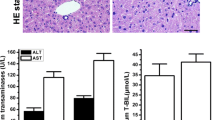
The induction of several microsomal cytochromes P450 was demonstrated by SDS–PAGE and Western blotting (Fig. 1). APFO clearly induced CYP2B1/2, CYP3A1, and CYP4A1 (Fig. 1a, b, and c, respectively). Wy 14,643 only induced CYP4A1 (Fig. 1c).

Sodium dodecyl sulphate–polyacrylamide gel electrophoresis and Western blotting of hepatic microsomal proteins from male control rats and male rats fed APFO or Wy 14,643 for up to 28 days for CYP2B1/2 (Fig. 1a), CYP3A1 (Fig. 1b), and CYP4A1 (Fig. 1c). Lane numbers correspond to individual rats. Treatment with APFO increased expression of all three CYPs when compared to control expression levels, while treatment with Wy 14,643 only increased expression of CYP4A1. Based on these data, APFO appears to be an activator of CAR, PXR, and PPARα nuclear receptors in the rat
Histopathology of the liver
In study 1, liver sections from control rats indicated no microscopic abnormalities. Both treatment with APFO and Wy 14,643 were found to decrease periportal hepatocellular glycogen at all time points, increase hepatocellular hypertrophy on days 8 and 29, increase hepatocellular hyperplasia on day 29, and increase fatty vacuolation on day 8. Histopathology was assessed as grades 1, 2, 3, or 4; equivalent to minimal, mild, moderate, or marked. Hepatocellular glycogen depletion was more pronounced with Wy 14,643 on day 2 (8 of 10 rats, grades 2-3) than with APFO (2 of 10 rats, grade 1). However, on days 8 and 29, grade 3 decreased glycogen was present in all APFO and Wy 14,643 rats with the exception of one grade 2 in an APFO-treated rat. On day 29, 2 of 10 control rats had glycogen depletion of either a grade 2 or grade 3. Hepatocellular hypertrophy and hyperplasia were not evident in APFO- and Wy 14.643-treated rats on day 2, but grade 1–2 hypertrophy was present in all APFO- and WY 14,643-treated rats on day 8, and progressed to grades 2–3 for APFO on day 29, at which time all Wy 14,643-treated rats were grade 1. Hyperplasia was only evident on day 29 and was present in all APFO-treated rats at grade 2 and all Wy 14,643-treated rats at grade 3. Fatty vacuolation was only observed in APFO- and Wy 14,643-treated rats on day 8 (3 of 9 rats, grades 1–2 for APFO and 5 of 10 rats, grades 1–2 for Wy 14,643).
In study 2, microscopic findings similar to those in study 1 were described. Liver sections from control rats again indicated no microscopic abnormalities. Wy 14,643 administration produced a depletion of hepatic glycogen one day after commencing dosing. This change increased in intensity at 8 and 29 days. Hepatocellular hypertrophy was seen at the 2 later time points. Wy 14,643 administration for 28 days induced marked hepatocellular hyperplasia (observed as an increase in the number of hepatocyte nuclei per unit area). APFO administration also resulted in a loss of hepatic glycogen, however, the process was slower than that observed in the Wy 14,643-treated rats. APFO-induced hepatocellular hypertrophy was seen at the later 2 time points. APFO administration for 28 days produced marked hepatocellular hyperplasia, although the severity was less than that seen with Wy 14,643.
Plasma APFO levels
Background levels of APFO (less than quantifiable) were present in the plasma of the control and Wy 14,643 rats at all 3 time points studied. Plasma levels of approximately 250 μg/mL were seen in the APFO-treated rats at all 3 blood sampling intervals (259 ± 39, 234 ± 33, and 252 ± 45 μg/mL, days 2, 8, and 29, respectively).
Discussion
The objective of the work reported herein was to characterize PFOA-induced hepatomegaly in male rats, particularly with respect to the potential role of PPARα-mediated cell proliferation and possible decreased apoptosis. Previous studies with APFO in male rats have shown that peroxisomal proliferation, hepatocellular hypertrophy, and hepatomegaly are sensitive outcomes of treatment (Kennedy et al. 2004). Two chronic (two-year duration) dietary studies with APFO have been conducted in which male Sprague–Dawley rats were fed 300 ppm (Biegel et al. 2001; Sibinski et al. 1983). Of these two, one demonstrated an increase in hepatocellular hyperplasia that was associated with an increase in benign hepatocellular adenoma (Biegel et al. 2001). As stated in the introduction, PFOA has been confirmed to activate and to be a ligand for the xenosensor nuclear receptor PPARα (Bjork and Wallace 2009; Maloney and Waxman 1999; Nakamura et al. 2009; Rosen et al. 2008a; Takacs and Abbott 2007; Vanden Heuvel et al. 2006; Wolf et al. 2008). It has been suggested that the hepatocellular tumours observed by Biegel et al. were the result of APFO-mediated PPARα activation, and most of the criteria for establishing activation of PPARα as the mode of action for the hepatocellular adenoma have been previously demonstrated (Klaunig et al. 2003). The key events involve activation of the nuclear receptor leading to hypertrophy and increased hepatocyte proliferation leading to hyperplasia, which, if sustained, leads to selective clonal expansion of pre-neoplastic cells, resulting in tumour formation (Klaunig et al. 2003; Lake 2009).
The one criterion that has not been evident was that of increased hepatocellular proliferation in response to APFO treatment. Biegel et al. (2001) included serial sacrifices in their design of the chronic dietary study with APFO to observe for increased replicative DNA synthesis. However, the earliest time point included was after 1 month on diet. At that time point, and in all subsequent time points, cell proliferation in APFO-treated rats was similar to that of control rats. In contrast, the potent and specific PPARα agonist, Wy 14,643, which was also included in the study design by Biegel et al. increased cell proliferation at all time points.
In the work reported herein, the response to Wy 14,643, the positive control for PPARα activation, was as expected for a specific PPARα agonist, and APFO treatment also produced most of these responses. Specifically, on treatment with Wy 14,643, both CYP4A1 and peroxisomal β-oxidation, markers for PPARα activation, were increased. Hepatic cell proliferation was increased and apoptosis was decreased, with a concomitant increase in liver DNA content. In addition, clinical chemistry findings were consistent with those associated with PPARα activation, notably decreased serum total cholesterol and triglycerides. Wy14,643, which is a specific PPARα ligand, did not induce CYP2B1/2 or CYP3A1, which are markers for the xenosensor nuclear receptors CAR and CAR/PXR, respectively. Prototypical inducers of CYP2B1/2 and CYP3A1 are phenobarbital and dexamethasone, respectively.
On treatment with APFO, the PPARα-associated responses observed with Wy 14,643 were also observed with the exception of decreased apoptosis and increased liver DNA content. The increase in hepatocellular proliferation without either a significant increase in total liver DNA or a decrease in apoptosis likely is a reflection of the prominent hepatocellular hypertrophic action of the combined activation of the three xenosensor nuclear receptors, PPARα and CAR/PXR, together with the weaker and less prolonged hyperplastic activity, when compared with Wy 14,643. With APFO, expression of CYP2B1/2 and CYP3A1, respectively, were also increased, indicating activation of CAR and PXR. Thus, the work reported herein has confirmed that APFO-mediated hypertrophic changes in the liver are the result of increased peroxisomal proliferation, expansion of smooth endoplasmic reticulum proliferation, and increased cell proliferation.
The hyperplastic properties characteristic of PPARα and CAR/PXR activators were demonstrated by increased cell proliferation as determined by increases in BrdU incorporation into hepatocyte nuclei. Both APFO and Wy 14,643 increased the hepatocellular labelling index (S-phase) by approximately 3- and 9-fold, respectively, 1 day after commencement of treatment. At day 8, this level of S-phase was maintained in the treated groups. At 29 days, the hepatocellular labelling index of livers from APFO-treated rats appeared to be approaching control levels. The labelling indices of livers from Wy14,643-treated rats were maintained at elevated levels on day 29.
In these studies, no signs of overt hepatotoxicity were observed. Neither APFO nor Wy 14,643 increased activities of either ALT or AST, consistent with a lack of histologically identified hepatocellular damage. Histopathology demonstrated that both APFO and Wy 14,643 caused hepatocellular glycogen loss, hypertrophy, and hyperplasia. The hypertrophy was characterized by increased cyanide-insensitive palmitoyl CoA oxidation (a marker of peroxisome proliferation) and the induction of cytochrome P450 CYP4A1 (accompanied by smooth endoplasmic reticulum proliferation). The induction of CYP4A1 isozyme is a characteristic of peroxisome proliferators. As noted earlier, the hepatocellular hyperplasia was confirmed to result from increased replicative DNA synthesis (labelling index, S-phase). At the dose levels used, Wy14,643 was more effective than APFO as a liver enlarging agent. On a mg/kg body weight basis, Wy 14,643 was about five times more potent than APFO. These changes were also reflected by relevant biochemical changes.
Taken together, the data obtained in this study clearly demonstrate that APFO exhibits the prototypical properties of a mixed PPARα and CAR/PXR agonist. The administration of APFO to rats leads to hepatomegaly characterized by hypertrophy and hyperplasia, as a result of early increases in cell proliferation, which ultimately leads to liver tumour formation. Several studies using receptor KO mice have shown the requirement for active CAR, PXR, or PPARα for the expression of the complete hypertrophic and hyperplastic changes characteristic of liver growth–inducing agents (Lee et al. 1995; Wei et al. 2000; Huang et al. 2005; Scheer et al. 2008). Furthermore, a limited number of studies with Car-null and Pparα-null mice have demonstrated that an active receptor is required for liver tumour promotion by phenobarbital and the induction of hepatic tumours by Wy 14,643 (Huang et al. 2005; Morimura et al. 2006; Yamamoto et al. 2004).
Although CAR is present in human liver and phenobarbital can increase liver size in rodents (Lake 2009) and humans (Pirttiaho et al. 1978; Pirttiaho et al. 1982), significant species differences in both the hyperplasia-inducing properties of phenobarbital and other compounds have been demonstrated. In contrast to effects in primary cultures of rodent hepatocytes, phenobarbital does not appear to induce replicative DNA synthesis in cultured human hepatocytes (Parzefall et al. 1991; Hirose et al. 2009; Lake 2009). Hence, in a limited number of studies to date, the hyperplastic component of the response to inducers appears to be largely confined to rodents. This apparent insensitivity of human hepatocytes to xenobiotic-mediated stimulation of replicative DNA synthesis is reflected by the lack of hyperplastic response seen in mice humanized simultaneously for CAR and PXR (Ross et al. 2010) to phenobarbital and chlordane, two well-characterized rodent non-genotoxic hepatocarcinogens.
Specific evidence exists for decreased sensitivity of human hepatocytes to the APFO-mediated effects of PPARα activation. Human hepatocytes in primary culture and HepG2 cells did not demonstrate activation of PPARα at concentrations of PFOA up to 200 μM in media (Bjork and Wallace 2009). In addition, SV/129 mice humanized for PPARα and treated with APFO by gavage for 2 weeks at daily doses of 0.1 and 0.3 mg/kg did not have elevated mRNA and protein levels of hepatic markers of PPARα target gene activation compared to untreated controls and wild-type mice similarly treated with APFO (Nakamura et al. 2009).
If chemically stimulated cell proliferation is pivotal for development of tumours, in the light of the inability of the human receptors to support the stimulation of the cell proliferation and hyperplasia, then the exposure of humans to such PPARα and CAR/PXR agonists is unlikely to pose a hepatocarcinogenic hazard. This conclusion is supported by cancer mortality studies on therapeutically administered agents such as phenobarbital, and clofibrate and bezafibrate, CAR/PXR and PPARα agonists, respectively (Bentley et al. 1993; Doull et al. 1999; Olsen et al. 1989, 1995; Whysner et al. 1996; IARC 2001). Specific to APFO, occupational cancer mortality studies have not found increased risk of death from liver cancer among workers occupationally exposed to APFO (Leonard et al. 2008; Lundin et al. 2009).
References
Bentley P, Calder I, Elcombe C, Grasso P, Stringer D, Wiegand H-J (1993) Hepatic peroxisome proliferation in rodents and its significance for humans. Food Chem Toxicol 31:857–907
Berthiaume J, Wallace KB (2002) Perfluorooctanoate, perflourooctanesulfonate, and N-ethyl perfluorooctanesulfonamido ethanol; peroxisome proliferation and mitochondrial biogenesis. Toxicol Lett 129:23–32
Biegel LB, Hurtt ME, Frame SR, O’Connor JC, Cook JC (2001) Mechanisms of extrahepatic tumor induction by peroxisome proliferators in male CD rats. Toxicol Sci 60:44–55
Bjork JA, Wallace KB (2009) Structure-activity relationships and human relevance for perfluoroalkyl acid-induced transcriptional activation of peroxisome proliferation in liver cell cultures. Toxicol Sci 111:89–99
Bronfmann M, Inestrosa NC, Leighton F (1979) Fatty acid oxidation by human liver peroxisomes. Biochem Biophys Res Commun 88:1030–1036
Burton K (1956) A study of the conditions and mechanism of the diphenylamine reaction for the colorimetric estimation of deoxyribonucleic acid. Biochem J 62:315–323
Butenhoff J, Costa G, Elcombe C, Farrar D, Hansen K, Iwai H, Jung R, Kennedy G Jr, Lieder P, Olsen G, Thomford P (2002) Toxicity of ammonium perfluorooctanoate in male cynomolgus monkeys after oral dosing for 6 months. Toxicol Sci 69:244–257
Cheung C, Akiyama TE, Ward JM, Nicol CJ, Feigenbaum L, Vinson C, Gonzalez FJ (2004) Diminished hepatocellular proliferation in mice humanized for the nuclear receptor peroxisome proliferator-activated receptor alpha. Cancer Res 64:3849–3854
Doull J, Cattley R, Elcombe C, Lake BG, Swenberg J, Wilkinson C, Williams G, van Gemert M (1999) A cancer risk assessment of di(2-ethylhexyl)phthalate: application of the new U.S. EPA risk assessment guidelines. Regul Toxicol Pharmacol 29:327–357
Gonzalez FJ, Shah YM (2008) PPARalpha: mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology 246:2–8
Hirose Y, Nagahori N, Yamada T, Deguchi Y, Tomigahara Y, Nishioka K, Uwagawa S, Kawamura S, Isobe N, Lake BG, Okuno Y (2009) Comparison of the effects of the synthetic pyrethroid Metofluthrin and phenobarbital on CYP2B form induction and replicative DNA synthesis in cultured rat and human hepatocytes. Toxicology 258:64–69
Huang W, Zhang J, Washington M, Liu J, Parant JM, Lozano G, Moore DD (2005) Xenobiotic stress induces hepatomegaly and liver tumors via the nuclear receptor constitutive androstane receptor. Mol Endocrinol 19:1646–1653
International Agency for Research on Cancer (IARC) (2001) IARC monographs on the evaluation of carcinogenic risks to humans. Some thyrotropic agents. Vol. 79. IARC Press, Lyon
Kennedy GL Jr, Butenhoff JL, Olsen GW, O’Connor JC, Seacat AM, Perkins RG, Biegel LB, Murphy SR, Farrar DG (2004) The toxicology of perfluorooctanoate. Crit Rev Toxicol 34:351–384
Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David RM, DeLuca JG, Lai DY, McKee RH, Peters JM, Roberts RA, Fenner-Crisp PA (2003) PPARalpha agonist-induced rodent tumors: modes of action and human relevance. Crit Rev Toxicol 33:655–780
Lake BG (2009) Species differences in the hepatic effects of inducers of CYP2B and CYP4A subfamily forms: relationship to rodent liver tumour formation. Xenobiotica 39:582–596
Lau C, Anitole K, Hodes C, Lai D, Pfahles-Hutchens A, Seed J (2007) Perfluoroalkyl acids: a review of monitoring and toxicological findings. Toxicol Sci 99:366–394
Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ (1995) Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol 15:3012–3022
Leonard RC, Kreckmann KH, Sakr CJ, Symons JM (2008) Retrospective cohort mortality study of workers in a polymer production plant including a reference population of regional workers. Ann Epidemiol 18:15–22
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:265–275
Lundin JI, Alexander BH, Olsen GW, Church TR (2009) Ammonium perfluorooctanoate production and occupational mortality. Epidemiology 20:921–928
Maloney EK, Waxman DJ (1999) Trans-Activation of PPARalpha and PPARgamma by structurally diverse environmental chemicals. Toxicol Appl Pharmacol 161:209–218
Morimura K, Cheung C, Ward J, Reddy JK, Gonzalez FJ (2006) Differential susceptibility of mice humanized for peroxisome proliferator-activated receptor a to Wy-14, 643-induced liver tumorigenesis. Carcinogenesis 27:1074–1080
Nakamura T, Ito Y, Yanagiba Y, Ramdhan DH, Kono Y, Naito H, Hayashi Y, Li Y, Aoyama T, Gonzalez FJ, Nakajima T (2009) Microgram-order ammonium perfluorooctanoate may activate mouse peroxisome proliferator-activated receptor α, but not human PPARα. Toxicology 265:27–33
Olsen JH, Boice JD Jr, Jensen JPA, Fraumeni JF Jr (1989) Cancer among epileptic patients exposed to anticonvulsant drugs. J Natl Cancer Inst 81:803–808
Olsen JH, Schulgen G, Boice JD Jr, Whysner J, Travis LB, Williams GM, Johnson FB, McGee JO (1995) Antiepileptic treatment and risk for hepatobiliary cancer and malignant lymphoma. Cancer Res 55:294–297
Parzefall W, Erber E, Sedivy R, Schulte-Hermann R (1991) Testing for induction of DNA synthesis in human hepatocyte primary cultures by rat liver tumor promoters. Cancer Res 51:1143–1147
Pastoor TP, Lee KP, Perri MA, Gillies PJ (1987) Biochemical and morphological studies of ammonium perfluorooctanoate-induced hepatomegaly and peroxisome proliferation. Exp Molec Path 47:98–109
Pirttiaho HI, Sotaniemi EA, Ahokas JT, Pitkänen U (1978) Liver size and indices of drug metabolism in epileptics. Br J Clin Pharmacol 6:273–278
Pirttiaho HI, Sotaniemi EA, Pelkonen RO, Pitkänen U (1982) Hepatic blood flow and drug metabolism in patients on enzyme-inducing anticonvulsants. Eur J Clin Pharmacol 22:441–445
Rosen MB, Lee JS, Ren H, Vallanat B, Liu J, Waalkes MP, Abbott BD, Lau C, Corton JC (2008a) Toxicogenomic dissection of the perfluorooctanoic acid transcript profile in mouse liver: evidence for the involvement of nuclear receptors PPAR alpha and CAR. Toxicol Sci 103:46–56
Rosen MB, Abbott BD, Wolf DC, Corton JC, Wood CR, Schmid JE, Das KP, Zehr RD, Blair ET, Lau C (2008b) Gene profiling in the livers of wild-type and PPAR{alpha}-null mice exposed to perfluorooctanoic acid (PFOA). Toxicol Pathol 36:592–607
Ross J, Plummer SM, Rode A, Scheer N, Bower CC, Vogel O, Henderson CJ, Wolf CR, Elcombe CR (2010) Human constitutive androstane receptor (CAR) and pregnane X receptor (PXR) support the hypertrophic but not the hyperplastic response to the murine non-genotoxic hepatocarcinogens phenobarbital and chlordane in vivo. Toxicol Sci. doi:10.1093/toxsci/kfq118
Scheer N, Ross J, Rode A, Zevnik B, Niehaves S, Faust N, Wolf CR (2008) A novel panel of mouse models to evaluate the role of human pregnane X receptor and constitutive androstane receptor in drug response. J Clin Invest 118:3228–3239
Shah YM, Morimura K, Yang Q, Tanabe T, Takagi M, Gonzalez FJ (2007) Peroxisome proliferator-activated receptor alpha regulates a microRNA-mediated signaling cascade responsible for hepatocellular proliferation. Mol Cell Biol 27:4238–4247
Sibinski LJ, Allen JL, Erickson EE (1983) Two year oral (diet) toxicity/carcinogenicity study of fluorochemical FC-143 in rats. Expt. No. 0281CR0012. Available on USEPA Docket AR226–0437-0440. Riker Laboratories, Inc., St. Paul
Takacs ML, Abbott BD (2007) Activation of mouse and human peroxisome proliferator-activated receptors (alpha, beta/delta, gamma) by perfluorooctanoic acid and perfluorooctane sulfonate. Toxicol Sci 95:108–117
Vanden Heuvel JP, Thompson JT, Frame SR, Gillies PJ (2006) Differential activation of nuclear receptors by perfluorinated fatty acid analogs and natural fatty acids: a comparison of human, mouse, and rat peroxisome proliferator-activated receptor-α, -β, and -γ, liver X receptor-β, and retinoid X receptor-α. Toxicol Sci 92:476–489
Walters MW, Bjork JA, Wallace KB (2009) Perfluorooctanoic acid stimulated mitochondrial biogenesis and gene transcription in rats. Toxicology 264:10–15
Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD (2000) The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature 407:920–923
Whysner J, Ross PM, Williams GM (1996) Phenobarbital mechanistic data and risk assessment: enzyme induction, enhanced cell proliferation, and tumor promotion. Pharm Ther 71:153–191
Wolf CJ, Takacs ML, Schmid JE, Lau C, Abbott BD (2008) Activation of mouse and human peroxisome proliferator-activated receptor alpha (PPARα) by perfluoroalkyl acids (PFAAs) of different functional groups and chain lengths. Toxicol Sci 106:162–171
Yamamoto Y, Moore R, Goldsworthy TL, Negishi M, Maronpot RR (2004) The orphan nuclear receptor constitutive active/androstane receptor is essential for liver tumor promotion by phenobarbital in mice. Cancer Res 64:7197–7200
Acknowledgments
This work was supported by Plastics Europe. We wish to thank Robert Powrie (CXR Biosciences) for the analyses of plasma PFOA concentrations.
Conflict of interest statement
With the exception of JRF, all authors represent organizations that have a current or former financial interest in ammonium perfluorooctanoate.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Elcombe, C.R., Elcombe, B.M., Foster, J.R. et al. Hepatocellular hypertrophy and cell proliferation in Sprague–Dawley rats following dietary exposure to ammonium perfluorooctanoate occurs through increased activation of the xenosensor nuclear receptors PPARα and CAR/PXR. Arch Toxicol 84, 787–798 (2010). https://doi.org/10.1007/s00204-010-0572-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-010-0572-2