
Abstract
Abstract HMG CoA reductase inhibitiors (statins) have been shown to be effective lipid lowering agents and are beneficial in the primary and secondary prevention of coronary heart disease. However, the overall benefits observed with statins appear to be greater than what might be expected from changes in lipid levels alone and the positive effects have only partially been reproduced with other lipid lowering drugs, suggesting effects in addition to cholesterol lowering. In experimental models, many of the cholesterol-independent effects of statins are mediated by inhibition of isoprenoids, which serve as lipid attachments for intracellular signalling molecules such as small Rho guanosine triphosphate-binding proteins, whose membrane localization and function are dependent on isoprenylation. This review summarizes the effects of statins on endothelial function and oxidative stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The primary mechanism of HMG CoA reductase inhibitors (statins) is the lowering of serum cholesterol levels via inhibition of hepatic cholesterol synthesis and subsequent upregulation of low-density lipoprotein (LDL)-receptors in the liver (Goldstein and Brown 1990). However, recent evidence suggests that statins have beneficial effects beyond cholesterol lowering and in extra-hepatic tissues. Experimental and clinical evidence revealed that several important regulators of the cardiovascular system can be regulated by statins. Prominent candidates are the endothelial NO synthase (eNOS), endothelin, free oxygen radicals, MHC-II, protein kinase Akt and the metalloproteinases. This article reviews the effects of statins on endothelial dysfunction and oxidative stress.
Properties of HMG-CoA reductase inhibitors
The rate-limiting enzyme in cholesterol biosynthesis in the liver is HMG-CoA reductase (Goldstein and Brown 1990) (Fig. 1). HMG-CoA reductase catalyses the four-electron reductive deacylation of HMG-CoA to CoA and mevalonate which is the committed and rate-limiting step in cholesterol biosynthesis (Rodwell et al. 1976). All HMG-CoA reductase inhibitors (statins) share an HMG-like moiety and inhibit the reductase by the same mechanism. The bulky, hydrophobic compounds of statins blocks the access of the substrate HMG-CoA to the reductase by occupying the HMG-binding pocket and part of the binding surface for CoA (Istvan and Deisenhofer 2001). The tight binding of statins is due to the large number of van der Waals interactions between inhibitors and HMG-CoA reductase. The liver is the primary place of action of all statins. The extra-hepatic plasma concentration and permeability, e.g. into vascular cells, differs between statins and depends mainly on their lipophilicity (McTaggart et al. 2001; Lea and McTavish 1997; Corsini et al. 1995; Blum 1994). Inhibition of cholesterol synthesis in hepatocytes upregulates the expression of the hepatic LDL receptor. As a consequence, LDL and its precursors are cleared from the circulation (Goldstein and Brown 1990). Furthermore, statin treatment also increases the plasma concentration of antiatherogenic HDL and apo A-I (Vega and Grundy 1998). In addition to lowering cholesterol synthesis, inhibition of the HMG-CoA reductase reduces the synthesis of intermediates of the mevalonate pathway (Goldstein and Brown 1990).

Inhibition of mevalonate synthesis not only blocks the synthesis of cholesterol but also the isoprenoid-intermediates of the cholesterol pathway. The isoprenoid geranylgeranylpyrophosphate plays an important role for the posttranslational modification of proteins. The membrane translocation and activity of the small GTP-binding proteins Rho and Rac depend on their geranylgeranylation. Ras translocation from the cytoplasm to the plasma membrane is dependent on farnesylation. Statins inhibit small G protein isoprenylation and function
Mechanism mediating cholesterol-independent effects of HMG-CoA reductase inhibitors
An important mechanism underlying cholesterol independent effects relates to the inhibition of isoprenoid intermediates of the cholesterol synthesis pathway (Liao and Laufs 2005). Statins work by reversibly inhibiting HMG-CoA reductase through side chains that bind to the enzyme’s active site and block the substrate-product transition state of the enzyme (Istvan and Deisenhofer 2001). Thereby statins competitively inhibit the synthesis of l-mevalonic acid, the immediate product of HMG-CoA reductase. At the same time, statins prevent the synthesis of other important isoprenoid intermediates of the cholesterol biosynthetic pathway, such as farnesylpyrophosphate and geranylgeranylpyrophosphate (Goldstein and Brown 1990) (Fig. 1). Isoprenoids are important lipid attachments for the posttranslational modification of a variety of proteins, like small guanosine triphosphate (GTP)-binding protein Ras; and Ras-like proteins, such as Rho, Rab, Rac, Ral, or Rap for example (Van and D’Souza-Schorey 1997). Thus, protein isoprenylation permits the covalent attachment, subcellular localization, and intracellular trafficking of membrane-associated proteins. Members of the Ras and Rho GTPase family are major substrates for posttranslational modification by prenylation (Hall 1998; Van and D’Souza-Schorey 1997). Rac, Ras and Rho are small GTP-binding proteins, which cycle between the inactive GDP-bound state and active GTP-bound state. In endothelial cells, Rho translocation is dependent on geranylgeranylation, whereas Ras translocation from the cytoplasm to the plasma membrane is dependent on farnesylation (Laufs et al. 1998; Laufs and Liao 1998). Statins inhibit Rac, Ras and Rho isoprenylation, leading to the accumulation of inactive Rac, Ras and Rho in the cytoplasm. Because Rho is major target of geranylgeranylation, inhibition of Rho and its downstream target, Rho-kinase, is a likely mechanism mediating some of the cholesterol-independent effects of statins on the vascular wall (Laufs et al. 2000). The members of the Rho GTPase family, which consists of RhoA, Rac and Cdc42, serve specific functions in terms of cell shape, motility, secretion and proliferation, although overlapping functions between the members could be observed in overexpressed systems. The distinct but complementary functions of Rho family members also extend to their effects on cell signalling. It is therefore not surprising to find that Rho-induced changes in the actin cytoskeleton and gene expression are related (Tapon and Hall 1997).
Endothelial function
Hypercholesterolemia causes endothelial dysfunction, which is an early manifestation of atherosclerosis and occurs even in the absence of angiographic evidence of disease (Libby et al. 1991, Libby 1995). Rapid lowering of plasma LDL cholesterol by apheresis improves endothelium-dependent vasodilatation (Tamai et al. 1997). These observations along with many other studies show that lowering of serum cholesterol levels is an important mechanism of the beneficial vascular effects of statins. Interestingly, in some studies with statins, restoration of endothelial function occurs before significant reduction in serum cholesterol levels (O’Driscoll et al. 1997; Anderson et al. 1995; Treasure et al. 1995), suggesting that there are additional effects on endothelial function beyond cholesterol reduction. Improvement of NO-dependent endothelial function is one of the clinical hallmarks of statin treatment and can be observed very rapidly. E.g., treatment with statins has been shown to improve coronary endothelial function within one day, significantly before serum cholesterol starts to fall (Liao and Laufs 2005; Wassmann et al. 2003; Laufs et al. 2001). An important characteristic of endothelial dysfunction is the impaired synthesis, release and activity of endothelial-derived nitric oxide (NO). Endothelial NO has been shown to inhibit several components of the atherogenic process. For example, endothelium-derived NO mediates vascular relaxation and inhibits platelet aggregation, vascular smooth muscle proliferation and endothelial-leukocyte interactions. Furthermore, inactivation of NO by superoxide radical (O2 −) limits the bioavailability of NO and leads to nitrate tolerance, vasoconstriction and hypertension (Harrison 1997; Munzel et al. 1995).
Endothelial dysfunction is an important and early marker of atherosclerosis (Liao 1998). The loss of NO contributes to the atherogenic process. On the other hand, hypercholesterolemia plays an important role in decreasing endothelial production and in increasing degradation of NO (Hernandez-Perera et al. 1998; Laufs et al. 1997; Liao 1994; Liao et al. 1995). One of the earliest effects of statin treatment is a rapidly increased bioavailability of NO (John et al. 2001; Tsunekawa et al. 2001; Kaesemeyer et al. 1999; O’Driscoll et al. 1997; Anderson et al. 1995) mediating improvement of endothelium-dependent vasodilatation. Increased endothelial NO release has been reported as early as 3 days after initiation of treatment (John et al. 2001; Tsunekawa et al. 2001). Upregulation of eNOS expression is independent of cholesterol levels (Feron et al. 2001; Amin-Hanjani et al. 2001; Wagner et al. 2000; Laufs et al. 1999; Tannous et al. 1999; Kaesemeyer et al. 1999; Hernandez-Perera et al. 1998; Williams et al. 1998; Laufs et al. 1998). On the molecular level, upregulation of endothelial NO production is mediated by several complementary mechanisms: reduction of LDL improves endothelial NO, in addition, statins increase eNOS mRNA stability by inhibiting the rhoA GTPase-dependent actin cytoskeleton (Laufs and Liao 1998, 2000). Additional important effects of statin treatment on eNOS function include the activation of protein kinase Akt (Kureishi et al. 2000), inhibition of calveolin (Laufs 2003) and activation of the phosphatidylinositol 3-kinase/protein kinase Akt (PI3K/Akt) pathway (Kureishi et al. 2000; Fulton et al. 1999). The important role of NO on endothelial dysfunction is also influenced by superoxide radicals. An imbalance between the production of superoxide radicals and NO in the vessel wall leads to an attenuated vasodilatator response and is known to promote the development of endothelial dysfunction (Harrison 1997).
Oxidative stress
Reactive oxygen species (ROS) include superoxide, hydroxyl (OH.) and hydrogen peroxide (H2O2). Free radicals can induce oxidation and damage to DNA, membranes, proteins and other macromolecules, if they are present in excess. Diverse specific and non-specific antioxidant defence systems therefore exist to scavenge and degrade ROS to non-toxic molecules (Li and Shah 2004). The “redox state” of a cell describes the balance between ROS production and their removal by antioxidant systems; a pathological imbalance in favour of excess ROS is termed oxidative stress. A small amount of O2 − is normally produced as a by product of the use of molecular oxygen during mitochondrial oxidative phosphorylation. A family of superoxide dismutase (SOD) enzymes rapidly converts O2 − to H2O2, which is itself broken down by glutathione peroxidase (GPX) and catalase (CAT) to water. The pathophysiological effects of ROS depend on the type, concentration and specific site of production. At lower concentrations local targeted production of ROS serves as a second-messenger system that transmits biological information through the highly specific modulation of intracellular signalling molecules, enzymes and proteins which is called redox signalling. These processes are involved in the activation of many signal transduction protein kinases and transcription factors, the stimulation of DNA synthesis and expression of growth-related genes, (Li and Shah 2004; Finkel 1999) and the regulation of myocardial excitation–contraction coupling (Gao et al. 1996). High levels of ROS tend to react with numerous protein centres, DNA, cell membranes and other molecules, causing considerable cellular damage as well as generating other more reactive radicals. Another ROS-related pathophysiological mechanism involves the reaction of O2 − with the signalling molecule nitric oxide, which in health has a central role in vascular homeostasis as well as in modulating cardiac function. The reaction between O2 − and nitric oxide leads to inactivation of nitric oxide and loss of its biological activity (Li and Shah 2004). Nitric oxide synthase enzymes normally generate nitric oxide, but may instead generate O2 − if they become “uncoupled”, a state that is especially likely to occur in the setting of deficiency of the NOS cofactor BH4 or the NOS substrate l-arginine (Verhaar et al. 2004). NOS uncoupling and subsequent O2 − production are implicated in the genesis of vascular endothelial dysfunction in patients with heart failure (Dixon et al. 2003).
Antioxidative effects of statins in the vascular system
Statins may also improve endothelial function through their antioxidant effects. For example, statins enhance endothelium-dependent relaxation by inhibiting production of ROS from aortas of cholesterol-fed rabbits (Rikitake et al. 2001). Importantly, lipid lowering by itself lowers vascular oxidative stress (Cai and Harrison 2000). In addition, other antioxidant effects of statins appear to be cholesterol independent. The major source of ROS in the vascular wall is the NAD(P)H oxidase complex. The small GTP binding protein Rac1 is important for the assembly of the NAD(P)H oxidase enzyme complex. Inhibition of Rac1 isoprenylation by statin treatment prevents the activation of NAD(P)H oxidase and ROS release (Wassmann et al. 2001a, 2001b; Endres et al. 1998; Williams et al. 1998; Laufs et al. 1997, 1998). These enzymes catalyse electron transfer from NADPH to molecular oxygen, resulting in the formation of O2 −. NADPH oxidase activity has been found to be increased in experimental models of LVH and CHF as well as in end-stage failing human myocardium (Dixon et al. 2003; Heymes et al. 2003; Maack et al. 2003; Li et al. 2002). Interestingly, ROS produced by NADPH oxidases can promote ROS generation by other sources, thereby amplifying total levels of ROS. For example, O2 − from NADPH oxidase may oxidize and degrade BH4, thereby leading to NOS uncoupling (Verhaar et al. 2004). Similarly, NADPH oxidase derived ROS may also activate xanthine oxidase (Li and Shah 2004). Wassmann et al. (Wassmann et al. 2001b) evaluated the effect of atorvastatin on the vascular production of ROS. After treatment of spontaneously hypertensive rats with atorvastatin, carbachol-induced vasorelaxation in aortic segments was significantly improved. Furthermore, vascular production of ROS was reduced. Interestingly, statin therapy reduced blood pressure in this rat model and downregulated the angiotensin II type 1 (AT1) receptor expression. Moreover, the expression of the eNOS expression and activity was enhanced.
Besides ROS generating enzymes, antioxidative defense systems are important for the oxidative stress that ultimately results. The SOD isoforms, GPX and CAT are enzymes residing within the vasculature that finally lead to the elimination of free radicals by the generation of water and oxygen (Strehlow et al. 2003; Andreoli 2000; Gutteridge and Halliwell 2000). The ultimate oxidative stress within vascular cells is determined by ROS production and corresponding elimination processes. The latter are realized by the radical scavenging enzymes GPX, the SOD isoforms and CAT. Whereas atorvastatin had no influence on the expression of GPX and SODs, CAT expression and activity were profoundly upregulated in vitro and in vivo. Physiologically, the upregulation of CAT can be observed after an increase of hydrogen peroxide concentrations. However, reduced superoxide production by NAD(P)H oxidase after decreased expression of essential subunits leads to reduced concentrations of hydrogen peroxide when SOD levels are not altered (Lassegue et al. 2001; Ushio-Fukai et al. 1996). Because CAT is used in the elimination of hydrogen peroxide (Andreoli et al. 2000; Gutteridge and Halliwell 2000), increased levels of CAT further reduce the concentrations of this radical, thereby accelerating the turnover of superoxide to hydrogen peroxide. Finally, this leads to a decrease of the overall intracellular free radical load in VSMCs. Therefore, upregulation of CAT may represent another antioxidative action of statins (Wassmann et al. 2003).
Antioxidative effects of statins in the myocardium
Although the main impact of statin therapy in cardiovascular disease appears to be predominantly vascular, recent animal and human studies suggest that statins may also have direct beneficial effects on the myocardium. Animal studies suggest that a phagocyte-type NADPH oxidase may be a relevant source of ROS in the ventricular myocardium (Bendall et al. 2002; MacCarthy et al. 2001; Aikawa et al. 2000). In the cardiomyocytes, three of its five components, p40phox PHOX (for phagocyte oxidase), p47phox, and p67phox, exist in the cytosol, forming a complex. The other two components, p22phox and gp91phox, are bound to the membranes. Various stimuli lead to the phosphorylation of the cytosolic components, and the entire cytosolic complex then migrates to the membrane. Importantly, not only the core subunits but also two low-molecular-weight guanine nucleotide-binding proteins, Rac1 and Rap, are required for activation. During activation, Rac1 binds GTP and migrates to the membrane with the core cytosolic complex. Therefore, Rac1 is critically involved in the activation of cardiovascular NADPH oxidase. NADPH oxidase-dependent ROS production is involved in cardiac hypertrophy in response to pressure overload (Li et al. 2002; MacCarthy et al. 2001), stretch (Aikawa et al. 1999), angiotensin II-infusion (Bendall et al. 2002), and α-adrenergic stimulation (Xiao et al. 2002).
Because Rac1 is required for NADPH oxidase activity and cardiac hypertrophy is mediated, in part, by myocardial oxidative stress, it is likely that statins could inhibit cardiac hypertrophy through an antioxidant mechanism involving inhibition of the association of RhoGDIα with Rac1 which is mediated by phosphatidylinositol-3 kinase and depends on geranylgeranylation (Custodis et al. 2006) (Fig. 2). Indeed, statins inhibit angiotensin II-induced oxidative stress and cardiac hypertrophy in rodents (Takemoto et al. 2001). This has also been observed in clinical studies where statins inhibit cardiac hypertrophy in humans with hypercholesterolemia (Lee et al. 2002). NADPH-oxidase-mediated ROS are increased in left ventricular myocardium from patients with heart failure and correlate with an increased activity of Rac1 GTPase, and oral statin treatment is able to decrease Rac1 function in the human heart (Maack et al. 2003).

Antioxidative mechanism of statins. Activation of NAD(P)H oxidase by angiotensin II via Rac1 GTPase. Statins could inhibit NAD(P)H oxidase through inhibition of the association of RhoGDIα with Rac1 which depends on geranylgeranylation
Furthermore plays increased atrial oxidative stress an important role in inducing and maintaining atrial fibrillation (AF) (Cai et al. 2002; Nattel et al. 2002; Mihm et al. 2001; Carnes et al. 2001). AF induced by rapid atrial pacing in pigs is characterized by increased NAD(P)H oxidase activity and superoxide production in the left atrium (Dudley et al. 2005). In isolated atrial myocytes from human right atrial appendages, NADPH oxidase is the main source of atrial superoxide production (Kim et al. 2005). NADPH-stimulated superoxide release was higher in patients with AF. NO synthase contributed to atrial superoxide production in fibrillating atria, suggesting that increased oxidative stress may lead to NOS “uncoupling”. These findings indicate that NADPH oxidase significantly contributes to superoxide production in AF (Kim et al. 2005). Indeed, left atrial tissue of patients with AF is characterized by upregulation of Rac1-GTPase and the superoxide-producing NADPH-oxidase compared to patients with sinus rhythm (Adam et al. 2007). In mice with cardiac specific overexpression of Rac1 under the control of the αMHC promoter (RacET), we observed AF with aging, which was associated with an increased NADPH oxidase activity. Notably, treatment with HMG-CoA reductase inhibitors inhibits Rac1 activation by inhibiting its geranylgeranylation and membrane translocation (Adam et al. 2007; Laufs et al. 2002). Indeed oral treatment of the Rac1-overexpressing mice with statins inhibited Rac1, thereby lowered NADPH-oxidase activity and markedly reduced the incidence of AF (Adam et al. 2007).
Open questions
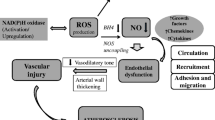
In experimental models, statins improve endothelial function through increased NO production and decreased oxidative stress (Fig. 3) in addition to cholesterol lowering. However, in clinical studies the quantitative contribution of cholesterol-independent compared to cholesterol-dependent effects is difficult to determine. Indirect evidence from small studies comparing statins with ezetimibe suggest a potential clinical relevance of these effects, however, the data are not fully conclusive (Landmesser et al. 2004). Another issue is the evidence suggesting that an abrupt discontinuation of statin medication may exert negative effects in patients with acute coronary syndromes or stroke while in stable vascular patients discontinuation may be safe. Withdrawal of statin treatment confers overshoot activation of small G-proteins Rho and Rac causing production of ROS and suppression of NO bio-availability (Endres and Laufs 2006).

Effects of statins on endothelial function and oxidative stress
Based on the data showing a rapid beneficial effect and the evidence for a potential harmful effect of withdrawal for certain patients, it may be of interest to evaluate an intravenous application of statins. Potential indications for an i.v. statin formulation could include the administration in patients with acute vascular syndromes such as ischaemic stroke and acute coronary syndromes as well as critically-ill patients pre-treated with statins whenever enteral administration is not possible (e.g., patients with vascular disease, those undergoing major surgery or trauma patients). Additional indications for i.v. treatment that have to be tested are the pre-treatment of patients undergoing surgery with high-vascular risk and peri-operative treatment of patients with high risk of AF [for review see (Endres and Laufs 2008)].
Summary
In summary, evidence from a variety of experimental and clinical studies show that statins have the potential to exert effects in addition to the lowering of serum cholesterol levels. These additional properties include beneficial effects on endothelial function and oxidative stress. Recent evidence show that many of these effects are mediated inhibition of isoprenoid synthesis, in particular Rho and Rac GTPases in vascular and myocardial cells. Despite the broad experimental evidence for beneficial vasoprotective cholesterol-independent and extra-hepatic actions of statins, the clinical importance of the pleiotropic effects of statins in addition to cholesterol-lowering remains to be clarified in more detail.
References
Adam O, Frost G, Custodis F, Sussman MA, Schafers HJ, Bohm M, Laufs U (2007) Role of Rac1 GTPase activation in atrial fibrillation. J Am Coll Cardiol 50:359–367
Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Zhu W, Kadowaki T, Yazaki Y (1999) Rho family small G proteins play critical roles in mechanical stress- induced hypertrophic responses in cardiac myocytes. Circ Res 84:458–466
Aikawa R, Nawano M, Gu Y, Katagiri H, Asano T, Zhu W, Nagai R, Komuro I (2000) Insulin prevents cardiomyocytes from oxidative stress-induced apoptosis through activation of PI3 kinase/Akt. Circulation 102:2873–2879
Amin-Hanjani S, Stagliano NE, Yamada M, Huang PL, Liao JK, Moskowitz MA (2001) Mevastatin, an HMG-CoA reductase inhibitor, reduces stroke damage and upregulates endothelial nitric oxide synthase in mice. Stroke 32:980–986
Anderson TJ, Meredith IT, Yeung AC, Frei B, Selwyn AP, Ganz P (1995) The effect of cholesterol-lowering and antioxidant therapy on endothelium-dependent coronary vasomotion. N Engl J Med 332:488–493
Andreoli TE (2000) Free radicals and oxidative stress. Am J Med 108:650–651
Bendall JK, Cave AC, Heymes C, Gall N, Shah AM (2002) Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 105:293–296
Blum CB (1994) Comparison of properties of four inhibitors of 3-hydroxy-3- methylglutaryl-coenzyme A reductase. Am J Cardiol 73:3D–11D
Cai H, Harrison DG (2000) Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87:840–844
Cai H, Li Z, Goette A, Mera F, Honeycutt C, Feterik K, Wilcox JN, Dudley SC Jr, Harrison DG, Langberg JJ (2002) Downregulation of endocardial nitric oxide synthase expression and nitric oxide production in atrial fibrillation: potential mechanisms for atrial thrombosis and stroke. Circulation 106:2764–2766
Carnes CA, Chung MK, Nakayama T, Nakayama H, Baliga RS, Piao S, Kanderian A, Pavia S, Hamlin RL, McCarthy PM, Bauer JA, Van Wagoner DR (2001) Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ Res 89:E32–E38
Corsini A, Maggi FM, Catapano AL (1995) Pharmacology of competitive inhibitors of HMG-CoA reductase. Pharmacol Res 31:9–27
Custodis F, Eberl M, Kilter H, Böhm M, Laufs U (2006) Association of RhoGDIalpha with Rac1 GTPase mediates free radical production during myocardial hypertrophy. Cardiovasc Res 71:342–351
Dixon LJ, Morgan DR, Hughes SM, McGrath LT, El Sherbeeny NA, Plumb RD, Devine A, Leahey W, Johnston GD, McVeigh GE (2003) Functional consequences of endothelial nitric oxide synthase uncoupling in congestive cardiac failure. Circulation 107:1725–1728
Dudley SC Jr, Hoch NE, McCann LA, Honeycutt C, Diamandopoulos L, Fukai T, Harrison DG, Dikalov SI, Langberg J (2005) Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: role of the NADPH and xanthine oxidases. Circulation 112:1266–1273
Endres M, Laufs U (2006) Discontinuation of statin treatment in stroke patients. Stroke 37:2640–2643
Endres M, Laufs U (2008) The medical case for the development of an intravenous statin formulation—beyond ischemic stroke. Cerebrovasc Dis 25:593–594
Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, Liao JK (1998) Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci USA 95:8880–8885
Feron O, Dessy C, Desager JP, Balligand JL (2001) Hydroxy-methylglutaryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation 103:113–118
Finkel T (1999) Signal transduction by reactive oxygen species in non-phagocytic cells. J Leukoc Biol 65:337–340
Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC (1999) Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399:597–601
Gao WD, Liu Y, Marban E (1996) Selective effects of oxygen free radicals on excitation-contraction coupling in ventricular muscle. Implications for the mechanism of stunned myocardium. Circulation 94:2597–2604
Goldstein JL, Brown MS (1990) Regulation of the mevalonate pathway. Nature 343:425–430
Gutteridge JM, Halliwell B (2000) Free radicals and antioxidants in the year 2000. A historical look to the future. Ann NY Acad Sci 899:136–147
Hall A (1998) Rho GTPases and the actin cytoskeleton. Science 279:509–514
Harrison DG (1997) Cellular and molecular mechanisms of endothelial cell dysfunction. J Clin Invest 100:2153–2157
Hernandez-Perera O, Perez-Sala D, Navarro-Antolin J, Sanchez-Pascuala R, Hernandez G, Diaz C, Lamas S (1998) Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorvastatin and simvastatin, on the expression of endothelin-1 and endothelial nitric oxide synthase in vascular endothelial cells. J Clin Invest 101:2711–2719
Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, Shah AM (2003) Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol 41:2164–2171
Istvan ES, Deisenhofer J (2001) Structural mechanism for statin inhibition of HMG-CoA reductase. Science 292:1160–1164
John S, Delles C, Jacobi J, Schlaich MP, Schneider M, Schmitz G, Schmieder RE (2001) Rapid improvement of nitric oxide bioavailability after lipid-lowering therapy with cerivastatin within two weeks. J Am Coll Cardiol 37:1351–1358
Kaesemeyer WH, Caldwell RB, Huang J, Caldwell RW (1999) Pravastatin sodium activates endothelial nitric oxide synthase independent of its cholesterol-lowering actions. J Am Coll Cardiol 33:234–241
Kim YM, Guzik TJ, Zhang YH, Zhang MH, Kattach H, Ratnatunga C, Pillai R, Channon KM, Casadei B (2005) A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ Res 97:629–636
Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K (2000) The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med 6:1004–1010
Landmesser U, Engberding N, Bahlmann FH, Schaefer A, Wiencke A, Heineke A, Spiekermann S, Hilfiker-Kleiner D, Templin C, Kotlarz D, Mueller M, Fuchs M, Hornig B, Haller H, Drexler H (2004) Statin-induced improvement of endothelial progenitor cell mobilization, myocardial neovascularization, left ventricular function, and survival after experimental myocardial infarction requires endothelial nitric oxide synthase. Circulation 110:1933–1939
Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK (2001) Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res 88:888–894
Laufs U (2003) Beyond lipid-lowering: effects of statins on endothelial nitric oxide. Eur J Clin Pharmacol 58:719–731
Laufs U, Liao JK (1998) Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem 273:24266–24271
Laufs U, La Fata L, Liao JK (1997) Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. J Biol Chem 272:31725–31729
Laufs U, La Fata V, Plutzky J, Liao JK (1998) Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation 97:1129–1135
Laufs U, Marra D, Node K, Liao JK (1999) 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPase-induced down- regulation of p27(Kip1). J Biol Chem 274:21926–21931
Laufs U, Endres M, Stagliano N, Amin-Hanjani S, Chui DS, Yang SX, Simoncini T, Yamada M, Rabkin E, Allen PG, Huang PL, Böhm M, Schoen FJ, Moskowitz MA, Liao JK (2000) Neuroprotection mediated by changes in the endothelial actin cytoskeleton. J Clin Invest 106:15–24
Laufs U, Wassmann S, Hilgers S, Ribaudo N, Böhm M, Nickenig G (2001) Rapid effects on vascular function after initiation and withdrawal of atorvastatin in healthy, normocholesterolemic men. Am J Cardiol 88:1306–1307
Laufs U, Kilter H, Konkol C, Wassmann S, Böhm M, Nickenig G (2002) Impact of HMG CoA reductase inhibition on small GTPases in the heart. Cardiovasc Res 53:911–920
Lea AP, McTavish D (1997) Atorvastatin. A review of its pharmacology and therapeutic potential in the management of hyperlipidaemias. Drugs 53:828–847
Lee TM, Chou TF, Tsai CH (2002) Association of pravastatin and left ventricular mass in hypercholesterolemic patients: role of 8-iso-prostaglandin f2alpha formation. J Cardiovasc Pharmacol 40:868–874
Li JM, Shah AM (2004) Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol 287:R1014–R1030
Li JM, Gall NP, Grieve DJ, Chen M, Shah AM (2002) Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 40:477–484
Liao JK (1994) Inhibition of Gi proteins by low density lipoprotein attenuates bradykinin-stimulated release of endothelial-derived nitric oxide. J Biol Chem 269:12987–12992
Liao JK (1998) Endothelium and acute coronary syndromes. Clin Chem 44:1799–1808
Liao JK, Laufs U (2005) Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol 45:89–118
Liao JK, Bettmann MA, Sandor T, Tucker JI, Coleman SM, Creager MA (1991) Differential impairment of vasodilator responsiveness of peripheral resistance and conduit vessels in humans with atherosclerosis. Circ Res 68:1027–1034
Liao JK, Zulueta JJ, Yu FS, Peng HB, Cote CG, Hassoun PM (1995) Regulation of bovine endothelial constitutive nitric oxide synthase by oxygen. J Clin Invest 96:2661–2666
Libby P (1995) Molecular bases of the acute coronary syndromes. Circulation 91:2844–2850
Maack C, Kartes T, Kilter H, Schäfers HJ, Nickenig G, Böhm M, Laufs U (2003) Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation 108:1567–1574
MacCarthy PA, Grieve DJ, Li JM, Dunster C, Kelly FJ, Shah AM (2001) Impaired endothelial regulation of ventricular relaxation in cardiac hypertrophy: role of reactive oxygen species and NADPH oxidase. Circulation 104:2967–2974
McTaggart F, Buckett L, Davidson R, Holdgate G, McCormick A, Schneck D, Smith G, Warwick M (2001) Preclinical and clinical pharmacology of Rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol 87:28B–32B
Mihm MJ, Yu F, Carnes CA, Reiser PJ, McCarthy PM, Van Wagoner DR, Bauer JA (2001) Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation 104:174–180
Munzel T, Sayegh H, Freeman BA, Tarpey MM, Harrison DG (1995) Evidence for enhanced vascular superoxide anion production in nitrate tolerance. A novel mechanism underlying tolerance and cross-tolerance. J Clin Invest 95:187–194
Nattel S, Khairy P, Roy D, Thibault B, Guerra P, Talajic M, Dubuc M (2002) New approaches to atrial fibrillation management: a critical review of a rapidly evolving field. Drugs 62:2377–2397
O’Driscoll G, Green D, Taylor RR (1997) Simvastatin, an HMG-coenzyme A reductase inhibitor, improves endothelial function within 1 month. Circulation 95:1126–1131
Rikitake Y, Kawashima S, Takeshita S, Yamashita T, Azumi H, Yasuhara M, Nishi H, Inoue N, Yokoyama M (2001) Anti-oxidative properties of fluvastatin, an HMG-CoA reductase inhibitor, contribute to prevention of atherosclerosis in cholesterol-fed rabbits. Atherosclerosis 154:87–96
Rodwell VW, Nordstrom JL, Mitschelen JJ (1976) Regulation of HMG-CoA reductase. Adv Lipid Res 14:1–74
Strehlow K, Rotter S, Wassmann S, Adam O, Grohe C, Laufs K, Bohm M, Nickenig G (2003) Modulation of antioxidant enzyme expression and function by estrogen. Circ Res 93:170–177
Takemoto M, Node K, Nakagami H, Liao Y, Grimm M, Takemoto Y, Kitakaze M, Liao JK (2001) Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J Clin Invest 108:1429–1437
Tamai O, Matsuoka H, Itabe H, Wada Y, Kohno K, Imaizumi T (1997) Single LDL apheresis improves endothelium-dependent vasodilatation in hypercholesterolemic humans. Circulation 95:76–82
Tannous M, Cheung R, Vignini A, Mutus B (1999) Atorvastatin increases ecNOS levels in human platelets of hyperlipidemic subjects. Thromb Haemost 82:1390–1394
Tapon N, Hall A (1997) Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol 9:86–92
Treasure CB, Klein JL, Weintraub WS, Talley JD, Stillabower ME, Kosinski AS, Zhang J, Boccuzzi SJ, Cedarholm JC, Alexander RW (1995) Beneficial effects of cholesterol-lowering therapy on the coronary endothelium in patients with coronary artery disease. N Engl J Med 332:481–487
Tsunekawa T, Hayashi T, Kano H, Sumi D, Matsui-Hirai H, Thakur NK, Egashira K, Iguchi A (2001) Cerivastatin, a hydroxymethylglutaryl coenzyme a reductase inhibitor, improves endothelial function in elderly diabetic patients within 3 days. Circulation 104:376–379
Ushio-Fukai M, Zafari AM, Fukui T, Ishizaka N, Griendling KK (1996) p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J Biol Chem 271:23317–23321
Van AL, D’Souza-Schorey C (1997) Rho GTPases and signaling networks. Genes Dev 11:2295–2322
Vega GL, Grundy SM (1998) Effect of statins on metabolism of apo-B-containing lipoproteins in hypertriglyceridemic men. Am J Cardiol 81:36B–42B
Verhaar MC, Westerweel PE, van Zonneveld AJ, Rabelink TJ (2004) Free radical production by dysfunctional eNOS. Heart 90:494–495
Wagner AH, Kohler T, Ruckschloss U, Just I, Hecker M (2000) Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol 20:61–69
Wassmann S, Laufs U, Bäumer AT, Müller K, Ahlbory K, Linz W, Itter G, Rösen R, Böhm M, Nickenig G (2001a) HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension 37:1450–1457
Wassmann S, Laufs U, Bäumer AT, Müller K, Konkol C, Sauer H, Böhm M, Nickenig G (2001b) Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol Pharmacol 59:646–654
Wassmann S, Faul A, Hennen B, Scheller B, Böhm M, Nickenig G (2003) Rapid effect of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition on coronary endothelial function. Circ Res 93:e98–e103
Williams JK, Sukhova GK, Herrington DM, Libby P (1998) Pravastatin has cholesterol-lowering independent effects on the artery wall of atherosclerotic monkeys. J Am Coll Cardiol 31:684–691
Xiao L, Pimentel DR, Wang J, Singh K, Colucci WS, Sawyer DB (2002) Role of reactive oxygen species and NAD(P)H oxidase in alpha(1)-adrenoceptor signaling in adult rat cardiac myocytes. Am J Physiol Cell Physiol 282:C926–C934
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Adam, O., Laufs, U. Antioxidative effects of statins. Arch Toxicol 82, 885–892 (2008). https://doi.org/10.1007/s00204-008-0344-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-008-0344-4