
Abstract
It has been reported that sorbitol induces apoptosis in several cancer cell lines. However, the molecular mechanism underlying the sorbitol-induced apoptotic process is not yet clearly understood. In the present study, the intracellular signaling pathways of sorbitol-induced apoptosis in human K562 cells were investigated using both morphological analysis and DNA fragmentation technique. In this study, we demonstrated that sorbitol-induced apoptosis in human K562 cells is a concentration- and time-dependent manner. This sorbitol-induced apoptosis in human K562 cells was also accompanied by the up-regulation of Bax, and down-regulation of p-Bcl-2, but no effect on the levels of Bcl-XL. Moreover, the sorbitol treatment resulted in a significant reduction of mitochondria membrane potential, increase in the release of mitochondrial cytochrome c (cyt c), and activation of caspase 3. Furthermore, treatment with caspase 3 inhibitor (z-DEVD-fmk) was capable of preventing the sorbitol-induced caspase 3 activity and cell death. These results clearly demonstrate that the induction of apoptosis by sorbitol involves multiple cellular/molecular pathways and strongly suggest that pro- and anti-apoptotic Bcl-2 family proteins, mitochondrial membrane potential, mitochondrial cyt c, and caspase 3, they all participate in sorbitol-induced apoptotic process in human K562 cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sorbitol is a hydrogenated form of carbohydrate obtained by the reduction of the carbonyl group of the glucose molecule to the hydroxyl group. It has been demonstrated that sorbitol is able to efficiently and rapidly induce apoptosis when provided at high concentrations, as a part of the mechanisms related to hyperosmotic stress. In fact, hyperosmotic challenge has been shown to induce apoptosis in several cell lines such as human neuroblastoma (Stoothoff and Johnson 2001), cardiac fibroblasts (Galvez et al. 2003) myocites (Murata et al. 2002), Hep-2 cells and human gastric cells (Teramachi and Izawa 2000). Moreover, among the polyhydric alcohols, it has been demonstrated that xylitol (five hydroxyl groups) and erythritol (four hydroxyl groups) are also able to induce chromosomal DNA fragmentation but less efficiently than mannitol (six hydroxyl groups), which behaves as sorbitol. In contrast, neither glycerol (three hydroxyl groups) nor ethylene glycol (two hydroxyl groups) are able to induce DNA fragmentation (Koyama et al. 2000). These data indicate that polyhydric alcohols, with at least four hydroxyl groups in the molecule, have an increasing ability to induce apoptosis correlated to the length of the molecule
Apoptosis is an important continuous process of destruction of undesirable cells during development or homeostasis in multi-cellular organisms. This process is characterized by distinct morphological changes including plasma membrane bleb, cell shrinkage, depolarization of mitochondria, chromatin condensation and DNA fragmentation (Kaufmann and Hengartner 2001; Reed 2001).
Many proteins are known to be involved in the process of apoptosis. Caspases are essential for the execution of cell death by various apoptotic stimuli (Cohen 1997). Caspase activation is often regulated by various proteins, including the member of inhibitors of apoptosis (IAP) and Bcl-2 family (Deveraux et al. 1998).
In the latter, the mitochondrial pathway is dependent upon the release of cyt c from mitochondria into the cytosol. This process is initiated by the interaction of mitochondria with one or more of the Bcl-2 family proteins. Such family, which comprises pro-apoptotic (e.g., Bax, Bak) as well as anti-apoptotic (e.g., Bcl-2, Bcl-xL) members (Antonsson and Martinou 2000), are major regulators of the intrinsic pathway. They form homo- and heterooligomers, which act directly at the outer mitochondrial membrane. The ratio of pro- to anti-apoptotic oligomers has been suggested to play an important role in determining commitment to cell death (Kuwana and Newmeyer 2003).
In this investigation, we studied the quantitative and qualitative changes of several known effectors in sorbitol-induced apoptotic process in human K562 cells.
Materials and methods
Cell cultures and treatments
Human chronic myelogenous leukemia cell line K562 was purchased from American Type Culture Collection (Rockville, MD) and grown in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum at 37°C in an atmosphere containing 5% CO2. Cells were routinely collected by centrifugation at 700×g and resuspended in fresh medium at a concentration of 2 × 105/ml. Cell viability was assessed by trypan blue exclusion.
In the first series of experiments, K562 cells were incubated with different concentrations of sorbitol in the range of 0.1–1.0 M for 1 h at 37°C, briefly washed with RPMI 1640, and then reincubated for an additional 3 h at 37°C in RPMI 1640 containing 10% FCS, and then only the concentration of 1.0 M of sorbitol was selected to induce apoptosis The cells not treated with sorbitol were considered as controls.
In a second experimental phase, K562 cells, pretreated with quercetin and rutin at the concentration 25 μM for 1 h, were incubated with 1 M sorbitol for 60 min. The cells not treated were considered as controls.
DNA fragmentation assay
Cells were washed twice with phosphate-buffered saline (PBS) and lysed by addition of a hypotonic solution (1% NP-40 in 20 mM EDTA, 50 mM Tris–HCl pH 7.5). The supernatant was collected and was prepared as reported earlier (Herrmann et al. 1994).
Analysis of mitochondrial membrane potential
For mitochondrial membrane potential analysis, cells were incubated with 40 nM TMRE for 30 min at 37°C before the addition of sorbitol and subjected to FACS analysis, as described previously (Scaduto and Grotyohann 1999). TMRE fluorescence was detected in viable cells using forward-scatter and side-scatter criteria. The mitochondrial uncoupling agent FCCP (10 μM) was added 15 min before TMRE staining to depolarize mitochondria and used as an indicator that staining was proportional to mitochondrial membrane potential.
Cytochrome c release and measurement of caspase 3 activity
After the treatment with indicated agent, human K562 cells were harvested, washed with PBS and prepared resuspending in buffer A (250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiolthione, 17 μg/mL phenylmethylsulfonyl fluoride, 8 μg/mL aprotinin, 2 μg/mL leupeptin pH 7.4) and then kept on ice for 30 min. Cells were passed through a needle 20 times. Unlysed cells, and nuclei were pelleted by centrifugation at 750×g for 10 min. The supernatant was then spun at 10,000×g for 15 min. The resulting pellet, the mitochondrial fraction, was resuspended in buffer A. The supernatant was centrifuged at 100,000×g for 30 min. The supernatant from this final centrifugation represents the cytosolic fraction. Cyt c release was determined by Western blot as described later. Furthermore, the cell pellets were resuspended in lysis buffer (caspase colorimetric assay kits; Bivision, Inc.) and left on ice for 30 min. The lysates were centrifuged at 10,000×g for 10 min and the supernatant (20 μl) was collected for caspase 3 activity assay in the lysis buffer containing DEVD–pNA, a specific substrate to caspase 3. The concentration of pNA, as the product from enzymatic converting of DEVD–pNA by caspase 3, was measured at 405 nm and used as an indicative of caspase 3 activity.
Western blot analysis
After exposed to the indicated concentration sorbitol, human K562 cells were washed with cold PBS. Whole cell extracts were prepared by incubating the cells with cold lysis buffer (20 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, and 1 mM PMSF). The protein content of the lysates was determined using the DC protein assay kit (Bio-Rad). The cell lysates (25 μg protein/lane) were electrophoresized on 12% SDS-polyacrylamide gels. The cellular proteins were then transferred to PVDF membranes by electroblotting for 2 h and Western blot analysis was carried out as previously described (Jow et al. 2004; Cheng et al. 2007). The protein levels were visualized with an enhanced chemiluminescence detection kit (Amersham).
Electron microscopy
Ultrastructural analysis of morphological changes in K562 cells was performed after fixing in conventional fixing solutions (2.5% glutaraldhyde in 0.1 M PBS pH 7.3). For processing the specimens were post-fixed for 1 h in 1.33% osmium tetroxide in 0.1 M PBS. Samples were processed (post-fixation and dehydration) for embedding in epoxy resin. Ultra-thin sections, stained with uranyl acetate and lead hydroxide, were examined by using a Philips CM-10 trasmission electron microscope. Ultra-thin sections for transmission electron microscopy (TEM) were also studied by morphometry to evaluate the ultrastructure and quantitative changes in the mitochondrial, nuclear and plasmamembrane compartments.
Transmission electron microscopy analysis
Cells were fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer pH 7.3, maintained for 1 h at 4°C, washed and incubated for 1 h in 1.33% osmium tetroxide. Dehydration and resin embedding were performed following a standard schedule. Ultra-thin sections (60–70 nm) were stained with uranyl acetate and lead hydroxide, and then analyzed with a CM-12 Philips transmission electron microscope (TEM). Ultra-thin sections were also studied by morphometry to evaluate the ultra structure and quantitative changes in the mitochondrial, nuclear and plasma membrane compartments. The measurements included the percentage of nuclear and mitochondrial areas as related to the cytoplasmic area. Morphometric analysis was carried out by a computer assisted image analyzer, utilizing the KS-300 software, directly on TEM photographic negatives. Twenty micrographs (×4,900), taken from three different serial sections from each TEM sample were digitalised and analysed.
Statistic analysis
Caspase 3 activity was presented as the mean and standard error (SEM) from four to five experiments. The statistical analysis of data was performed by one-way ANOVA, followed by the Schefft test and P-values less than 0.05 were considered significant.
Results
Sorbitol induced DNA fragmentation and morphological changes in K562 cells
In preliminary studies, we established the effects of the dose and the time of incubation of sorbitol treatment in K562 cells on induction of apoptosis. In these experiments, the criterion for induction of apoptosis was the fragmentation of chromosomal DNA resulting from random cleavage at intranucleosomal intervals evaluated by agarose gel electrophoresis. Titrations of sorbitol in the range from 0.1 to 1 M, in K562 cells incubated for 1 h at 37°C, indicated that concentrations from 0.6 to 1 M showed a major amount of fragmented chromosomal DNA than the ones treated at concentrations from 0.1 to 0.4 M sorbitol (Fig. 1). Therefore, we chose to study sorbitol’s effect on apoptotic cell death and its effects on caspases and Bcl-2 members. To characterize the cell death induced by sorbitol, we examined the morphological changes of sorbitol treated K562 cells by scanning electron microscopy. As illustrated in Fig. 2, cell shrinking and blebbing could be seen on the surface of K562 cells when treated with 1 M sorbitol for 1 h.

Sorbitol induces apoptosis in K562 cells. a Dose-dependent DNA fragmentation. Cells were exposed to the indicated concentrations of sorbitol for 1 h. Cells were harvested by centrifugation and DNA was extracted. The DNA fragments were separated on 1.8% agarose gel electrophoresis and were visualized under ultraviolet light after staining with ethidium bromide. M, size marker (100 base pair DNA ladder); Lane 1, control culture; Lane different concentration of sorbitol treatment

Sorbitol induces the morphological changes typical of apoptosis. b Chromatin alteration, large bleb formation appear after 30 min of sorbitol treatment. c Blebbing activity of the plasmamembrane increases especially after 60 min of sorbitol treatment and includes typical large blebs with cytoplasmic content and membrane fine shedding. d Cells appear in advanced phase of degradation after 60 min of sorbitol treatment
Determination of the involvement of caspase 3 activation
In the present study, we examined whether caspase 3 activation is involved in the apoptotic process triggered by sorbitol. In order to investigate the mechanism involved caspase activation, we analyzed the caspase cascade by immunoblot analysis for procaspase 8, procaspase 9 and procaspase 3. Western blot analysis of whole cell lysates obtained from K562 cells treated with 1 M of sorbitol revealed an increase in caspase 9 and decrease in procaspase 3 activities at a time point of 30 min (Fig. 3a). Furthermore, human K562 cells were pretreated with 50 μM caspase 3 inhibitors (z-DEVD-fmk) for 2 h, and then incubated with sorbitol for 1 h. The results clearly showed that administration of caspase 3 inhibitor alone did not affect caspase 3 activity (Fig. 3b). However, caspase 3 activation in K562 cells, treated with sorbitol, was significantly inhibited by z-DEVD-fmk (specific caspase 3 inhibitor) (Fig. 3b).

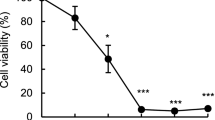
Sorbitol induces caspase 9 and caspase 3 activation. a Western blot analysis of procaspase 9, procaspase 3 and procaspase 8 in sorbitol treated K562 cells. Cells were treated with 1 M sorbitol for the indicated time periods. After treatment, the cell lysates were resolved by SDS-PAGE, transferred onto cellulose membranes, and then probed with specific antibodies. The amount of α-tubulin was measured as an internal control. Each blot is representative of three independent experiments. b Inhibition of caspase 3 activity and attenuation of sorbitol-induced cell death by caspase 3 inhibitor (z-DEVD-fmk). Human K562 cells were treated with 50 μM specific caspase 3 inhibitor (z-DEVD-fmk) 2 h prior to 1 h of 1 M sorbitol treatment. After incubation, caspase 3 activity was examined as described in Materials and Methods. All values are mean ± SEM of 4–5 determinations, *P < 0.001 compared with the respective sorbitol and z-DEVD-fmk free control and **P < 0.001 comparison between the absence and presence of z-DEVD-fmk in the same sorbitol treatment group
Changes of mitochondrial membrane potential and release of cytochrome c from mitochondria
In the present study, we analyzed the cyt c release and mitochondrial membrane potential. As shown in Fig. 4a, we noticed a time-dependent decrease in the amount of mitochondrial membrane potential after treatment with 1 M sorbitol. Concomitantly, we examined whether mitochondrial cyt c was released into cytosol. As shown in Fig. 4b, sorbitol treatment caused an accumulation of cyt c in the cytosolic fraction, compared to that of the control cells, together with a reduction in the mitochondrial fraction in a time-dependent manner. After 1 h treatment, we clearly observed a decrease in cyt c in the mitochondria and an accumulation in the cytosol. These data suggest that loss of mitochondrial membrane potential may be required for sorbitol-induced cyt c release into cytosol, that later triggered the cleavage and activation of mitochondrial downstream caspases and onset of apoptosis

The effect of sorbitol on mitochondrial impairment and cyt c release. a K562 cells were incubated for 30 min with 40 nM of the mitochondrial specific dye, TMRE and then transferred in media containing 1 M sorbitol cells were then harvested and analyzed as described in materials and methods. Data are from a typical experiment out of three giving comparable results. Fluorescence was analyzed by a FACScalibur instrument, and percentages of cells with collapsed m were calculated using WinMDI version 2.8 software. b K562 cells were treated with sorbitol (1 M) for the indicated time periods. Equal amounts of cytochrome protein from mitochondrial fraction (lower panel) and cytosolic fraction (upper panel) were analyzed by Western blotting using an anti-cytochrome c antibody. β-Actin was used as an equal loading control
Regulation of Bcl-2 family proteins in sorbitol-treated leukemia cancer cells
To determine whether Bcl-2 family proteins were modulated in sorbitol-induced apoptosis in human K562 cells, the expression of several members of Bcl-2 family proteins was examined by Western blot analysis. As shown in Fig. 5, exposure of human K562 cells to 1 M sorbitol resulted in a marked decrease of p-Bcl-2 protein expression along with a drastic increase of Bax protein expression. However, the levels of Bcl-XL proteins were not affected by sorbitol treatment. Moreover, sorbitol significantly up-regulated the expression of p-Bad, while it did not affect the expression of Bad protein (Fig. 5).

Expression of Bcl-2 family proteins in sorbitol-treated human K562 cells. These cells were treated with 1 M sorbitol for 30 and 60 min. After the treatment, cell lysates were extracted, and the levels of Bcl-2 family proteins were analyzed by Western blot analysis
Antioxidant effect on sorbitol-induced apoptotis K562 cells
Our previous study demonstrated that sorbitol induced-apoptosis was mediating by production of reactive oxygen species (ROS) (Sinibaldi-Salimei et al. 2007; Aquilano et al. 2007). To measure the effect of two antioxidants on sorbitol induction of apoptosis of K562 cells, we pre-treated the cells with quercetin and rutin for 1 h before treating the K562 cells with 1 M sorbitol. Both antioxidants were found able to inhibit sorbitol-induced apoptosis of the K562 cells. The effect of these antioxidants in sorbitol treatment on the integrity of genomic DNA shows in Fig. 6.

Protective effect of quercetin and rutin in sorbitol-induced apoptosis. K562 cells, at a density of 2 × 105/ml, were pre-incubated with quercetin and rutin at 25 μM concentration for 60 min and then treated with 1 M sorbitol. Cells were then harvested and DNA extracts were obtained as described in Materials and methods. DNA ladder was quantified using Quantity One soft. DNA fragmentation is not evident when the cell line was pre-incubated with antioxidants (M, marker; CN, control)
Discussion
In the present study, we investigated the possible mechanisms via which sorbitol should induce apoptosis in human K562 cells. First, we demonstrated that sorbitol induced cell death in a concentration range from 0.6 to 1 M in K562 cells. These cells treated with sorbitol exhibited characteristic morphological features of apoptosis, such as membrane shrinkage, chromosomal condensation. The several mechanisms of apoptosis activation in different either physiological or pathological conditions in cells have been proposed and studied intensively (Vaux and Korsmeyer 1999). Numerous factors, such as cytosolic cyt c release, caspase 3 activation, and the expression of Bcl-2 family proteins, have been suggested to play an essential role in the apoptotic process in cancer cells. Our study have demonstrated that sorbitol-treated K562 cells underwent apoptosis in dose and time-dependent manner. In addition, progressive decrease of the mitochondrial membrane potential as well as release of cyt c into the cytosol were also observed. In many in vitro systems apoptosis has been noticed to be associated with a loss of mitochondrial membrane potential, which may correspond to the opening of an outer membrane pore (permeability transition pore). Thus, this event has been suggested to be responsible for cyt c release into cytosol from mitochondria (Kantrow and Piantasdosi 1997). In our study, the cytosolic cyt c accumulation in sorbitol-treated human K562 cells is probably the consequence of the loss of mitochondrial membrane potential, which finally leads to the cell death. The Bcl-2 family is composed of a number of genes that play critical roles in the control of mitochondrial integrity. The reduction, or total diminution, of mitochondrial membrane potential leads to the release of intermembrane proteins, such as cyt c and apoptosis-inducing factors, into the cytosol and consequently induces apoptotic cell death (Yang et al. 1997). Several studies have shown that over-expression of Bcl-2 and Bcl-XL prevents the mitochondrial release of cyt c, thereby inhibiting the activation of caspases cascade and apoptosis (Solange and Martinou 2000; Gross et al. 1999; Budihardjo et al. 1999; Salvesen and Dixit 1999). Bax, a proapoptotic member of Bcl-2 family, can dimerize with either itself or Bcl-2 or Bcl-XL (Gross et al. 1999). In the present study, sorbitol-induced apoptosis in human K562 cells was accompanied by up-regulation of Bax, p-Bad, and down-regulation of p-Bcl-2, without effect on Bcl-XL. Other studies have demonstrated that Bcl-2, Bcl-XL, Bax, can act as channel proteins within the mitochondrial membrane (Gross et al. 1999; Eskes et al. 2000; Wood and Newcomb 2000). It is conceivable that the channel property of Bax may control the mitochondrial permeability transition and other early mitochondrial perturbation. Thus, Bax may facilitate the passage of some important proteins, such as cyt c or other apoptosis-inducing factors that trigger the activation of caspase cascade and apoptosis. Previous reports have also documented that the ratio of pro- and anti-apoptotic proteins determines, at least in part, the susceptibility of cells to a death signal (Gross et al. 1999; Vander Heiden and Thompson 1999; Zhang et al. 2000). Our results showed that expression of Bcl-2 family proteins p-Bcl-2, Bcl-XL, Bax, p-Bad can be differently regulated by sorbitol, suggesting that this mechanism is controlled by a balanced expression between those apoptosis-inducing and apoptosis- suppressing molecules. Thus, enforced dimerization or inducible expression of Bax may result in altering permeability, triggering mitochondrial cyt c release into cytosol, activating caspase 3 cascade, and eventually promoting cell death. Moreover, K562 cells were preincubated with specific caspase 3 inhibitor (z-DEVD-fmk) before being treated with sorbitol and the caspase 3 activity was analyzed. Results showed that pre-incubation of cells with z-DEVD-fmk effectively inhibited the caspase 3 activity. In summary, the treatment of human K562 cells with sorbitol activates a cell death pathway that regulates the mitochondrial membrane permeability by down-regulation of p-Bcl-2 and up-regulation of Bax, and p-Bad, triggering the cyt c release from mitochondria into cytosol. In addition, sorbitol could induce the caspase 3 activation, and consequently cleaves specific substrates leading to process apoptotic changes, such as nuclear condensation, cell shrinkage, and DNA fragmentation. Taken all together, our studies indicate that down-regulation of p-Bcl-2 and up-regulation of Bax, and p-Bad triggering the accumulation of cytosolic cyt c, may play an important role in the regulation and activation of the executioner phase of sorbitol-induced apoptosis.
References
Antonsson B, Martinou IC (2000) The Bcl-2 protein family. Exp Cell Res 256:50–57
Aquilano K, Filomeni G, Di Renzo L, Vito M, Stefano C, Salimei PS, Ciriolo MR, Marfè G (2007) Reactive oxygen and nitrogen species are involved in sorbitol-induced apoptosis of human erithroleukaemia cells K562. Free Radic Res 41:452–460
Budihardjo I, Oliver H, Lutter M, Luo X, Wang X (1999) Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol 15:269–290
Cheng AC, Jian CB, Huang YT, Lai CS, Hsu PC, Pan MH (2007) Induction of apoptosis by Uncaria tomentosa through reactive oxygen species production, cytochrome c release, and caspases activation in human leukemia cells. Food Chem Toxicol 45:2206–2218
Cohen GM (1997) Caspases: the executioner of apoptosis. Biochem J 326:1–16
Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC (1998) IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J 17:2215–2223
Eskes R, Desagher S, Antonsson B, Martinou JC (2000) Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol 20:929–935
Galvez AS, Ulloa JA, Chiong M, Criollo A, Eisner V, Barros LF, Lavandero S (2003) Aldose reductase induced by hyperosmotic stress mediates cardiomyocyte apoptosis: differential effects of sorbitol and mannitol. J Biol Chem 278:38484–38494
Gross A, McDonnell JM, Korsmeyer SJ (1999) Bcl-2 family members and the mitochondrial in apoptosis. Genes Dev 13:1899–1911
Herrmann M, Lorenz HM, Voll R, Grunke M, Woith W, Kalden JR (1994) A rapid and simple method for the isolation of apoptotic DNA fragments. Nucleic Acids Res 22:5506–5507
Jow GM, Wu YC, Guh JH, Teng CM (2004) Armepavine oxalate induces cell death on CCRF-CEM leukemia cell line through an apoptotic pathway. Life Sci 75:549–557
Kantrow SP, Piantasdosi CA (1997) Release of cytochrome c from liver mitochondria during permeability transition. Biochem Biophys Res Commun 232:669–671
Kaufmann SH, Hengartner MO (2001) Programmed cell death: alive and well in the new millennium. Trends Cell Biol 11:526–534
Koyama AH, Arakawa T, Adachi A (2000) Characterization of apoptosis induced by sorbitol: a unique system for the detection of antiapoptotic activities of viruses. Microbes Infect 2:599–606
Kuwana T, Newmeyer DD (2003) Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol 15:691–699
Murata T, Goshima F, Yamauchi Y, Koshizuka T, Takakuwa H, Nishiyama Y (2002) Herpes simplex virus type 2 US3 blocks apoptosis induced by sorbitol treatment. Microbes Infect 4:707–712
Reed JC (2001) Apoptosis-regulating proteins as targets for drug discovery. Trends Mol Med 7:314–319
Salvesen G, Dixit V (1999) Caspase activation: the induced-proximity model. Proc Natl Acad Sci USA 96:10964–10967
Scaduto RC Jr, Grotyohann LW (1999) Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J 76:469–477
Sinibaldi-Salimei P, Marfe G, Di Renzo L, Di Stefano C, Giganti MG, Filomeni G, Ciriolo MR (2007) The interference of rosmarinic acid in the DNA fragmentation induced by osmotic shock. Front Biosci 12:1308–1317
Solange D, Martinou JC (2000) Mitochondria as the central control point of apoptosis. Trends Cell Biol 10:369–377
Stoothoff WH, Johnson GV (2001) Hyperosmotic stress-induced apoptosis and tau phosphorylation in human neuroblastoma cells. J Neurosci Res 65:573–582
Teramachi K, Izawa M (2000) Rapid induction of apoptosis in human gastric cancer cell lines by sorbitol. Apoptosis 5:181–187
Vander Heiden MG, Thompson CB (1999) Bcl-2 proteins: regulators of apoptosis or of mitochondrial homeostasis. Nat Cell Biol 1:E209–E216
Vaux DL, Korsmeyer SJ (1999) Cell death in development. Cell 96:245–254
Wood DE, Newcomb EW (2000) Cleavage of Bax enhances its cell death function. Exp Cell Res 256:375–382
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X (1997) Prevention of apoptosis by Bcl-2 release of cytochrome c from mitochondria blocked. Science 275:1129–1132
Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B (2000) Role of Bax in the apoptotic response to anticancer agents. Science 290:989–992
Author information
Authors and Affiliations
Corresponding author
Additional information
The present study focused on K562 cells, a bcr-abl-exspressing human chronic myelogenous leukemia line that has been reported to resistent induction of apoptosis by many of same stimuli. Sorbitol can induce apoptosis in cultured cells: (1) apoptosis is induced simply by the addition of the reagent to the culture medium and following withdrawal from the medium, (2) apoptotic response is quick and efficient.
Rights and permissions
About this article
Cite this article
Marfè, G., Morgante, E., Di Stefano, C. et al. Sorbitol-induced apoptosis of human leukemia is mediated by caspase activation and cytochrome c release. Arch Toxicol 82, 371–377 (2008). https://doi.org/10.1007/s00204-007-0261-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-007-0261-y